

Abstract
Xanthogranulomatous pyelonephritis (XGP) and malakoplakia (MKP) are chronic inflammatory condition of kidney characterized by infiltration of inflammatory cells. We are presenting a rare case of concomitant XGP and MKP in the same kidney. This signifies that the two are different spectrums of the same disease process.
Keywords: Chronic inflammation, malakoplakia, Michaelis–Gutmann, xanthogranulomatous pyelonephritis
INTRODUCTION
Xanthogranulomatous pyelonephritis (XGP) and malakoplakia (MKP) are both chronic infective condition of the kidney. Host responses to chronic infection result in characteristic pathological lesions. MKP is characterized by periodic acid-Schiff (PAS) positive Michaelis–Gutmann bodies, whereas XGP is characterized by PAS negative, otherwise both the disease share similar gross and microscopic features. Rarely, they may co-exist.[1,2,3,4] We present such an unusual case of MKP and XGP co-existing in the same kidney. This indicates that it may be the different spectrums of the same disease process.
CASE REPORT
A 65-year-old female presented with right flank pain for 3 months with recurrent attacks of fever with chills and dysuria and a painful lump in the right flank for 1 month. She was a known diabetic for 3 years and was on oral hypoglycemic drugs.
On examination, she was found to be anemic. She had a tender renal lump in the right flank 5 cm × 4 cm. Blood picture showed hemoglobin was 9 g/dl, total leukocyte count was 21,900/cm3 with 87% neutrophil count. Blood sugar was within normal range with drugs. Serum urea was 27 mg/dl and creatinine was 1.4 mg/dl. Routine and microscopic urine examination showed 20–25 pus cell/hpf and culture was positive for proteus. Ultrasonography (USG) showed a grossly hydronephrotic right kidney with thick material inside [Figure 1] with a normal left kidney. Contrast-enhanced computerized tomography (CECT) abdomen suggested right sided renal enlargement and gross dilatation of pelvicalyceal system with pyonephrosis [Figure 2]. There was no evidence of obstructing calculus. Diethylene triamine pentaacetic acid (DTPA) renogram showed estimated glomerular filtration rate (GFR) of the right kidney - 6.18 ml/min and of the left kidney - 34.18 ml/min. USG guided percutaneous nephrostomy (PCN) of the right kidney drained 200 ml of pus and then PCN output was reduced to <5 ml/day. A repeat DTPA showed no improvement of renal function in the right side even after 1 month. The patient underwent a simple nephrectomy. The postoperative period was uneventful. On the 3rd month of postoperative follow-up, the patient was asymptomatic with normal renal biochemical parameters and a GFR on the left side 39.68 ml/min.
Figure 1.
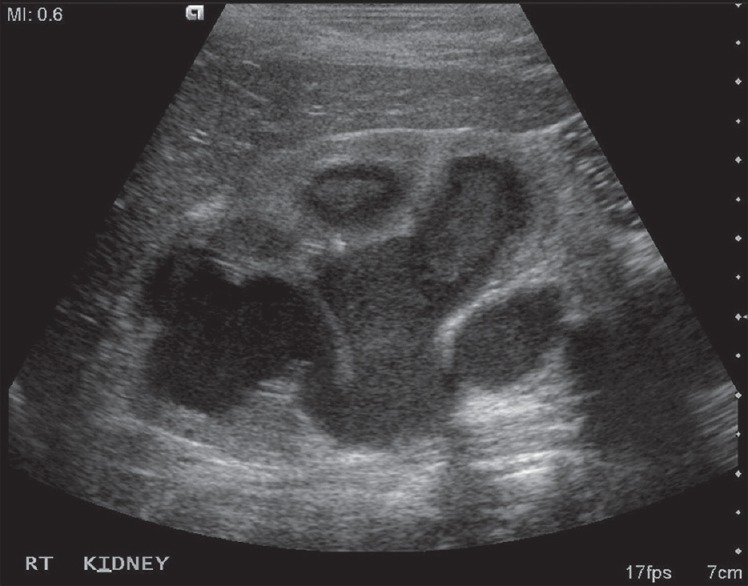
Ultrasonography showing grossly hydronephrotic right kidney with thick materials inside
Figure 2.
