

Abstract
Recent results from large-scale genomic projects suggest that allele frequencies, which are highly relevant for medical purposes, differ considerably across different populations. The need for a detailed catalog of local variability motivated the whole-exome sequencing of 267 unrelated individuals, representative of the healthy Spanish population. Like in other studies, a considerable number of rare variants were found (almost one-third of the described variants). There were also relevant differences in allelic frequencies in polymorphic variants, including ∼10,000 polymorphisms private to the Spanish population. The allelic frequencies of variants conferring susceptibility to complex diseases (including cancer, schizophrenia, Alzheimer disease, type 2 diabetes, and other pathologies) were overall similar to those of other populations. However, the trend is the opposite for variants linked to Mendelian and rare diseases (including several retinal degenerative dystrophies and cardiomyopathies) that show marked frequency differences between populations. Interestingly, a correspondence between differences in allelic frequencies and disease prevalence was found, highlighting the relevance of frequency differences in disease risk. These differences are also observed in variants that disrupt known drug binding sites, suggesting an important role for local variability in population-specific drug resistances or adverse effects. We have made the Spanish population variant server web page that contains population frequency information for the complete list of 170,888 variant positions we found publicly available (http://spv.babelomics.org/), We show that it if fundamental to determine population-specific variant frequencies to distinguish real disease associations from population-specific polymorphisms.
Keywords: population variability, exome sequencing, disease variants, pharmacogenomic variants.
Introduction
Recent large-scale population genomic projects (Durbin et al. 2010; Fu et al. 2013) have revealed the existence of an enormous amount of rare variation at the genome level in human populations (Coventry et al. 2010; Li et al. 2010; Gravel et al. 2011; Marth et al. 2011; Keinan and Clark 2012; Nelson et al. 2012; Tennessen et al. 2012). In addition to the anticipated neutral variation, apparently normal healthy individuals present a considerable number of deleterious variants with a putative effect on the function of human protein-coding genes (Kryukov et al. 2007; MacArthur and Tyler-Smith 2010; Marth et al. 2011; Nelson et al. 2012; Tennessen et al. 2012; Xue et al. 2012; Garcia-Alonso et al. 2014) and functional noncoding genomic elements, including miRNAs (Carbonell et al. 2012) and other regulatory regions (Dunham et al. 2012; Spivakov et al. 2012). Moreover, recent studies have described a remarkable local component (Kryukov et al. 2007; Marth et al. 2011; Nelson et al. 2012) and a high stratification level (Mathieson and McVean 2012; Moreno-Estrada et al. 2013) in many rare variants with uncertain functional consequences. It is likely that these rare variants help explaining the differential risk of many diseases in distinct human populations (Corona et al. 2013; Fernandez et al. 2013). All of these observations highlight the need for population-specific catalogs of genetic variation (Bustamante et al. 2011).
Despite being systematically studied at the single nucleotide polymorphism (SNP) level (Bustamante et al. 2011), sample sizes for individual European populations (amounting to only 50–90 individuals per population) in large-scale genome sequencing projects (Durbin et al. 2010) limit the precision at which their genetic variation at nucleotide resolution level can be assessed. Extensive variability surveys of European populations based on SNPs show a clear correspondence between genetic and geographic distances (Novembre et al. 2008). On the other hand, previous studies reported that the proportion of damaging substitutions is appreciably higher in individuals with European ancestry than in those with African ancestry (Barreiro et al. 2008; Lohmueller et al. 2008; Vasseur and Quintana-Murci 2013), which reinforces the need to gain insight into the degree of genetic variation at the population level. To our knowledge, only two initiatives that produced population-specific catalogs of genetic variation have been published to date: a whole-genome sequence (WGS) study of 100 Malays (Wong et al. 2013) and the recent Genome of the Netherlands with low-resolution (∼13×) WGS data of 250 trio-families from across the entire country (The_Genome_of_the_Netherlands_Consortium 2014). Another recent study of 109 exomes from French-Canadians, descendants of a small number of French settlers, show how the frequency of rare variants increases after population bottlenecks (Casals et al. 2013).
Genomic data are providing an increasingly detailed perspective of the landscape of human variability at the nucleotide resolution level (Goldstein et al. 2013). Precision medicine, an emerging paradigm of medicine oriented to maximizing individual care and disease prevention rather than merely treating disease (Hood and Friend 2011), requires knowledge about the genetic structure of human populations, particularly in its preventive aspects (Khoury et al. 2012). Such knowledge is also essential for identifying genetic factors contributing to variation in disease risk as well as to drug pharmacokinetics, treatment efficacy, and adverse drug reactions (Dopazo 2014). Attempts to map the genetic basis of any of these disease traits will likely produce spurious associations if the genetic structure of the population is not properly accounted for (Price et al. 2006; Goldstein et al. 2013; Boomsma et al. 2014). Despite the extensive use of targeted exome sequencing to discover disease genes in Mendelian disorders (Ng et al. 2010; Bamshad et al. 2011) or cancer (Garraway and Lander 2013; Vogelstein et al. 2013), lack of information about local genetic variation can severely hinder the discrimination of real disease variants from local polymorphisms and rare variants.
The classical consensus about the existence of relative homogeneity within European populations started to be questioned by genome-wide association studies (GWAS) reporting a correspondence between genetic and geographic distances (Novembre et al. 2008). Our observations support this view and suggest that the level of variation in local populations, even in the absence of geographic barriers, is higher than expected from previous variability studies based on polymorphisms (Novembre et al. 2008; Bustamante et al. 2011). A large proportion of this variability corresponds to private variants, as previously reported in several studies (Coventry et al. 2010; Li et al. 2010; Marth et al. 2011; Keinan and Clark 2012; Nelson et al. 2012; Tennessen et al. 2012). Here, we have analyzed whole-exome sequencing (WES) data from a sample of 267 healthy individuals of Spanish origin, which allowed us to carry out an exhaustive study of variability in the healthy Spanish population. The individuals were used as controls in the Medical Genome Project (MGP; http://www.medicalgenomeproject.com, last accessed January 28, 2016), a public–private partnership which aims to discover disease genes and mutations. Our analysis discovered a high degree of local variability private to the Spanish population, and a considerable difference in allele frequencies in polymorphic variants relative to geographically close populations (e.g., Tuscans from Italy). These observations extend to disease susceptibility or causal disease variants. Although allele frequencies for complex diseases in the Spanish population seem to be similar to other populations, the scenario for Mendelian and rare diseases seems to be the opposite, with significant differences in allele frequencies in the Spanish population (at least for several paradigmatic cases, including hereditary cardiomyopathies, degenerative retinal dystrophies, and others). Interestingly, differences in the allelic distributions of variants affecting known drug binding sites seem to be frequent, which suggests that drug resistance, adverse effects, etc., may have an important population-specific component. Finally, we report an example illustrating the importance of local variation to distinguish disease variants from local polymorphisms which could otherwise be taken as rare variants.
Results
Collection of Healthy Individuals
Blood samples from a total of 267 unrelated individuals of Spanish origin (obtained mainly in the North West—in Galicia—and in the South—in Andalusia—), who were phenotyped as healthy (i.e., with no known diseases or genetic conditions in the family history), were collected (see Materials and Methods).
Sequencing Results
The pipeline for primary data analysis (quality control and mapping) is described in Materials and Methods. The observed average coverage was satisfactory in all the samples analyzed (above 40×, which approximately corresponds to the expected coverage). The frequency of the alternative allele when the variant call was heterozygous was over 30% of the reads. These results ensured the quality of the variant calling process in this population.
Sequence data has been deposited at the European Genome-phenome Archive (EGA, see http://ega.crg.eu, last accessed January 28, 2016), under accession number EGAS00001000938.
Variability Distribution in the Spanish Population
A summary of the variability corresponding to the exonic regions of the Spanish population is shown in table 1. Almost one-third of the variants found had not been previously described in dbSNP (Sherry et al. 2001), 1000 Genomes populations (1000G) (Durbin et al. 2010), or the National Heart, Lung, and Blood Institute Exome Sequencing Project (Fu et al. 2013). This level of discovery is similar to that previously observed in other sequencing projects (Fu et al. 2013). A large proportion of variants were found in only one individual in the Spanish population (85%), which also agrees with previous observations of rare variant frequencies in different human populations (Coventry et al. 2010; Li et al. 2010; Marth et al. 2011; Keinan and Clark 2012; Nelson et al. 2012; Tennessen et al. 2012; Casals et al. 2013). The average number of variants per individual in the coding regions of the genome analyzed was ∼19,000. Among these variants, the observed number of nonsynonymous changes per individual was 9,194. In particular, an average of 95.8 stop gains and 29.4 stop losses per individual was observed. There was also an average of 417.2 variants which affect splicing. As observed in other large-scale genomic projects (MacArthur and Tyler-Smith 2010; Xue et al. 2012), there was an average of 352 likely deleterious nonsynonymous single nucleotide variants (SNVs) (those which meet at least two of the three pathogenicity indexes indicative of a potential deleterious effect; see Materials and Methods) per individual. Among these, more than 50 variants per individual were homozygotes, therefore representing the presence of a considerable amount of potentially deleterious variation in the Spanish population. Only 27.5% of the variants were already present in the IBS population of 16 individuals, making 60.3% of the variability we describe here new. The Spanish population variant server web page contains the complete list of 170,888 variant positions found in the Spanish MGP population sequenced in this study, which can be interactively queried (http://spv.babelomics.org/, last accessed January 28, 2016).
Table 1.
