

Abstract
Rationale
Mutations in several genes have been identified that are responsible for approximately 25% of families with familial thoracic aortic aneurysms and dissections (TAAD). However, the causative gene remains unknown in 75% of families.
Objectives
To identify the causative mutation in families with autosomal dominant inheritance of TAAD.
Methods and Results
Exome sequencing was used to identify the mutation responsible for a large family with TAAD. A heterozygous rare variant, c.839G>T (p.Ser280Arg), was identified in LOX, encoding a lysyl oxidase, that segregated with disease in the family. Sanger and exome sequencing was performed to investigate mutations in candidate genes in an additional 410 probands from unrelated families. Additional LOX rare variants that segregated with disease in families were identified, including c.125G>A (p.Trp42*), c.604G>T (p.Gly202*), c.743C>T (p.Thr248Ile), c.800A>C (p.Gln267Pro), and c.1044T>A (p.Ser348Arg). The altered amino acids cause haploinsufficiency for LOX or are located at a highly conserved LOX catalytic domain, which is relatively invariant in the population. Expression of the LOX variants p.Ser280Arg and p.Ser348Arg had significantly lower lysyl oxidase activity when compared with the wild type protein. Individuals with LOX variants had fusiform enlargement of the root and ascending thoracic aorta, leading to ascending aortic dissections.
Conclusions
These data, along with previous studies showing the deficiency of LOX in mice or inhibition of lysyl oxidases in turkeys and rats causes aortic dissections, support the conclusion that rare genetic variants in LOX predispose to thoracic aortic disease.
Keywords: Aortic aneurysm, aortic dissection, lysyl oxidase, gene mutation, aortic disease, exome sequencing
INTRODUCTION
Thoracic aortic aneurysms, involving the aortic root, the ascending aorta, or fusiform dilatation of both segments, are typically asymptomatic as they progressively enlarge. If undiagnosed, these aneurysms can cause an acute dissection of the ascending aorta. Family studies indicate that up to 20% of individuals with thoracic aortic aneurysms leading to acute dissections (TAAD) who do not have a known genetic syndrome (e.g., Marfan syndrome [MIM: 154700]) have a family history of TAAD, termed familial TAAD (FTAAD).1, 2 The 11 genes identified to date for FTAAD are only responsible for disease in 25% of FTAAD families, thus demonstrating further genetic heterogeneity for this disorder. The identified genes predisposing to TAAD encode proteins involved in the following: the extracellular matrix (ECM) of the aortic wall (FBN1, MFAP5), smooth muscle cell (SMC) contraction or metabolism (ACTA2, MYH11, MYLK, PRKG1, MAT2A) and canonical TGF-β signaling (TGFBR1, TGFBR2, TGFB2, SMAD3).3–10
The pathologic changes associated with TAAD primarily involve the thick medial layer of the aortic wall, which is composed of alternating layers of SMCs and elastic lamellae. With thoracic aortic disease, there is fragmentation and loss of the elastic lamellae, focal areas lacking SMCs, and accumulation of proteoglycans. The layers of elastin in the aorta are laid down during development and have an estimated half-life of approximately 40 years.11 Additionally, there is a collagen deposition between the SMCs and the elastin lamellae. The proper construction of the elastic lamellae and collagen fibers is dependent on lysyl oxidases, which are extracellular copper enzymes that catalyze the formation of lysine-derived crosslinks in elastin and hydroxylysine-derived crosslinks in collagens. Mammalian genomes have 5 lysyl oxidase gene family members, including the prototypic LOX and LOX-like proteins 1 through 4, all of which have highly similar catalytic domains. We describe here variants in one of the lysyl oxidase genes, LOX, that disrupt enzyme function and predispose to TAAD.
METHODS
Exome sequencing was used to identify the mutation responsible for a large family with TAAD. Sanger and exome sequencing was performed to investigate rare variants in candidate genes in an additional 410 probands from unrelated families and tested the segregation of these variant with aortic diseases in families. Expression of the LOX variants and lysyl oxidase activities were measured to investigate biological function of rare LOX variants. An expanded Methods section is available in the Online Data Supplement at http://circres.ahajournals.org.