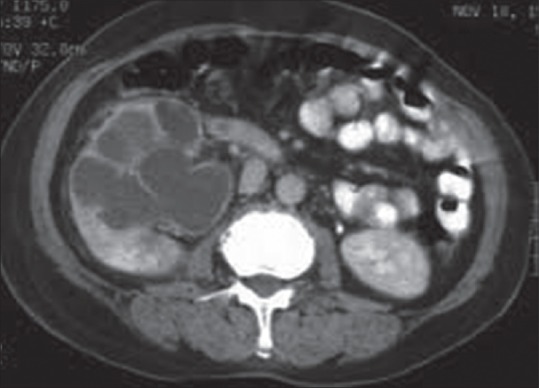
Contrast-enhanced computerized tomography showing right renal enlargement and gross dilatation of pelvicalycial system with pyonephrosis
On gross examination, whitish multiple cystic areas with solid intervening parts were seen. Microscopically, sheets of foamy histiocytes, mixed inflammatory cell infiltrations and scattered giant cells and distorted tubule were also noted. In addition, PAS-positive cytoplasmic bodies, i.e. Michales–Guttmenn bodies were seen suggesting MKP of right kidney in the setting of XGP [Figure 3a and b].
Figure 3.
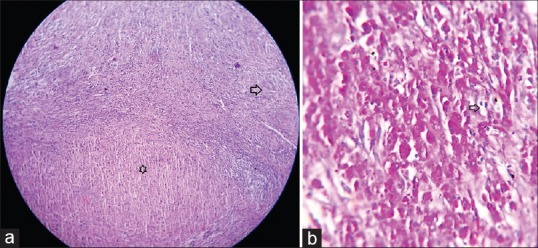
(a) Histopathology combined feature of malakoplakia and xanthogranulomatous pyelonephritis in same slide. Arrow head: Granuomatous lesions. Star: Sheets of histiocytes (H and E, ×40). (b) Histopathology showing Michaelis–Gutmann body (arrow head) with foamy histiocytes (PAS, ×400)
DISCUSSION
The term MKP is derived from Greek word malakos (soft) and plakos (plaque). The term was coined by von Hansmann. MKP is an unusual chronic granulomatous inflammatory disease, characterized by soft, yellow-brown plaques with granulomatous lesions. The first pathologic description of MKP was reported by Michaelis and Gutmann.[1] It was first described in the urinary bladder, but it can also occur in gastrointestinal tract, skin, lung bones, and lymph nodes.[2] The kidney is affected with MKP in only 15% of patients.[2] Females are more commonly affected than males with a 4:1 ratio. Patients usually present in >50 years of age though it can occur in any age group. The presentation is nonspecific with flank pain, fever, features of urinary tract infection (UTI), or signs of perinephric abscess. Sometimes it may affect both the kidneys. It is more common in immunocompromised patients such as immunodeficiency syndrome, autoimmune disease, carcinoma, or another systemic disorder.[3] The exact pathogenesis is not clear, but MKP probably results from abnormal macrophage function in response to a bacterial infection when the immune system is suppressed. A defect in intraphagosomal bacterial digestion leading to accumulation of bacterial fragments and deposition of calcium in monocyte or macrophage producing basophilic inclusion body.[4] The most common bacteria involved is Escherichia coli. It is a histological diagnosis, macroscopically characterized by yellowish or tan nodules of variable size, discrete or coalesced involving much of the renal substance, or may undergo suppuration with abscess formation. Microscopically, the lesion consists of clusters of moderately large polygonal cells with a foamy eosinophilic cytoplasm and densely staining nuclei, known as von Hansemann cells. Within the cytoplasm of these cells there are granules that stain positively with the PAS method and also larger inclusions, 4–10 µm in diameter, that stain strongly with hematoxylin. These larger inclusions which may be homogeneous or laminated are called Michaelis–Gutmann bodies. Diagnosis can only be confirmed by histopathology.