Variants in the Exonic Regions of the MGP Spanish Population.
| All Variants |
Private MGP Variants |
|||||
|---|---|---|---|---|---|---|
| Total Variants | Average Variants per Individual | Average Variants per Individual (homozygous) | Total Variants | Average Variants per Individual | Average Variants per Individual (homozygous) | |
| Exome positions with SNV | 170,888 | 18,875.8 | 6,906 | 63,243 | 835.8 | 59.4 |
| Exome monoallelic positions | 170,370 | 18,871.6 | 6,906 | 63,143 | 835.9 | 59.4 |
| Exome multiallelic positions | 518 | 4.2 | 0 | 100 | 0.8 | 0 |
| Exome SNV | 171,406 | 18,880.1 | 6,906 | 63,343 | 836.7 | 59.4 |
| Singletons | 54,214 | 202 | 59.4 | 54,214 | 202 | 59.4 |
| Nonsynonymous SNV | 97,589 | 9,193.7 | 3,335.5 | 40,564 | 538.6 | 41 |
| Synonymous SNV | 73,011 | 9,734 | 3,596.5 | 21,857 | 287.2 | 18 |
| Stop gain SNV | 1,852 | 95.8 | 22 | 1,060 | 15.9 | 0.4 |
| Stop loss SNV | 178 | 29.4 | 12 | 71 | 0.6 | 0.1 |
| Splicing SNV | 4,217 | 417.2 | 154.8 | 1,842 | 25.1 | 2 |
| LoF SNV | 32,736 | 1,163.8 | 211.2 | 17,314 | 141.8 | 3.3 |
| LoF stricta SNV | 12,639 | 352.6 | 51.4 | 7,136 | 51 | 0.3 |
aAll three pathogenicity predictors (SIFT, Polyphen, and conservation score) reported these SNVs as pathogenic, in contrast with loss-of-function (LoF) in which only two pathogenicity predictions were required to consider the variant as pathogenic.
Figure 1 depicts the extent of the variability of the Spanish population captured by this study. The total number of new variants present only in the Spanish population grew linearly with the number of individuals analyzed and seemed to be far from reaching a plateau in our study. However, when new variants were decomposed into rare variants (singletons) and polymorphic variants (those shared by several individuals) it was apparent that the main contribution to the private Spanish variability comes from rare variants, while polymorphic variants soon reached a plateau. This suggests that most of the polymorphisms within coding regions, observed only in the Spanish population, were apparently discovered in this work and seem to be restricted to ∼10,000 positions. Approximately, one-third of the variants found in the Spanish population are homozygous. This proportion decreases to 7% if only Spanish-specific variants are considered. The heterogeneity in the population can be viewed in supplementary figure S1, Supplementary Material online, and probably corresponds to different geographic locations. Unfortunately, anonymization and randomization of the samples (see Materials and Methods) precludes the assignation of specific samples to precise geographic locations.
Fig. 1.

Accumulative number of new variants contributed by individuals. The red line represents the number of variants found as the number of sequenced individuals increase. The green line represents the number of already known variants among all the variants found. The blue line represents the number of new variants not present in the 1000G populations. New variants are decomposed into polymorphic variants (present in more than one individual in the MGP population) represented by the blue dashed line, and rare variants (present in only one MGP individual), represented by the blue dotted line.
The distribution pattern of homozygotes and heterozygotes is consistent with a scenario in which most of the variants are in Hardy–Weinberg equilibrium (Stern 1943). Thus, at low allelic frequencies of the alternative allele, heterozygotes are prevalent, while the situation is the opposite at high allelic frequencies, where many alternative alleles are fixed in the population.
In summary, we observed an excess of low-frequency nonsynonymous coding variants, most of them heterozygotes, thus confirming the observations made in other populations (Coventry et al. 2010; Li et al. 2010; Marth et al. 2011; Keinan and Clark 2012; Nelson et al. 2012; Tennessen et al. 2012; Casals et al. 2013).
The Relationship of Spanish Populations to Other Populations
Variants located in coding regions, with a minor allele frequency (MAF) > 0.01 were used to carry out a principal components analysis (PCA) analysis using the SNPRelate program. Supplementary figure S2, Supplementary Material online represents the two main axes of the PCA which depicts the relationship between the MGP population and the different 1000G populations. As expected, the Spanish population closely related to other European populations, with the Italian (from Tuscany) population (TSI) being the closest. Labels in the plot are located at the average of the coordinates for each individual. As expected as well, the location of the Spanish population in the plot coincides with the Spanish individuals included in the 1000G project (IBS population).
Disease Variants and Disease Risk in the Spanish Population
All 170,888 variant positions found in the 267 exomes of the MGP population were screened for known disease variants present in the Human Genome Mutation Database (HGMD, commercial release 2011.4). We identified the presence of 3,069 variants annotated in the disease database. Among them, 193 had MAFs in the Spanish population which exceeded those found in the 1000G populations by 2-fold (supplementary table S1, Supplementary Material online). When compared with the 1000G subpopulation with European ancestry (the TSI, FIN, GBR, and CEU populations), 69 disease variants still showed MAFs in the MGP population which were at least 2-fold larger than those observed in European ancestry populations (table 2). Some examples of familial diseases with variants with remarkably high (between 4- and 18-fold) allelic frequencies in Spain are: Marfan syndrome, Von Willebrand syndrome, Ellis-van Creveld syndrome, Wilson disease, cystinuria, Crohn’s disease, or Charcot-Marie-Tooth disease, just to cite a few. In particular, several degenerative retinal dystrophies seem to have associated mutations at unusually high frequencies in the Spanish population, such as the autosomal dominant cone dystrophy (heterozygous in 4 out of the 267 Spanish samples but absent in the 1000G populations), retinitis pigmentosa, Leber congenital amaurosis, Bardet–Biedl syndrome, and other ocular diseases such as primary open angle glaucoma or Stargardt. All these diseases showed significant differences in allele frequencies among the populations compared when taking adjusted P values <0.05 (supplementary table S1, Supplementary Material online).
Table 2.
Variants Associated with Diseases That Have Allele Frequencies in the Spanish Population at Least 2-Fold Higher Than in the 1000G Populations.
| MGP |
1000G |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| chr | Start | CT | R | A | R/R | R/A | A/A | MAF | R/R | R/A | A/A | MAF | Ratio | Ratio E | HGMD_disease |
| 22 | 36688178 | ns | G | A | 263 | 4 | 0 | 0.0075 | 1,078 | 0 | 0 | 0 | NA | NA | Epstein syndrome? |
| 6 | 42141500 | ns | C | T | 263 | 4 | 0 | 0.0075 | 1,078 | 0 | 0 | 0 | NA | NA | Cone dystrophy, autosomal dominant |
| 22 | 36688178 | syn | G | A | 263 | 4 | 0 | 0.0075 | 1,078 | 0 | 0 | 0 | NA | NA | Epstein syndrome? |
| 17 | 36104650 | ns | C | A | 264 | 3 | 0 | 0.0056 | 1,078 | 0 | 0 | 0 | NA | NA | Diabetes, Maturity onset diabetes of the young (MODY) |
| 15 | 48704816 | ns | G | A | 262 | 5 | 0 | 0.0094 | 1,077 | 1 | 0 | 5.00E-04 | 18.8000 | NA | Marfan syndrome |
| 6 | 162206852 | ns | G | A | 262 | 5 | 0 | 0.0094 | 1,077 | 1 | 0 | 5.00E-04 | 18.8000 | NA | Parkinsonism. juvenile. autosomal recessive |
| 1 | 216420460 | ns | C | A | 263 | 4 | 0 | 0.0075 | 1,077 | 1 | 0 | 5.00E-04 | 15.0000 | NA | Retinitis pigmentosa. recessive. no hearing loss |
| 17 | 41199716 | sg | A | T | 264 | 3 | 0 | 0.0056 | 1,077 | 1 | 0 | 5.00E-04 | 11.2000 | NA | Ovarian cancer |
| 19 | 8436373 | ns | C | T | 263 | 4 | 0 | 0.0075 | 1,076 | 2 | 0 | 9.00E-04 | 8.33333 | NA | Lower plasma triglyceride level |
| 11 | 18050850 | ns | C | T | 263 | 4 | 0 | 0.0075 | 1,076 | 2 | 0 | 9.00E-04 | 8.33333 | NA | Attention deficit hyperactivity disorder |
| 3 | 123376066 | ns | C | T | 263 | 4 | 0 | 0.0075 | 1,076 | 2 | 0 | 9.00E-04 | 8.33333 | NA | Aortic dissections? |
| 7 | 107329557 | ns | T | C | 262 | 5 | 0 | 0.0094 | 1,075 | 3 | 0 | 0.0014 | 6.71429 | NA | Pendred syndrome |
| 7 | 99032559 | ns | G | A | 250 | 17 | 0 | 0.0318 | 1,064 | 13 | 1 | 0.007 | 4.54286 | 12.23076 | Complex I deficiency |
| 18 | 2937867 | ns | C | A | 260 | 7 | 0 | 0.0131 | 1,075 | 3 | 0 | 0.0014 | 9.35714 | 10.07692 | Psoriasis |
| 15 | 58957371 | ns | C | G | 261 | 6 | 0 | 0.0112 | 1,076 | 2 | 0 | 9.00E-04 | 12.4444 | 8.615384 | Alzheimer disease. late onset |