Family recruitment and characterization
Blood or saliva samples were collected from affected individuals and family members after obtaining informed consent and approval from all participating institutions, including the University of Texas Health Science Center at Houston, Cleveland Clinic, and the Centre de Reference pour les Syndromes de Marfan et Apparente’s in France. Medical records, including imaging studies of the aorta, surgical reports, hospital records, death certificates and physicians’ notes were reviewed.
RESULTS
Identification of LOX rare variants associated with familial thoracic aortic aneurysms and dissections
To identify additional novel genes that cause an inherited predisposition to TAAD, exome sequencing was pursued using DNA from three affected individuals from family TAA602 with autosomal dominant inheritance of thoracic aortic disease but no mutations in the known TAAD genes (Figure 1A).8 Gene variants identified by whole exome sequencing were filtered based on the following: 1) variants that altered the amino acid sequence of proteins, including nonsynonymous, stop-lost, stop-gain, coding indels, frameshift, or splice site variants; 2) variants that had minor allele frequency (MAF) of less than 0.05% in the NHLBI Exome Sequencing Project (ESP) database; and 3) variants shared between the three affected relatives.9 The variants that met these criteria were assessed for segregation with aortic disease in TAA602, decreasing the number of genes with candidate rare variants down to 10 (Supplemental Table I). Among these genes, LOX (NM_002317.5) was the best candidate gene for causing aortic disease because of the established role of lysyl oxidases in connective tissue mechanical stability, the relatively high expression in SMCs, and the fact that Lox−/− mice die of thoracic aortic rupture shortly after birth.12 The LOX variant identified in TAA602, p.Ser280Arg, falls in the catalytic domain of the enzyme and is predicted to be damaging by five of seven functional prediction programs. The C score of combined annotation dependent depletion prediction (http://cadd.gs.washington.edu/score) is 17.36. This variant is absent in 13,000 chromosomes in the ESP database and only present in two out of 121,214 chromosomes, i.e., MAF 1.65 × 10−5 in the ExAC (Exome Aggregation Consortium) database. Additionally, serine 280 and its flanking amino acids are highly conserved from human through zebrafish (Figure 1B).
Figure 1. LOX rare variants identified in FTAAD families.
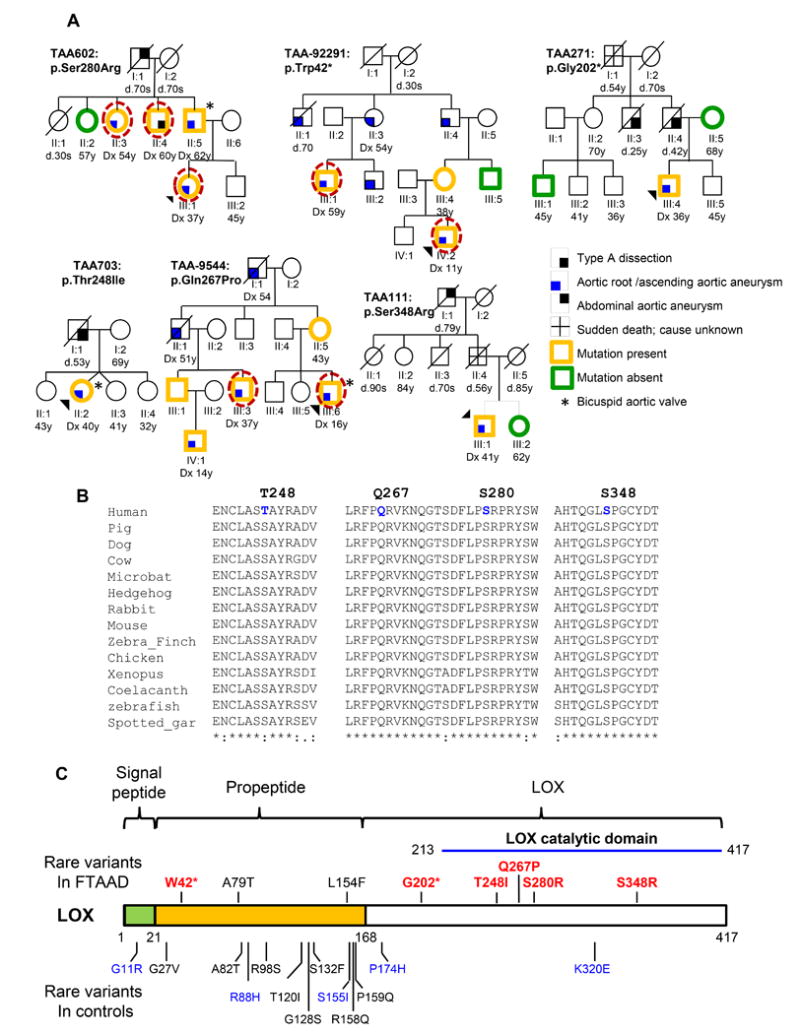
A. Pedigrees of families with LOX rare variants. The legend indicates the disease and mutation status of the family members. The age at diagnosis of aortic aneurysm or dissection (“dx”) and age at death (“d”) are shown in years. An asterisk indicates individuals with a bicuspid aortic valve. A dashed circle around a symbol indicates individuals whose DNA was used for exome sequencing. B. Orthologue conservation of LOX variants identified in individuals with TAAD and surrounding amino acids. C. Schematic representation of lysyl oxidase encoded by LOX with the domains of the proteins indicated. The LOX rare variants identified in this study are on the top of the protein diagram and the rare variants identified in European Americans in the NHLBI ESP database are indicated below. Red or blue font designates variants predicted to be possibly or probably damaging, respectively, and black font indicates variants predicted to be benign by PolyPhen-2 analysis.