XGP is a rare chronic infective condition of the kidney leading to diffuse destruction of the renal parenchyma. It is characterized by the accumulation of lipid-laden foamy macrophages, that starts at the pelvis and calyces, but extending to and destroys the renal parenchymal and sometimes the adjacent tissues. Urinary obstruction is an almost invariable feature, and stones are found in almost 83% cases.[5] The most common organism is proteus, E. coli is next most frequent. Usual presented is in the fifth or sixth decade of age but can be present at any age. Both the sexes are equally involved. Clinically XGP should be suspected when a patient is presented with UTI and a unilateral enlarged, nonfunctioning with a stone or a mass lesion. The most common symptoms are a flank pain, fever with chills, malaise, or renal abscess. USG is useful in an unstable patient showing enlarged kidney with multiple hypoechoic fluid-filled masses that are debris-filled, dilated calyces or foci of parenchymal destruction. XGP can be confused with other chronic inflammatory condition of the kidney as well as malignancy. Definitive diagnosis is confirmed only by histopathological examination. CECT is investigation of choice showing the classical triad of XGP: Unilateral renal enlargement, no or little function and a large stone in the pelvis.
Macroscopically the kidney is massively enlarged with a normal contour. The calyces are dilated and filled with purulent material. Often it is associated with thinned cortex and replaced by xanthogranulomatous tissue. On microscopic examination, the yellowish nodules are found to contain dark sheets of lipid-laden macrophages intermixed with lymphocytes, giant cells, and plasma cells. The pathogenesis of the XGP is multifactorial. Infection in a primarily obstructed kidney may lead to tissue destruction and collection of lipid-laden macrophages. Other possible interrelated factors include venous occlusion and hemorrhage, abnormal lipid metabolism, lymphatic blockage, failure of antimicrobial therapy in UTI, altered immunologic competence, and renal ischemia.[5,6] Renal MKP and XGP have similarity, and they may overlap each other.[6,7,8]
As in our case the patient was presented with chronic inflammation of the kidney and on histology, we found features of both XGP and MKP. We can hypothesize that these two are two different spectrum of the same disease process that starts with infection with incomplete digestion of the bacteria may because of virulence or altered host response and leads to chronic inflammatory cellular infiltrate and destruction of the renal parenchyma. This association is also found by Esparza et al. in one case of their five cases.[8]
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Michaelis L, Gutmann C. Ueber einschlusse in blasentumoren. Z Klin Med. 1902;47:208–15. [Google Scholar]
- 2.Singh PB, Gupta RC, Dwivedi US, Rastogi BL. Renal parenchymal malakoplakia. Br J Urol. 1989;63:99–100. doi: 10.1111/j.1464-410x.1989.tb05135.x. [DOI] [PubMed] [Google Scholar]
- 3.Weiss M, Liapis H, Tomaszewski JE, Arend LJ. Pyelonephritis and other infections, reflux nephropathy, hydronephrosis, and nephrolithiasis. In: Charles JJ, Olson JL, Schwartz MM, Silva FG, editors. Hepinstall's Pathology of the Kidney. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 992–1066. [Google Scholar]
- 4.Dobyan DC, Truong LD, Eknoyan G. Renal malacoplakia reappraised. Am J Kidney Dis. 1993;22:243–52. doi: 10.1016/s0272-6386(12)70313-x. [DOI] [PubMed] [Google Scholar]
- 5.Smith BH. Malacoplakia of the urinary tract: A study of twenty-four cases. Am J Clin Pathol. 1965;43:409–17. doi: 10.1093/ajcp/43.5.409. [DOI] [PubMed] [Google Scholar]
- 6.Cohen MS. Granulomatous nephritis. Urol Clin North Am. 1986;13:647–59. [PubMed] [Google Scholar]
- 7.Kelly DR, Murad TM. Megalocytic interstitial nephritis, xanthogranulomatous pyelonephritis, and malakoplakia: An ultrastructural comparison. Am J Clin Pathol. 1981;75:333. doi: 10.1093/ajcp/75.3.333. [DOI] [PubMed] [Google Scholar]
- 8.Esparza AR, McKay DB, Cronan JJ, Chazan JA. Renal parenchymal malakoplakia. Histologic spectrum and its relationship to megalocytic interstitial nephritis and xanthogranulomatous pyelonephritis. Am J Surg Pathol. 1989;13:225–36. [PubMed] [Google Scholar]