| 15 | 42684875 | nc | C | T | 261 | 6 | 0 | 0.0112 | 1,072 | 6 | 0 | 0.0028 | 4.00000 | 8.615384 | Muscular dystrophy. limb girdle |
| 19 | 4159747 | ns | G | A | 262 | 5 | 0 | 0.0094 | 1,076 | 2 | 0 | 9.00E-04 | 10.4444 | 7.230769 | Hypertriglyceridemia |
| 12 | 6143978 | ns | C | T | 262 | 5 | 0 | 0.0094 | 1,075 | 3 | 0 | 0.0014 | 6.71429 | 7.230769 | Von Willebrand. Normandy variant |
| 1 | 115221116 | ns | C | A | 257 | 10 | 0 | 0.0187 | 1,073 | 5 | 0 | 0.0023 | 8.13043 | 7.192307 | Adenosine monophosphate deaminase deficiency |
| 12 | 32994073 | ns | G | A | 257 | 10 | 0 | 0.0187 | 1,073 | 5 | 0 | 0.0023 | 8.13043 | 7.192307 | Arrhythmogenic right ventricular dysplasia/cardiomyopathy |
| 4 | 5627493 | syn | G | T | 258 | 9 | 0 | 0.0169 | 1,074 | 3 | 1 | 0.0023 | 7.34783 | 6.5 | Ellis-van Creveld syndrome |
| 2 | 71825797 | ns | C | G | 263 | 4 | 0 | 0.0075 | 1,077 | 1 | 0 | 5.00E-04 | 15.0000 | 5.769230 | Muscular dystrophy. limb girdle 2B |
| 13 | 52534410 | ns | C | T | 263 | 4 | 0 | 0.0075 | 1,077 | 1 | 0 | 5.00E-04 | 15.0000 | 5.769230 | Wilson disease |
| 8 | 145699735 | ns | G | C | 263 | 4 | 0 | 0.0075 | 1,077 | 1 | 0 | 5.00E-04 | 15.0000 | 5.769230 | Congenital heart defects |
| 1 | 196709833 | ns | C | T | 263 | 4 | 0 | 0.0075 | 1,076 | 2 | 0 | 9.00E-04 | 8.33333 | 5.769230 | Factor H deficiency |
| 1 | 94568686 | ns | C | T | 263 | 4 | 0 | 0.0075 | 1,076 | 2 | 0 | 9.00E-04 | 8.33333 | 5.769230 | Stargardt disease |
| 5 | 110454719 | ns | A | G | 255 | 12 | 0 | 0.0225 | 1,074 | 4 | 0 | 0.0019 | 11.8421 | 5.625 | Glaucoma. primary open angle |
| 11 | 67799622 | ns | C | T | 260 | 7 | 0 | 0.0131 | 1,076 | 2 | 0 | 9.00E-04 | 14.5555 | 5.038461 | Complex I deficiency |
| 8 | 100832259 | ns | A | G | 260 | 7 | 0 | 0.0131 | 1,074 | 4 | 0 | 0.0019 | 6.89474 | 5.038461 | Cohen síndrome |
| 1 | 183532364 | ns | T | A | 260 | 7 | 0 | 0.0131 | 1,071 | 7 | 0 | 0.0032 | 4.09375 | 5.038461 | Chronic granulomatous disease |
| 1 | 76211574 | ns | C | A | 264 | 3 | 0 | 0.0056 | 1,078 | 0 | 0 | 0 | NA | 4.307692 | Medium chain acyl CoA dehydrogenase deficiency |
| 9 | 120475248 | ns | G | A | 261 | 6 | 0 | 0.0112 | 1,076 | 2 | 0 | 9.00E-04 | 12.444 | 4.307692 | Meningococcal disease? |
| 22 | 45691554 | ns | C | T | 264 | 3 | 0 | 0.0056 | 1,077 | 1 | 0 | 5.00E-04 | 11.200 | 4.307692 | Renal adysplasia |
| 11 | 88924465 | ns | C | A | 264 | 3 | 0 | 0.0056 | 1,077 | 1 | 0 | 5.00E-04 | 11.200 | 4.307692 | Albinism. oculocutaneous 1 |
| 14 | 21811213 | ns | A | G | 264 | 3 | 0 | 0.0056 | 1,077 | 1 | 0 | 5.00E-04 | 11.200 | 4.307692 | Leber congenital amaurosis |
| 14 | 21811213 | ns | A | G | 264 | 3 | 0 | 0.0056 | 1,077 | 1 | 0 | 5.00E-04 | 11.200 | 4.307692 | Retinitis pigmentosa? |
| 13 | 48939088 | ns | C | T | 264 | 3 | 0 | 0.0056 | 1,077 | 1 | 0 | 5.00E-04 | 11.200 | 4.307692 | Retinoblastoma |
| 14 | 23862646 | ns | C | A | 264 | 3 | 0 | 0.0056 | 1,077 | 1 | 0 | 5.00E-04 | 11.200 | 4.307692 | Cardiomyopathy. dilated |
| 5 | 70945029 | ns | T | C | 261 | 6 | 0 | 0.0112 | 1,074 | 4 | 0 | 0.0019 | 5.89474 | 4.307692 | Complex I deficiency |
| 18 | 2925359 | ns | C | T | 245 | 22 | 0 | 0.0412 | 1,061 | 17 | 0 | 0.0079 | 5.21519 | 4.12 | Psoriasis |
| 6 | 31729925 | ns | C | T | 225 | 40 | 2 | 0.0824 | 980 | 87 | 11 | 0.0506 | 1.62846 | 4.12 | Leukemia. risk. association with |
| 17 | 56348226 | sp | T | G | 259 | 8 | 0 | 0.015 | 1,075 | 3 | 0 | 0.0014 | 10.7142 | 3.75 | Myeloperoxidase deficiency |
| 1 | 203194834 | ns | C | T | 262 | 5 | 0 | 0.0094 | 1,076 | 2 | 0 | 9.00E-04 | 10.444 | 3.615384 | Chitotriosidase deficiency |
| 16 | 16259579 | syn | G | A | 262 | 5 | 0 | 0.0094 | 1,075 | 3 | 0 | 0.0014 | 6.71429 | 3.615384 | Pseudoxanthoma elasticum |
| 2 | 44513202 | ns | T | C | 262 | 5 | 0 | 0.0094 | 1,075 | 3 | 0 | 0.0014 | 6.71429 | 3.615384 | Cystinuria |
| 2 | 71738977 | ns | G | A | 262 | 5 | 0 | 0.0094 | 1,074 | 4 | 0 | 0.0019 | 4.94737 | 3.615384 | Muscular dystrophy. limb girdle/Miyoshi myopathy |
| 13 | 32914592 | ns | C | T | 262 | 5 | 0 | 0.0094 | 1,074 | 4 | 0 | 0.0019 | 4.94737 | 3.615384 | Breast and/or ovarian cancer? |
| 1 | 2234791 | ns | C | T | 260 | 7 | 0 | 0.0131 | 1,073 | 5 | 0 | 0.0023 | 5.69565 | 3.275 | Cleft lip? |
| 15 | 75012987 | ns | G | T | 233 | 33 | 1 | 0.0655 | 1,048 | 29 | 1 | 0.0144 | 4.54861 | 3.275 | Colorectal cancer. reduced risk. association with |
| 17 | 73837042 | ns | T | C | 263 | 4 | 0 | 0.0075 | 1,076 | 2 | 0 | 9.00E-04 | 8.33333 | 2.884615 | Hemophagocytic lymphohistiocytosis. Familial |
| 1 | 12064892 | ns | G | A | 261 | 6 | 0 | 0.0112 | 1,073 | 4 | 1 | 0.0028 | 4.00000 | 2.8 | Charcot-Marie-Tooth disease 2a |
| 21 | 44317156 | ns | A | C | 254 | 13 | 0 | 0.0243 | 1,070 | 8 | 0 | 0.0037 | 6.56757 | 2.43 | Complex I deficiency |
| 21 | 44317156 | sp | A | C | 254 | 13 | 0 | 0.0243 | 1,070 | 8 | 0 | 0.0037 | 6.56757 | 2.43 | Complex I deficiency |
| 18 | 58038832 | ns | T | G | 254 | 13 | 0 | 0.0243 | 1,069 | 9 | 0 | 0.0042 | 5.78571 | 2.43 | Obesity. autosomal dominant? |
| 12 | 6103650 | ns | G | A | 254 | 13 | 0 | 0.0243 | 1,069 | 9 | 0 | 0.0042 | 5.78571 | 2.43 | Von Willebrand disease 1? |
| 17 | 33430313 | ns | T | C | 254 | 13 | 0 | 0.0243 | 1,065 | 13 | 0 | 0.006 | 4.05000 | 2.43 | Breast cancer. increased risk. association with |
| 13 | 49281554 | ns | A | G | 254 | 13 | 0 | 0.0243 | 1,061 | 17 | 0 | 0.0079 | 3.07595 | 2.43 | Atopy. association with |
| 12 | 6458350 | ns | A | G | 242 | 25 | 0 | 0.0468 | 1,055 | 22 | 1 | 0.0111 | 4.21622 | 2.34 | Ischemic cerebrovascular events. association with |
| 17 | 42463054 | ns | G | C | 255 | 12 | 0 | 0.0225 | 1,068 | 10 | 0 | 0.0046 | 4.89130 | 2.25 | Glanzmann thrombasthenia |
| 12 | 22017410 | sp | C | T | 255 | 12 | 0 | 0.0225 | 1,066 | 12 | 0 | 0.0056 | 4.01786 | 2.25 | Myocardial infarction. association with |
| 12 | 22017410 | ns | C | T | 255 | 12 | 0 | 0.0225 | 1,066 | 12 | 0 | 0.0056 | 4.01786 | 2.25 | Myocardial infarction. association with |
| 1 | 158624528 | ns | G | T | 232 | 34 | 1 | 0.0674 | 1,042 | 34 | 2 | 0.0176 | 3.82955 | 2.246666 | Spherocytosis. association with? |
| 22 | 46614274 | ns | C | G | 224 | 40 | 3 | 0.0861 | 1,027 | 49 | 2 | 0.0246 | 3.50000 | 2.1525 | Elevated plasma lipid conc. assoc. in diabetes |
| 5 | 82491674 | ns | T | C | 233 | 34 | 0 | 0.0637 | 1,042 | 36 | 0 | 0.0167 | 3.81437 | 2.123333 | Lung cancer. susceptibility to. association with |
| 19 | 36341311 | ns | T | A | 256 | 11 | 0 | 0.0206 | 1,072 | 6 | 0 | 0.0028 | 7.35714 | 2.06 | Focal segmental glomerulosclerosis |
| 5 | 151202476 | ns | C | T | 256 | 11 | 0 | 0.0206 | 1,066 | 12 | 0 | 0.0056 | 3.67857 | 2.06 | Hyperekplexia |
| 5 | 110428060 | ns | T | C | 256 | 11 | 0 | 0.0206 | 1,064 | 14 | 0 | 0.0065 | 3.16923 | 2.06 | Glaucoma. primary open angle. association with? |
| 13 | 78475230 | ns | C | T | 256 | 11 | 0 | 0.0206 | 1,064 | 14 | 0 | 0.0065 | 3.16923 | 2.06 | Hirschsprung disease |
| 1 | 227170648 | syn | C | T | 257 | 9 | 1 | 0.0206 | 1,063 | 15 | 0 | 0.007 | 2.94286 | 2.06 | Ubiquinone deficiency with cerebellar ataxia |
Note.—The first column indicates the chromosome; the second column indicates the position of the variant; the third column labeled CT, contains the consequence type, which are ns, nonsynonymous SNV; syn, synonymous; sg, stop gain; sp, variant affecting splicing; nc, ncRNA_exonic; the fourth column, labeled R, contains the reference allele in the position; the fifth column, labeled A, contains the alternative allele; the three following columns (sixth, seventh, and eight), labeled R/R, R/A, and A/A contain the number of individuals in which a reference homozygote (R/R), heterozygote (R/A) or an alternative homozygote (A/A) are found in the Spanish population, respectively; the ninth column, labeled MAF, contains the alternative allele frequency in the Spanish population; the three following columns (tenth, 11th, and 12th) contain the number of individuals in which a reference homozygote (R/R), heterozygote (R/A), or an alternative homozygote (A/A) are found in the 1000G populations; the 13 column, labeled Ratio, contains the ratio between the Spanish and the 1000G MAFs, the 14th column, labeled Ratio E, contains the ratio between the Spanish MAF and the 1000G MAFs of populations with European ancestry only; and finally, the 15th column, labeled as HGMD disease, contains the description of the disease caused by the variant, which can be a causal effect, or an association (when the description ends in “association with”) and can also be uncertain (then, the definition includes a question mark).