Exome and Sanger sequencing of an additional 410 unrelated FTAAD probands identified five additional LOX rare variants that disrupted the protein sequence: c.125G>A (p.Trp42*), c.604G>T (p.Gly202*), c.743C>T (p.Thr248Ile), c.800A>C (p.Gln267Pro), c.1044T>A (p.Ser348Arg), c.235G>A (p.Ala79Thr), and c.460C>T (p.Leu154Phe). The nonsense variant p.Trp42* identified in TAA-92291 is predicted to lead to nonsense mediated decay and co-segregated with thoracic aortic aneurysms in in the family. The p.Gly202* variant identified in TAA271 is predicted to lead to nonsense mediated decay. This LOX variant is not present in the proband’s unaffected mother; however, samples from the affected father and uncle were not available. Notably, no LOX nonsense or indel variants is found in the ESP database and only one frameshift variant is found in one of 121,182 chromosomes in the ExAC database. Variants, p.Thr248Ile, p.Gln267Pro, and p.Ser348Arg, are absent in the ESP and ExAC databases, are predicted to be damaging or deleterious by more than four of seven programs, have C scores of 11.7, 28.0 and 24.7, respectively, and are located in the highly conserved catalytic domain of the protein. Glutamine 267, serine 280, serine 348 and are conserved from human through zebrafish, and the amino acid at position 248 is threonine or serine among these species (Figure 1B and 1C). The ESP database has only one non-synonymous rare variant in European Americans, p.Lys320Glu, in the LOX catalytic domain. Additional samples were not available from other affected members of TAA111 and TAA703. The last two rare variants identified disrupt Ala79 and Leu154, both of which are located in the propeptide region of LOX and this region is removed after post-translational pocedure. This region of LOX contains multiple rare variants in the general population and LOX p.Ala79Thr and p.Leu154Phe are predicted to be benign or neutral by six of seven programs and are not conserved (Figure 1C and Table 1). Thus, these LOX rare variants are likely to benign.Additionally, LOX p.Ala79Thr was identified in a family with a disease-causing ACTA2 mutation. Therefore, we determined that possible disease-causing LOX variants are responsible for approximately 1% of familial thoracic aortic disease and associated with decreased penetrance.
TABLE 1.
Computational and predicted effect on function of rare variants in LOX
| Amino-acid substitution | Family ID | Conservationa | Cscore | Predicted effect on functionb | MAF in ExAC |
|---|---|---|---|---|---|
| p.Ser280Arg | TAA602 | 1 | 17.7 | 5/7 | 1.65E-05 |
| p.Trp42* | TAA-92291 | 1 | 22.2 | 1/1 | 0 |
| p.Gly202* | TAA271 | 1 | 22.5 | 1/1 | 0 |
| p.Thr248Ile | TAA703 | 1 | 24.6 | 4/7 | 0 |
| p.Gln267Pro | TAA-9544 | 1 | 28.0 | 7/7 | 0 |
| p.Ser348Are | TAA111 | 1 | 11.7 | 7/7 | 0 |
| p.Leu154Phe | TAA289 | 0.15 | 15.9 | 1/7 | 1.7E-05 |
| p.Ala79Thr | TAA296/TAA806 | 0.011 | 0.53 | 0/7 | 3.8E-04 |
PhastCons conservation score
LRT, MutationTaster, Polyphen2 HDIV, Polyphen2 HVAR, SIFT, PROVEAN, and MutationAssessor.
Clinical characterization of affected individuals with LOX rare variants
The proband of family TAA602 is a 37 year old European American female with fusiform dilatation of the aortic root and ascending aorta, measuring 3.8 cm and 4.1 cm, respectively. Mild mitral regurgitation was also noted, and her physical exam was negative for syndromic features. Her father was 62 years old when he was diagnosed with a 5.2 cm aortic root aneurysm that involved the ascending aorta (4.2 cm) and aortic arch (4.0 cm). He also has a bicuspid aortic valve, a three-vessel coronary artery disease and severe left ventricular hypertrophy. He underwent an aortic valve replacement and root and ascending aorta replacement, along with a coronary artery bypass graft. The proband’s paternal uncle had an ascending aortic dissection at 60 years of age. A paternal aunt was diagnosed with a 3.9 cm aortic root aneurysm and an ectatic ascending aorta at the age of 54 years.
The proband of family TAA111 is a European American male who had an incidental finding of a 12.5 cm ascending aortic aneurysm and underwent an aortic valve replacement and supracoronary ascending aortic replacement at the age of 41 years and aortic root aneurysm repair at the age of 51 years. His father died suddenly at the age of 56 years. The proband of family TAA271 is a 36 year old European American male with an unremarkable physical exam, who was diagnosed with a 4.2 cm aortic root with fusiform enlargement of the ascending aorta. Due to the dissection deaths of his father and paternal uncle, he underwent an elective aortic root and ascending aorta replacement and aortic valve repair. The proband of TAA703 is a European American female, who was diagnosed with a 5.2 cm ascending aortic aneurysm and transverse aortic arch aneurysm and bicuspid aortic valve at the age of 40 years. She has no features of a genetic syndrome. Her father died at the age of 53 years due to a ruptured ascending aorta. The proband of TAA-92291 underwent surgery for a thoracic aortic aneurysm at 11 years of age and has additional Marfan-like features, including dolichostenomelia, scoliosis and pectus excavatum. Three of the proband’s relatives with the LOX variant (II:3, II:4, III:1) underwent thoracic aortic aneurysm repair in their 50’s. Individual II:1 died at 70 years of age after thoracic aortic surgery and individual III:2 has a thoracic aortic aneurysm requiring surgery; DNA samples from these individuals were not available. Limited clinical information was available for this family. The proband of TAA-9544 had a bicuspid aortic valve and thoracic aortic aneurysm, and underwent a Bentall procedure at 16 years of age. He also has features of Marfan syndrome, including a highly arched palate, joint laxity and skin striae. A maternal uncle (II:1), who also underwent a Bentall procedure at the age of 51 years, and a first cousin (III:3) and first cousin once removed (IV:1) with aortic root enlargement carry the LOX variant. DNA samples were not available from the proband’s maternal grandparents, but the grandfather had a thoracic aortic surgery at the age of 54 years. Dural ectasia, pectus deformity, joint hypermobility and skin striae were variably present in the family members.
The aortic pathology in two patients, the proband’s father in TAA602 (II:5) with p.Ser280Arg and the proband in TAA271 (III:4) with p.Gly202*, both of whom underwent prophylactic repair of thoracic aortic aneurysms, was assessed. The control aorta shows the orderly structure of the alternating layers of wavy elastic fibers (black) and SMCs (red; Figure 2). The aortas from both patients with LOX variants showed mild medial degeneration characterized by focal loss of elastin fibers and smooth muscle cells, but limited deposition of proteoglycans (top panels, Figure 2). Perhaps more significant in the patients’ aortic pathology is the disorganization of the elastic fiber deposition when compared to the control aorta (middle panels, Figure 2). In addition, collagen deposition is increased in the patients’ aortas (lower panel, Figure 2).
Figure 2. Aortic pathology associated with aneurysms in individuals with LOX variants.