In contrast, the frequencies were more similar across all the populations for variants which conferred susceptibility to common diseases. There are several exceptions, in which a particular variant, among the many associated with the disease, displayed remarkably higher allelic frequency in the Spanish population when compared with the 1000G populations. Such cases include certain forms of diabetes, juvenile Parkinsonism or late-onset Alzheimer (table 2 and supplementary table S1, Supplementary Material online). Furthermore, a few variants which have been associated with different types of cancer also displayed comparatively high allelic frequencies in the Spanish population, including variants for ovarian cancer, breast and ovarian cancer, retinoblastoma, and increased melanoma risk, among others (table 2 and supplementary table S1, Supplementary Material online). Variants associated with other diseases with less severe symptoms also had comparatively high allelic frequencies in the Spanish population (e.g., psoriasis and a type of autosomal dominant obesity).
There were also relatively underrepresented variants in the Spanish population. As an anecdotal example, a variant associated with red hair (CM003595, gene MC1R, which causes both a nonsynonymous change and simultaneously affects an exomic ncRNA) occurs at a low frequency (0.0037) in the Spanish population compared with the relatively higher frequency in the 1000G populations with European ancestry (0.07). Similarly, variants associated with rare diseases also showed remarkable differences in allelic frequencies in the opposite direction. For example, the allele for the 2L form of Charcot-Marie-Tooth disease is underrepresented in the Spanish population (MAF 0.0037 versus 0.052 in 1000G, see supplementary table S1, Supplementary Material online), while the 2a form is overrepresented (see earlier). This is in agreement with the observation that some diseases are caused by different alleles in different populations (Fernandez et al. 2013) that can be relevant for diagnosis. Variants associated with several cardiovascular pathologies are also underrepresented in the Spanish population (supplementary table S1, Supplementary Material online).
In addition, there are 376 rare variants annotated to diverse diseases present only in one Spanish exome and absent in all the 1000G individuals (supplementary table S1, Supplementary Material online).
Allele Frequencies in Mendelian and Rare versus Complex Diseases
Population differences in allele frequencies behave different for complex than for mendelian and rare disease. Figure 2 and supplementary figure S3, Supplementary Material online provide comparative information about allele frequencies of all the diseases with more than five variants recorded in HGMD and reveal an interesting trend. Most associations with complex diseases show an almost identical distribution of frequencies for all the variants in all the genes, including associations with Alzheimer disease (fig. 2A), schizophrenia, myocardial infarction, type 2 diabetes, obesity, or essential hypertension, as well as most cancer associations (fig. 2B and supplementary fig. S3, Supplementary Material online). In sharp contrast, mendelian and rare diseases such as Marfan syndrome (fig. 2C), Wilson disease, phenylketonuria, several degenerative retinopathies such as age-related macular degeneration and many others (fig. 2D), have remarkably different allelic frequencies in the Spanish population compared with the 1000G populations.
Fig. 2.

Comparison of allelic frequencies described in HGMD between the MGP Spanish population and the 1000 genomes populations in four diseases with more than five variants. Upper left panel shows the frequencies in the Spanish MGP samples found for all the variants associated with Alzheimer disease in HGMD (X axis) versus the corresponding frequencies observed in all the individuals of the 1000 genomes populations (Y axis). Upper right panel presents a similar plot for variants described in HGMD as associated to leukemia risk. Lower left and right panels depict the same relationship for two rare diseases, Marfan syndrome, and age-related macular degeneration, respectively.
Relationship between Variant Frequencies and the Prevalence of the Disease in the Population
To test whether population differences in frequencies of risk alleles (for both, complex and mendelian diseases) result in difference in the prevalence of the corresponding diseases, we have collected data from the “Global Burden of Disease database” (see Materials and Methods). We have found data on “differences in disability-adjusted life years” (DALYs), a widely used proxy of disease prevalence (Murray et al. 2015), for several of the diseases analyzed here.
Interestingly, when the relative differences in allele frequencies found in the Spanish population with respect to European populations (TSI, FIN, GBR, and CEU) are compared with the corresponding relative differences in DALYs between Spain and the Central and East European populations, a remarkable correspondence between both parameters was found. Figure 3 depicts these relationships for the diseases showing the most extreme differences in allelic frequencies (Alzheimer, Attention deficit hyperactivity disorder, Parkinson, Psoriasis, and Cardiovascular diseases). Observed increases or decreases in allele frequencies in the Spanish population relative to European populations correspond to increases or decreases in the prevalence of the diseases, respectively.
Fig. 3.

Comparison of the relative prevalence and MAFs for several of the diseases showing the most extreme differences in allelic frequencies. The two first bars in each disease represent the log2 of the ratios of prevalence of the disease (DALYs) in Spain with respect to the corresponding prevalence in Central and East Europe, respectively, and the third bar represents the log2 of the ratio of the MAF of alleles of the disease in Spain and the corresponding MAF in the European populations of 1000G. The diseases are abbreviated as: Alzheimer (AD), Attention deficit hyperactivity disorder (ADHD), Parkinson (PD), Psoriasis (PSO), and Cardiovascular diseases (CAD).
Variants of Pharmacogenetic Relevance
The 267 exomes of the MGP population were screened for variants of pharmacogenetics relevance. In particular, we considered variants which affect drug binding sites, thus potentially disrupting the binding domain, without being deleterious to the protein. These variants will likely cause total or partial drug binding inhibition with potential effects such as resistance to treatments or may even cause adverse effects.
There are 112 variants affecting well defined drug binding domains (Hopkins and Groom 2002). Among these, 31 are predominant in the Spanish population, with MAFs 1.5-fold higher than those observed in the 1000G populations (supplementary table S2, Supplementary Material online). For example, the gene CYP11B2 from the Cytochrome P450 family is affected in the binding site of different drugs (Eplerenone, Etomidate, Hydrocortisone, Metoclopramide, Metyrapone) used to treat a variety of diseases (including heart failure or hypercortisolism) or symptoms (antiemetic), by a nonsynonymous mutation which has a MAF prevalence in the Spanish population 15 times higher than that observed in the 1000G populations. Binding sites for several statins, drugs for migraine treatment, analgesics, and others (up to a total of 31) were also found to be affected by nonsynonymous variants present in the Spanish population at higher frequencies (over 1.5 times higher) than in the 1000G populations.
Binding sites for several natural substances were also comparatively more affected by nonsynonymous variants in the MGP population than in the 1000G populations, including Ursodeoxycholic acid, Vitamin A, Choline (B-vitamin complex), L-Tryptophan, Glycine, and Tetrahydrobiopterin, among others.
On the other hand, there are also 46 drug and natural compound binding sites that were remarkably less affected by variants in the Spanish population than in the 1000G reference population (with MAFs which are less than one-half that observed in 1000G; see supplementary table S2, Supplementary Material online). The fact that different drug binding sites are affected at different frequencies in different populations could account for population-specific differences in sensitivity, efficiency, and even resistance to drugs or their adverse effects.