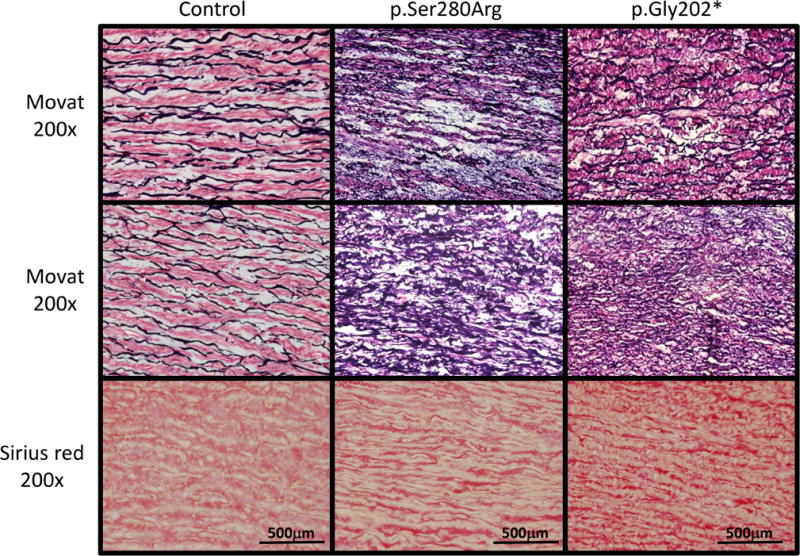
Movat pentachrome and picro sirius red staining was done on control aortic tissue and aortic tissue removed at the time of prophylactic repair of a thoracic aortic aneurysms from patients with the p.Gly202* and p.Ser280Arg mutations. Evidence of medial degeneration is evident by Movat staining in the patients’ aortas as indicated by focal fragmentation of elastic fibers (black) and a decreased number of SMCs (red) when compared to the control aorta. Movat staining also shows the disorganization of elastin deposition in the patients’ aortas compared with control aortas. Picro sirius red staining of collagen fibers shows increased collagen deposition in the patients’ aortas compared with the control aorta.
LOX missense variants in the enzymatic domain decrease LOX activity
The protein product of LOX (referred to as LOX) is synthesized as a 50-kDa inactive proenzyme (pro-LOX) that subsequently undergoes N- and O-glycosylation at sites in the propeptide domain, and then is secreted from the cells and cleaved extracellularly to form the functional C-terminal 30-kDa enzyme and 18-kDa N-terminal propeptide fragment (Figure 1C). Recombinant plasmids were constructed to express either the wild type (WT) LOX or each of the mutant LOX proteins (Thr248Ile, Ser280Arg and Ser348Arg) with either a human influenza hemagglutinin (HA) or FLAG tag at the C-terminus. The plasmids were transfected individually into HeLa cells and immunoblot analyses using antibodies against either LOX or the protein tags showed similar levels of the pro-LOX protein for all four constructs (Figure 3A). At the same time, immunoblot analysis using the LOX antibody showed that the levels of active mutant LOX (molecular weight 34 kDa) was lower than the WT, with the Thr248Ile mutation 4% lower, the Ser280Arg 27% lower and the Ser348Arg 14% lower (Figure 3A). Surprisingly, the antibodies directed against either the FLAG or HA tag failed to recognize the active protein. Lysyl oxidase activity was assayed at five time points by spectrophotometric detection of the byproduct of LOX enzymatic activity, hydrogen peroxide, and the enzymatic activity corrected for the decreased protein levels of the active LOX mutants relative to the WT level. The mutant LOX constructs, Thr248Ile, Ser280Arg and Ser348Arg, had lysyl oxidase activity levels that were 92%, 50%, and 79% that of WT LOX, respectively (p values 0.03 to 5.2 × 10−8; (Figure 3B).
Figure 3. Missense variants identified in FTAAD patients decrease the enzymatic activity of LOX.