Supplementary table S2, Supplementary Material online lists other natural substances of interest. Since the binding sites of these substances may have been under negative selective pressure we studied possible deviations from the Hardy–Weinberg equilibrium which could be caused by a deleterious allele. Only a few of them (32; see supplementary table S2, Supplementary Material online) deviated significantly from the equilibrium, which suggests that either the majority of variants do not deactivate the binding sites or that there are other binding sites which compensate for their putative loss.
Among the variants that deviate from the Hardy–Weinberg equilibrium there is one that affects the binding site for several compounds, including Glutamic Acid, in the gene GRIN3A. This gene is a glutamate receptor known to be under geographically localized positive selection, and to be related to obesity, coronary artery calcification, and Thiazide-induced adverse metabolic effects in hypertensive patients (Colonna et al. 2014). Moreover, four variants which affect three binding sites for NADH display significantly lower frequencies in the Spanish population when compared with the 1000G populations and one of them, located in the SORD gene, significantly deviates from the Hardy–Weinberg equilibrium (adjusted P value = 0.00005). The same occurs with a binding site for L-Phenylalanine, L-Tyrosine, and Tetrahydrobiopterin, to which the antihypertensive drug Metyrosine (a tyrosine hydroxylase enzyme inhibitor), also binds (supplementary table S2, Supplementary Material online). Interestingly, a variant which affects the binding site of two diuretic drugs (Amiloride, Triamterene) also significantly deviates from the Hardy–Weinberg equilibrium (adjusted P value < 0.00005). In these three cases, the 1000G population presented significantly higher MAFs and did not deviate from the Hardy–Weinberg equilibrium. We also observed that the corresponding frequencies in the Exome Variant Server (Fu et al. 2013) are similar to the 1000G population.
Selective Pressures
We used the McDonald–Kreitman test (MKT) (McDonald and Kreitman 1991) to search for signals of natural selection acting on genes. Although a total of 145 genes, corresponding to 365 different transcripts showed events of positive selection (supplementary table S3, Supplementary Material online lists genes with a nominal P value < 0.05), only MUC4 was still significant after multiple testing correction (adjusted P value = 1.7 × 10−6). Interestingly, MUC4 has a variant associated with Ulcerative colitis (HGMD ID CM066583) which is significantly underrepresented in the MGP population (adjusted P value = 0.00208) (supplementary table S1, Supplementary Material online).
To find signals of recent positive selection FST values were calculated as a measurement of population differentiation between the MGP population and all the European 1000G populations (TSI, FIN, GBR, and CEU, excluding IBS). As expected, the mean FST between these two groups was low (0.007) although several loci showed extreme values (supplementary fig. S4, Supplementary Material online for a histogram of the FST distribution). SNPs with exceptionally high FST values (FST > 0.2) were considered candidates for selection (152 SNPs, 0.41% of the SNPs, see supplementary table S4, Supplementary Material online). Interestingly, a SNP (rs2550270) in the gene MUC4 presented an extreme FST value (FST = 0.3; see supplementary table S4, Supplementary Material online) but it is not the same one associated with Ulcerative colitis.
Increased Resolution Using Local Variability to Find Disease Genes
Knowledge of local variability can also have a practical application in clinical research. The systematic use of WES for finding disease genes has proven to be very successful in discovering new disease genes (Bamshad et al. 2011). Since exomes contain a vast number of mutations the number of candidate disease variants must be reduced in a process of prioritization involving a series of filters to exclude variants that are not likely to cause disease. One of the most stringent filters involves discarding variants which are present in the population at frequencies similar to or above the prevalence of the disease itself (Goldstein et al. 2013). Since local population frequencies are not typically available, general repositories, such as 1000G and others, are used. As a practical demonstration of the importance of knowledge about local variability, here we describe a specific example of a large family affected by autosomal-dominant retinitis pigmentosa (adRP; OMIM 268000). The use of the conventional consecutive filtering approach, implemented in the BiERApp tool (Aleman et al. 2014) which includes a specific filtering step for local variants, enormously increases the discovery power of the methodology.
The family, of Spanish origin, comprises three generations and our study included seven affected members (fig. 4A), who were clinically diagnosed with adRP, following ophthalmic criteria as previously described (Mendez-Vidal et al. 2013). All the affected individuals were derived from the Ophthalmology Department of the Genetic, Reproduction and Fetal Medicine Department at the Hospital Virgen del Rocio (Seville, Spain). The family did not present any known mutation for adRP and none of their members was included in the 267 MGP samples analyzed. The KING program (Manichaikul et al. 2010) was used to confirm the absence of any possible kinship between the family studied and any of the samples in the MGP population.
Fig. 4.
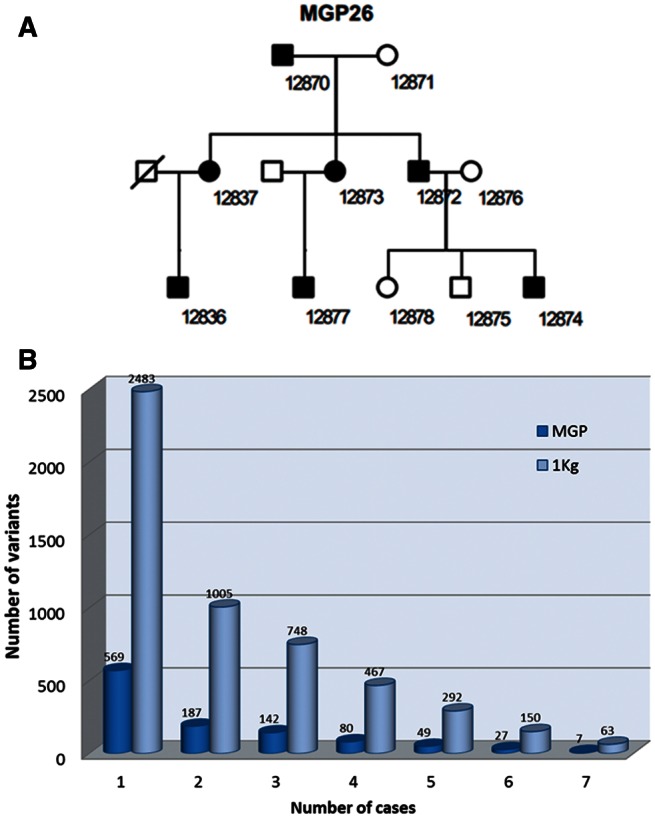
Effect of filtering out variants with high MAFs using frequency data inferred either from the available databases (1000G) or from the MGP Spanish population sequenced here. (A) Pedigree of the family studied with seven members affected by adRP. (B) Segregation analysis across the family was carried out, followed by a step filtering out the variants found in a reference population with a MAF incompatible with the observed prevalence of adRP. The plot represents the number of candidate variants that segregate with the family as a growing number of affected members were used to select the variants (from one to seven) and when two reference populations (1000G, pale blue and the MGP Spanish local population, dark blue) were used to filter out variants with MAFs that were too high to be compatible with the prevalence observed for the disease (>0.001 in 1000G and >0.004 in the MGP population, respectively). The filtering effect on the local Spanish population was drastically more stringent than for the 1000G population.
WES of all the affected patients as well as the grandmother (with a genetic background common to all of them) was carried out as described in Materials and Methods. The selection of heterozygous variants segregating with the pedigree in figure 4A raised many possible candidates. Since the incidence of the disease is below 1 in 4,000 (Ayuso and Millan 2010), variants present in normal populations at frequencies higher than 0.001, the lowest frequency that can be obtained from 1000G (Durbin et al. 2010) populations, were discarded as putative disease-causing variants. The dark blue bars in figure 4B correspond to the number of variants which do not appear in the 1000G populations but that still segregate with the family when a growing number of affected individuals (from one to seven) are used to filter out variants. When we instead used the local Spanish population (MGP; sequenced here) to filter out variants present in the healthy population (pale blue bars in fig. 4B) the filtering power was strongly increased. We used the BiERapp tool (Aleman et al. 2014) to apply consecutive filters (segregation along the family pedigree, predicted pathogenicity, and population frequency) to select potential disease variants and genes. The analysis of variants shared by the seven affected members after filtering out those present in the Spanish population rendered a total of 7 possible variants corresponding to 7 candidate genes. We then performed cosegregation analysis using DNA samples from available family members which confirmed the presence of the variant in the family. The novel variant identified was subsequently screened in 200 healthy matched control subjects by Sanger sequencing, confirming its absence and thus validating the c.937-2_944del variant as a novel causal RP mutation. This variant produces the loss of a cryptic splice acceptor site in intron 4–5 of the RHO gene, and therefore the use of a cryptic splice site upstream of the normal acceptor splice site results in a truncated protein which might be subject to nonsense-mediated decay in these patients. Sanger sequencing was used to validate the mutation found. For this purpose, specific primers encompassing the RHO intron 4–exon 5 junction were designed using the Primer3 software (Rozen and Skaletsky 2000) with sense and antisense sequences TACAGAACACCCTTGGCACA and AGGTGTAGGGGATGGGAGAC, respectively, rendering an amplicon length of 424 bp and a Tm of 62° C.
Discussion
Our work describes with precision the level of variation observed in the genomic coding regions of 267 unrelated healthy Spanish individuals, which makes it the largest study to date on local variation in a single population. Thanks to large sample size, the conclusions can be considered more significant than those obtained from general studies involving multiple populations with smaller numbers of individuals (Durbin et al. 2010; Li et al. 2010; Corona et al. 2013; Fu et al. 2013; Moreno-Estrada et al. 2013). However, it must also be taken into account that, while the population-specific results can be considered robust, the existence of a certain bias in the comparative analysis of the Spanish and the 1000G populations, derived from the fact that they are independent experiments, using different sequencing technologies and sampling strategies cannot be completely ruled out.
Here, we document that while the polymorphic variants private to the Spanish population are almost completely described in this work by analyzing only 267 individuals (fig. 1), the rate of discovery of new rare variants with increasing numbers of sequenced individuals was still far from reaching a plateau. Although many Spanish rare variants remain to be discovered, the use of the population frequencies obtained in this work does already afford increased ability (relative to the use of 1000G project data) to filter candidate variants in a Spanish family which could otherwise be interpreted as possible disease variants (fig. 4).
As expected given the magnitude of the rare variation discovered in the Spanish population, a significant number of variants were related to diseases (Kryukov et al. 2007; MacArthur and Tyler-Smith 2010; MacArthur et al. 2012; Xue et al. 2012). When the frequencies of disease variants or disease-risk variants in the Spanish population are compared with the corresponding frequencies observed in the 1000G populations an interesting trend emerges: complex disease variants seem to have similar allelic frequencies in both the MGP Spanish samples and 1000G populations. In contrast, mendelian or rare diseases tend to present dissimilar allelic frequency distributions (supplementary fig. S3, Supplementary Material online). In these, and other similar diseases, the most prevalent alleles are different in distinct populations. This observation agrees with the fact that, while high-frequency variants and variants underlying complex diseases tend to be shared across populations (Marigorta and Navarro 2013), low-frequency alleles tend to be private (Casals et al. 2013). This, together with recent discoveries of new population-specific variants causal of inherited diseases, such as retinopathies (Méndez-Vidal et al. 2014), strongly points at a crucial role of private mutations in the configuration of the mutational spectrum of certain diseases. In other words, geographic heterogeneity in the genetic architecture of disease, that is, the fact that different variants, often in different genes, can cause common multigenic diseases in different populations (Fernandez et al. 2013), may be more frequent than expected, again highlighting the need for local variation catalogs (Bustamante et al. 2011).
Since the use of drugs is very recent in evolutionary terms, it is expectable that the observed differences in frequencies in the variants located in a number of drug binding sites are due to population founder effects rather than any selective processes. However, once an area of a protein’s surface turns out to be a drug binding site it becomes relevant from a clinical perspective. We observed a total of 121 variants affecting the binding sites of different drugs. We also observed differences in the frequencies of variants affecting the binding sites of some natural products, such as Vitamin A, Choline, L-Tryptophan, Glycine, etc. In this case, some selective effect against mutations in the binding sites could be hypothesized. It is known that genes related to xenobiotic metabolism (Arbiza et al. 2006), pathogen adaptation to (Karlsson et al. 2014), or dietary change (Luca et al. 2010) are under selective pressures due to their relationship to disease in modern humans (Babbitt et al. 2011; Engelken et al. 2014). However, the study of Hardy–Weinberg equilibrium does not support the existence of selective pressure against any of the alleles for most of the cases of variants affecting the binding sites of natural products. Therefore, either the power of the test is too low or many of the studied variations are unlikely to inactivate these binding sites. In this study only three specific binding sites, corresponding to NADH, L-Phenylalanine, and L-Tyrosine, were affected by variants that significantly deviate from the Hardy–Weinberg equilibrium, thus suggesting the existence of some type of selection against such variants in the Spanish population which has not been detected in the 1000G project.
Our findings clearly highlight the importance of local variability in any study which attempts to relate genotype to phenotype, specifically when the phenotype is a disease. In the example given, the local variability filter discarded five times more candidate variants (false positives) than the filter based only on population allelic frequencies derived from foreign populations (1000G). Although the need for population-specific catalogs of genetic variation has been previously noted (26), our results clearly reveal the quantitative magnitude of the differences expected between the use of a general population and a local population, and its impact on clinically relevant human variation. To foster research about other pathologies, we have made publicly available through the Spanish population variant server web page (http://spv.babelomics.org/, last accessed January 28, 2016) all the relevant information on population frequencies for the 170,888 variant positions found in this study.
Materials and Methods
Human Subjects
Following informed consent, 267 unrelated samples of Spanish origin, which were phenotyped as healthy, were obtained and further anonymized and sequenced. The criteria followed for declaring them healthy were the absence of current known disease or genetic conditions in the family history, although diseases appearing at older ages cannot be completely ruled out. The samples were collected in 2004 and stored in the Biobank at the Hospital Virgen del Rocio (Seville, Spain), where they were routinely used as controls for genotype studies. Their geographical origin corresponds mainly to the North (Galicia and Catalonia), the center (Madrid), and the South of Spain (Andalucia). The sampling centers were the Camas and the Candelaria hospitals (Andalucia), the Dr Joan Vilaplana Hospital (Cataluña), the Hospital Clínico Universitario de Santiago (Galicia), and the Almodeva Hospital (Madrid). The number of individuals sampled in each location was approximately proportional to the populations of the corresponding regions. Because the samples were sequenced in the context of the Medical Genome Project, we called this population MGP. Samples were obtained in accordance with the approved protocols of the respective institutional review boards for the protection of human subjects. The study conformed to the tenets of the declaration of Helsinki.
Human Populations
A total of 13 human populations were used in this study which included: European populations TSI from Tuscany in Italy (98 samples), FIN Finnish from Finland (93 samples), GBR British from England and Scotland (89 samples), CEU residents of Utah (CEPH collection) with northern and western European ancestry (85 samples), and the IBS, from Spain (14 samples); Asian populations CHB Han Chinese in Beijing, China (97 samples), CHS southern Han Chinese (100 samples), and JPT Japanese in Tokyo, Japan (89 samples); American populations were MXL Mexican Ancestry in Los Angeles, CA (66 samples), PUR Puerto Rican in Puerto Rico (55 samples), and CLM Colombian in Medellin, Colombia (60 samples); and African populations were YRI Yoruba in Ibadan, Nigeria (88 samples), LWK Luhya in Webuye, Kenya (97 samples), and ASW African Ancestry in Southwest United States (61 samples). The exome sequences of all the individuals corresponding to the 13 populations were downloaded from the 1000 genomes web page (http://www.1000genomes.org/, last accessed January 28, 2016) in multisample variant calling format (VCF).
Finally, we used the MGP Spanish samples (367), totaling 1,359 studied individuals.
Construction of DNA Libraries and Sequencing
Library preparation and exome capture were carried out according to a protocol based on the Baylor College of Medicine protocol version 2.1 (with several minor modifications). First, 5 µg of input genomic DNA is sheared, end-repaired and ligated with specific adaptors. A fragment size distribution ranging from 160 to 180 bp after shearing and 200–250 bp after adaptor ligation was verified using a Bioanalyzer (Agilent). The library is amplified using a precapture linker-mediated polymerase chain reaction (LM-PCR) using a FastStart High Fidelity PCR System (Roche) and barcoded primers. After purification, 2 µg of LM-PCR product are hybridized to NimbleGen SeqCap EZ Exome libraries V3. After washing, amplification is performed by postcapture LM-PCR using a FastStart High-Fidelity PCR System (Roche). Capture enrichment is measured by qPCR according to the NimbleGen protocol. The successfully captured DNA is measured by Quant-iT PicoGreen dsDNA reagent (Invitrogen) and subjected to standard sample preparation procedures for sequencing on the SOLiD 5500xl platform as recommended by the manufacturer. Emulsion PCR is performed on the E80 scale (about 1 billion template beads) using a concentration of 0.616 pM that contains 4 equimolecular pooled libraries of enriched DNA. After breaking and enrichment, ∼276 million enriched template beads are sequenced per lane on a 6-lane SOLiD 5500xl slide.
Sequencing Data Analysis
A customized pipeline for processing the raw sequences (FastQ files) was applied. First, sequence reads were aligned to the reference human genome build GRCh37 (hg19) by using the SHRiMP tool (Rumble et al. 2009). Correctly mapped reads were further filtered with SAMtools (Li et al. 2009), which was also used for sorting and indexing mapping files. Only high quality sequence reads mapping to the reference human genome in unique locations were used for calling variants. The Genome Analysis Toolkit (GATK) (McKenna et al. 2010) was used to realign the reads around known indels and for base quality score recalibration. Identification of single nucleotide variants and indels was performed using GATK standard hard filtering parameters (DePristo et al. 2011). The result of this pipeline was a VCF file for each sequenced sample.
All the VCF files were then scanned for disease variants (variants with a reported association with a disease). The program VARIANT (Medina et al. 2012), which contains annotations from the latest versions of Ensembl Variation (Flicek et al. 2012), Uniprot (Magrane and Consortium 2011), dbSNP (Sherry et al. 2001), and HGMD (Stenson et al. 2009) was used for this purpose. Positions that were not determined (because of any quality control problems or lack of coverage) for more than the 75% of the samples analyzed were not considered in the study.
Tests for Selection
We used the MKT (McDonald and Kreitman 1991) to test for possible selective pressures in the MGP population. The MKT is based on the comparison of the ratio of nonsynonymous to synonymous SNPs between species (Dn/Ds) and within species (Pn/Ps). Assuming that synonymous mutations behave neutrally, a higher Dn/Ds than Pn/Ps ratio is expected in case of adaptive selection, because mutations that are positively selected in a population, and therefore rise quickly to fixation, would contribute more to divergence than to polymorphism. We estimated the per gene proportion of base substitutions fixed by natural selection, α (Smith and Eyre-Walker 2002) with:
where Pn and Ps are the total number of nonsynonymous and synonymous polymorphisms and Dn and Ds the number of nonsynonymous and synonymous divergent differences, respectively. Significant positive values of α (Dn/Pn > Ds/Ps) indicate an excess of fixation of nonneutral mutations suggesting that positive selection is driving a change in this gene. Measures of polymorphism in the MGP population and human–chimpanzee divergence were used to calculate α for all coding sequences. Sites diverging between panTro2 and hg19 were inferred on the Galaxy website (http://main.g2.bx.psu.edu/, last accessed January 28, 2016) using the regional variation/fetch substitutions from the pairwise alignments tool. For each identified nucleotide substitution, pairwise alignments of panTro2/hg19 were downloaded using the fetch alignments/fetch pairwise MAF blocks tool for our set of genomic intervals.
In addition, to detect signals of recent positive selection we calculated the FST values (Weir and Cockerham 1984) between the MGP population and all European 1000G populations (TSI, FIN, GBR, and CEU, excluding IBS) as a measure of population differentiation between two populations . Only polymorphic positions with maximum of 75% missing data and with a MAF of at least 10% were used. These SNPs were assigned to genes using the SNP Nexus webtool (Dayem Ullah et al. 2012). In the cases in which SNP Nexus could not assign genes to SNPs, they were assigned manually using the Ensembl Biomart (GRCh37 archive site) (Kinsella et al. 2011) and dbSNP (Sherry et al. 2001) websites.
Finally, as additional evidence for possible selection against the alternative allele homozygote, deviations from the Hardy–Weinberg proportions were tested for all the variants with a χ2 test (Wigginton et al. 2005).
Statistical Analysis
The program SNPRelate (Zheng et al. 2012) was used to carry out principal component analyses.
A χ2 test was used to assess the significance of the differences in allele frequencies in the MGP population when compared with the 1000G populations.
Multiple testing adjustments were performed using the False Discovery Rate method (Benjamini and Hochberg 1995).
The KING program (Manichaikul et al. 2010), with the parameters—unrelated—degree 2 (which extracts a list of individuals with no first- or second-degree relationship between any pairs), was used to confirm that all the samples used in the study were unrelated individuals. The VCF file from the sequencing data was converted into the PLINK binary format required by KING via VCFtools (Danecek et al. 2011).
Functional enrichment was assessed using the FatiGO (Al-Shahrour et al. 2004) as implemented in Babelomics (Alonso et al. 2015). We also applied a gene set enrichment analysis as described in Daub et al. (2013) using the sum of FST values as summary statistic of genes in a gene set.
Disease Variants, Genes, and Definitions
Disease annotations were taken from HGMD commercial release 2011.4 (Stenson et al. 2009). A total of 76,128 annotations, including 69,965 unique variants (denoted by the chromosome and start position in chromosomal coordinates) are contained in the database. HGMD incorporates different types of variants that include not only single base pair substitutions and indels affecting coding regions but also variants with consequences for mRNA splicing and regulatory abnormalities. HGMD stores only disease-causing mutations and disease-associated/functional polymorphisms but also includes a number of SNPs from GWAS, although only if there is evidence of an effect on function. Since not all the HGMD identifiers were still public at the time of writing this article, we included the source from which the evidence of the mutation was taken (a publication) in the tables presented here.
The DALYs were taken as an approximation to the prevalence of the diseases. DALYs from the “Global Burden of Disease” Study (Murray et al. 2015) were obtained from the repository of this study (http://www.healthdata.org/gbd, last accessed January 28, 2016).
Definition of Deleterious Variants
Variants with “synonymous” or “unknown” functional consequences were filtered out. Then, using the VARIANT software (Medina et al. 2012) the putative impact and damaging effect of the variants on protein function was predicted by computing both SIFT (Kumar et al. 2009) and Polyphen (Ramensky et al. 2002) damage scores and the phastCons (Siepel et al. 2005) conservation score. Since the conservation score is the only parameter applicable to any type of position, it was used as a primary filter. Variants with a phastCons conservation score higher than 200 were selected as damaging variants. SIFT and Polyphen scores were only used when available. SIFT scores lower than 0.05 and/or Polyphen scores higher than 0.95 indicate that a variant is most likely deleterious. Variants with at least two of the three pathogenicity predictors indicating a potential deleterious effect were considered deleterious.
Drug Targets
A list of 130 drug binding domains was extracted from “The druggable genome” publication (Hopkins and Groom 2002). The domains were mapped in the corresponding proteins using InterPro (version 47.0, 20 May 2014) (Hunter et al. 2012).
To define what domains were affected by substitutions that disrupt the domain without being deleterious for the protein only SNVs mapping to drug binding domains with SIFT and Polyphen values outside the deleteriousness range (i.e., SIFT scores higher than 0.05 and Polyphen scores lower than 0.95) were considered.
Supplementary Material
Supplementary tables S1–S4 and supplementary figures S1–S4 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The MGP is a joint initiative between the Consejería de Salud de la Junta de Andalucía and Roche, supported by the “Programa Nacional de Proyectos de investigación Aplicada,” I+D+i 2008, “Subprograma de actuaciones Científicas y Tecnológicas en Parques Científicos y Tecnológicos” (ACTEPARQ 2009), and European Regional Development Funds (ERDF). This work is also supported by grants BIO2014-57291-R and BFU2012-38236 from the Spanish Ministry of Economy and Competitiveness and “Plataforma de Recursos Biomoleculares y Bioinformáticos” PT 13/0001/0030 from the ISCIII, both cofunded with ERDF; grants PI1102923 and PI1001290 from the Fondo de Investigación Sanitaria, PROMETEOII/2014/025 from the Generalitat Valenciana (GVA-FEDER), FP7-PEOPLE-2012-ITN MLPM2012 318861 from the EU FP7, Fundació la Marató TV3 [20133134], and by Direcció General de Recerca, Generalitat de Catalunya (2014SGR1311). The CIBER de Enfermedades Raras is an Instituto de Salud Carlos III initiative. The authors express their gratitude to Carlos Freixas from Roche Diagnostics S.L., for his constant support of the MGP as well as to Javier Escalante, Anabel Lopez, and Federica Trombetta for their excellent work in the laboratory.
References
- Al-Shahrour F, Diaz-Uriarte R, Dopazo J. 2004. FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics 20:578–580. [DOI] [PubMed] [Google Scholar]
- Aleman A, Garcia-Garcia F, Salavert F, Medina I, Dopazo J. 2014. A web-based interactive framework to assist in the prioritization of disease candidate genes in whole-exome sequencing studies. Nucleic Acids Res. 42:W88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso R, Salavert F, Garcia-Garcia F, Carbonell-Caballero J, Bleda M, Garcia-Alonso L, Sanchis-Juan A, Perez-Gil D, Marin-Garcia P, Sanchez R, et al. 2015. Babelomics 5.0: functional interpretation for new generations of genomic data. Nucleic Acids Res. 43:W117–W121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbiza L, Dopazo J, Dopazo H. 2006. Positive selection, relaxation, and acceleration in the evolution of the human and chimp genome. PLoS Comput Biol. 2:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayuso C, Millan JM. 2010. Retinitis pigmentosa and allied conditions today: a paradigm of translational research. Genome Med. 2:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbitt CC, Warner LR, Fedrigo O, Wall CE, Wray GA. 2011. Genomic signatures of diet-related shifts during human origins. Proc Biol Sci. 278:961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. 2011. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 12:745–755. [DOI] [PubMed] [Google Scholar]
- Barreiro LB, Laval G, Quach H, Patin E, Quintana-Murci L. 2008. Natural selection has driven population differentiation in modern humans. Nat Genet. 40:340–345. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 57:289–300. [Google Scholar]
- Boomsma DI, Wijmenga C, Slagboom EP, Swertz MA, Karssen LC, Abdellaoui A, Ye K, Guryev V, Vermaat M, van Dijk F, et al. 2014. The Genome of the Netherlands: design, and project goals. Eur J Hum Genet. 22:221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante CD, Burchard EG, De la Vega FM. 2011. Genomics for the world. Nature 475:163–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonell J, Alloza E, Arce P, Borrego S, Santoyo J, Ruiz-Ferrer M, Medina I, Jimenez-Almazan J, Mendez-Vidal C, Gonzalez-Del Pozo M, et al. 2012. A map of human microRNA variation uncovers unexpectedly high levels of variability. Genome Med. 4:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casals F, Hodgkinson A, Hussin J, Idaghdour Y, Bruat V, de Maillard T, Grenier JC, Gbeha E, Hamdan FF, Girard S, et al. 2013. Whole-exome sequencing reveals a rapid change in the frequency of rare functional variants in a founding population of humans. PLoS Genet. 9:e1003815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna V, Ayub Q, Chen Y, Pagani L, Luisi P, Pybus M, Garrison E, Xue Y, Tyler-Smith C. 2014. Human genomic regions with exceptionally high levels of population differentiation identified from 911 whole-genome sequences. Genome Biol. 15:R88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona E, Chen R, Sikora M, Morgan AA, Patel CJ, Ramesh A, Bustamante CD, Butte AJ. 2013. Analysis of the genetic basis of disease in the context of worldwide human relationships and migration. PLoS Genet. 9:e1003447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coventry A, Bull-Otterson LM, Liu X, Clark AG, Maxwell TJ, Crosby J, Hixson JE, Rea TJ, Muzny DM, Lewis LR, et al. 2010. Deep resequencing reveals excess rare recent variants consistent with explosive population growth. Nat Commun. 1:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinformatics 27:2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub JT, Hofer T, Cutivet E, Dupanloup I, Quintana-Murci L, Robinson-Rechavi M, Excoffier L. 2013. Evidence for polygenic adaptation to pathogens in the human genome. Mol Biol Evol. 30:1544–1558. [DOI] [PubMed] [Google Scholar]
- Dayem Ullah AZ, Lemoine NR, Chelala C. 2012. SNPnexus: a web server for functional annotation of novel and publicly known genetic variants (2012 update). Nucleic Acids Res. 40:W65–W70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dopazo J. 2014. Genomics and transcriptomics in drug discovery. Drug Discov Today. 19:126–132. [DOI] [PubMed] [Google Scholar]
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Gibbs RA, Hurles ME, McVean GA. 2010. A map of human genome variation from population-scale sequencing. Nature 467:1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelken J, Carnero-Montoro E, Pybus M, Andrews GK, Lalueza-Fox C, Comas D, Sekler I, de la Rasilla M, Rosas A, Stoneking M, et al. 2014. Extreme population differences in the human zinc transporter ZIP4 (SLC39A4) are explained by positive selection in Sub-Saharan Africa. PLoS Genet. 10:e1004128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez RM, Bleda M, Luzon-Toro B, Garcia-Alonso L, Arnold S, Sribudiani Y, Besmond C, Lantieri F, Doan B, Ceccherini I, et al. 2013. Pathways systematically associated to Hirschsprung’s disease. Orphanet J Rare Dis. 8:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, et al. 2012. Ensembl 2012. Nucleic Acids Res. 40:D84–D90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, O’Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, Gabriel S, Rieder MJ, Altshuler D, Shendure J, et al. 2013. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493:216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alonso L, Jimenez-Almazan J, Carbonell-Caballero J, Vela-Boza A, Santoyo-Lopez J, Antinolo G, Dopazo J. 2014. The role of the interactome in the maintenance of deleterious variability in human populations. Mol Syst Biol. 10:752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Lander ES. 2013. Lessons from the cancer genome. Cell 153:17–37. [DOI] [PubMed] [Google Scholar]
- Goldstein DB, Allen A, Keebler J, Margulies EH, Petrou S, Petrovski S, Sunyaev S. 2013. Sequencing studies in human genetics: design and interpretation. Nat Rev Genet. 14:460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel S, Henn BM, Gutenkunst RN, Indap AR, Marth GT, Clark AG, Yu F, Gibbs RA, Bustamante CD. 2011. Demographic history and rare allele sharing among human populations. Proc Natl Acad Sci U S A. 108:11983–11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood L, Friend SH. 2011. Predictive, personalized, preventive, participatory (P4) cancer medicine. Nat Rev Clin Oncol. 8:184–187. [DOI] [PubMed] [Google Scholar]
- Hopkins AL, Groom CR. 2002. The druggable genome. Nat Rev Drug Discov. 1:727–730. [DOI] [PubMed] [Google Scholar]
- Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, Bernard T, Binns D, Bork P, Burge S, et al. 2012. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 40:D306–D312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson EK, Kwiatkowski DP, Sabeti PC. 2014. Natural selection and infectious disease in human populations. Nat Rev Genet. 15:379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keinan A, Clark AG. 2012. Recent explosive human population growth has resulted in an excess of rare genetic variants. Science 336:740–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury MJ, Gwinn ML, Glasgow RE, Kramer BS. 2012. A population approach to precision medicine. Am J Prev Med. 42:639–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella RJ, Kahari A, Haider S, Zamora J, Proctor G, Spudich G, Almeida-King J, Staines D, Derwent P, Kerhornou A, et al. 2011. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database 2011:bar030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryukov GV, Pennacchio LA, Sunyaev SR. 2007. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet. 80:727–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 4:1073–1081. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E, Jiang T, Jiang H, Albrechtsen A, Andersen G, Cao H, Korneliussen T, et al. 2010. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet. 42:969–972. [DOI] [PubMed] [Google Scholar]
- Lohmueller KE, Indap AR, Schmidt S, Boyko AR, Hernandez RD, Hubisz MJ, Sninsky JJ, White TJ, Sunyaev SR, Nielsen R, et al. 2008. Proportionally more deleterious genetic variation in European than in African populations. Nature 451:994–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca F, Perry GH, Di Rienzo A. 2010. Evolutionary adaptations to dietary changes. Annu Rev Nutr. 30:291–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB, et al. 2012. A systematic survey of loss-of-function variants in human protein-coding genes. Science 335:823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Tyler-Smith C. 2010. Loss-of-function variants in the genomes of healthy humans. Hum Mol Genet. 19:R125–R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane M, Consortium U. 2011. UniProt Knowledgebase: a hub of integrated protein data. Database 2011:bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. 2010. Robust relationship inference in genome-wide association studies. Bioinformatics 26:2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marigorta UM, Navarro A. 2013. High trans-ethnic replicability of GWAS results implies common causal variants. PLoS Genet. 9:e1003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marth GT, Yu F, Indap AR, Garimella K, Gravel S, Leong WF, Tyler-Smith C, Bainbridge M, Blackwell T, Zheng-Bradley X, et al. 2011. The functional spectrum of low-frequency coding variation. Genome Biol. 12:R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson I, McVean G. 2012. Differential confounding of rare and common variants in spatially structured populations. Nat Genet. 44:243–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351:652–654. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina I, De Maria A, Bleda M, Salavert F, Alonso R, Gonzalez CY, Dopazo J. 2012. VARIANT: command line, web service and web interface for fast and accurate functional characterization of variants found by next-generation sequencing. Nucleic Acids Res. 40:W54–W58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez-Vidal C, Bravo-Gil N, González-del Pozo M, Vela-Boza A, Dopazo J, Borrego S, Antinolo G. 2014. Novel RP1 mutations and a recurrent BBS1 variant explain the co-existence of two distinct retinal phenotypes in the same pedigree. BMC Genet 15:143. [DOI] [PMC free article] [PubMed]
- Mendez-Vidal C, Gonzalez-Del Pozo M, Vela-Boza A, Santoyo-Lopez J, Lopez-Domingo FJ, Vazquez-Marouschek C, Dopazo J, Borrego S, Antinolo G. 2013. Whole-exome sequencing identifies novel compound heterozygous mutations in USH2A in Spanish patients with autosomal recessive retinitis pigmentosa. Mol Vis. 19:2187–2195. [PMC free article] [PubMed] [Google Scholar]
- Moreno-Estrada A, Gravel S, Zakharia F, McCauley JL, Byrnes JK, Gignoux CR, Ortiz-Tello PA, Martinez RJ, Hedges DJ, Morris RW, et al. 2013. Reconstructing the population genetic history of the Caribbean. PLoS Genet. 9:e1003925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CJ, Barber RM, Foreman KJ, Ozgoren AA, Abd-Allah F, Abera SF, Aboyans V, Abraham JP, Abubakar I, Abu-Raddad LJ, et al. 2015. Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990-2013: quantifying the epidemiological transition. Lancet 386:2145–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MR, Wegmann D, Ehm MG, Kessner D, St Jean P, Verzilli C, Shen J, Tang Z, Bacanu SA, Fraser D, et al. 2012. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science 337:100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et al. 2010. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 42:30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novembre J, Johnson T, Bryc K, Kutalik Z, Boyko AR, Auton A, Indap A, King KS, Bergmann S, Nelson MR, et al. 2008. Genes mirror geography within Europe. Nature 456:98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 38:904–909. [DOI] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. 2002. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 30:3894–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 132:365–386. [DOI] [PubMed] [Google Scholar]
- Rumble SM, Lacroute P, Dalca AV, Fiume M, Sidow A, Brudno M. 2009. SHRiMP: accurate mapping of short color-space reads. PLoS Comput Biol. 5:e1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15:1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NG, Eyre-Walker A. 2002. Adaptive protein evolution in Drosophila. Nature 415:1022–1024. [DOI] [PubMed] [Google Scholar]
- Spivakov M, Akhtar J, Kheradpour P, Beal K, Girardot C, Koscielny G, Herrero J, Kellis M, Furlong EE, Birney E. 2012. Analysis of variation at transcription factor binding sites in Drosophila and humans. Genome Biol. 13:R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Howells K, Phillips AD, Mort M, Cooper DN. 2009. The Human Gene Mutation Database: providing a comprehensive central mutation database for molecular diagnostics and personalized genomics. Hum Genomics. 4:69–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern C. 1943. The Hardy-Weinberg Law. Science 97:137–138. [DOI] [PubMed] [Google Scholar]
- The_Genome_of_the_Netherlands_Consortium. 2014. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet. 46:818–825. [DOI] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, et al. 2012. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasseur E, Quintana-Murci L. 2013. The impact of natural selection on health and disease: uses of the population genetics approach in humans. Evol Appl. 6:596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. 2013. Cancer genome landscapes. Science 339:1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir B, Cockerham C. 1984. Estimating F-Statistics for the analysis of population structure. Evolution 38:1358–1370. [DOI] [PubMed] [Google Scholar]
- Wigginton JE, Cutler DJ, Abecasis GR. 2005. A note on exact tests of Hardy-Weinberg equilibrium. Am J Hum Genet. 76:887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LP, Ong RT, Poh WT, Liu X, Chen P, Li R, Lam KK, Pillai NE, Sim KS, Xu H, et al. 2013. Deep whole-genome sequencing of 100 southeast Asian Malays. Am J Hum Genet. 92:52–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Chen Y, Ayub Q, Huang N, Ball EV, Mort M, Phillips AD, Shaw K, Stenson PD, Cooper DN, et al. 2012. Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am J Hum Genet. 91:1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS. 2012. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28:3326–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.