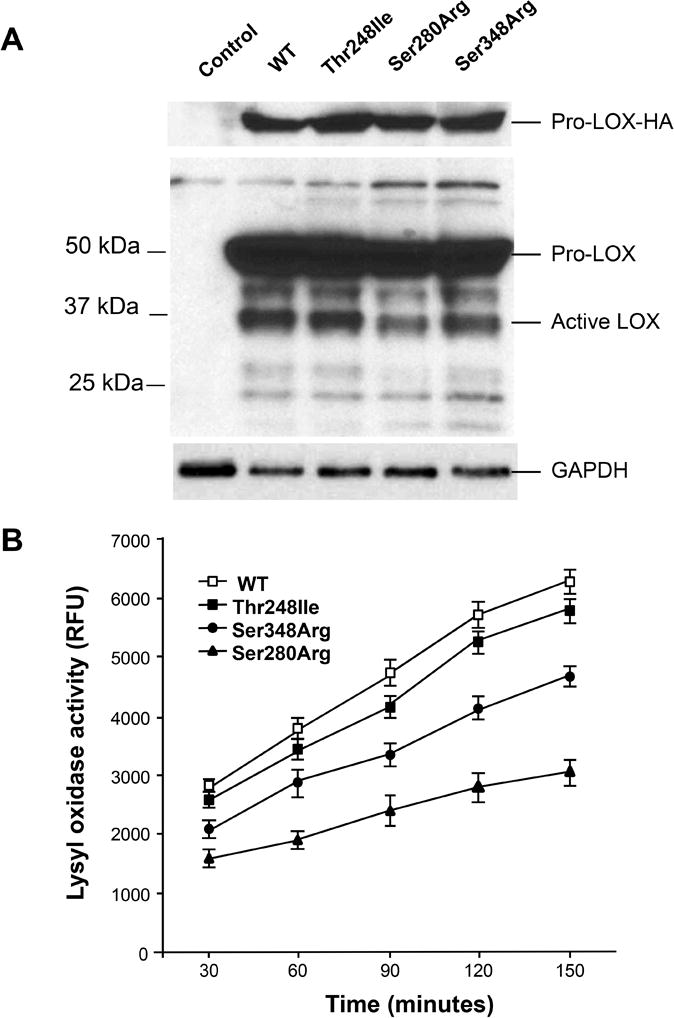
A. Immunoblot analyses of HeLa lysates transfected with plasmid containing WT LOX or the individual LOX variants using an antibody directed against HA tag (top) and the LOX protein (middle) show equal expression of a pro-LOX-HA with all plasmids. B. Lysyl oxidase enzymatic activity assays show that LOX activities were significantly decreased when compared with WT for Ser280Arg and Ser348Arg LOX variants, but not for Thr248Ile. All LOX activity data are resulted from two separate transfections, and triplicate analysis of the lysates from each transfection. Lysyl oxidase activities were measured at 30, 60, 90, 120, and 150 minutes, respectively. Data are normalized to activity of the WT LOX with the non-transfected cells activity subtracted. Activity is presented in relative fluorescent units with each data point, error bars indicate standard deviation. *p<0.05 for the comparison of mutant versus WT LOX activity levels.
DISCUSSION
The data presented here support the conclusion that heterozygous loss-of-function mutations in LOX, specifically variants that disrupt the catalytic activity or lead to haploinsufficiency, predispose to thoracic aortic aneurysms and acute aortic dissections. Overlapping syndromic features of Marfan syndrome, such as pectus deformities and striae, were reported in family members with LOX variants but these features were not sufficient to meet diagnostic criteria for Marfan syndrome.13 Thoracic aortic aneurysms in these individuals are either aortic root aneurysms or fusiform aneurysms, involving both the aortic root and ascending aorta. Although mutation carriers died of ascending aortic dissections, there were no reports of aortic dissections with minimal enlargement of the ascending aorta. None of the affected individuals presented with descending thoracic aortic aneurysms or dissections. Interestingly, a bicuspid aortic valve was identified in three out of 18 individuals with LOX mutations. It is also notable that probands from two families, TAA-92291 and TAA-9544, underwent thoracic aortic aneurysm repair at 11 and 16 years of age, respectively, which is younger than their affected family members and other LOX mutation carriers. This young age of onset could be due to environmental modifiers of the phenotype but is perhaps more likely due to a second genetic alteration leading to an early onset of the disease.
Lysyl oxidases are extracellular copper enzymes that initiate the formation of lysine-derived crosslinks in elastin and collagen. There are five lysyl oxidase gene family members with highly similar catalytic domains. The specific function of some of these genes has been determined through deletion of these genes in mice. Lox−/− mice die at the end of gestation or soon after parturition due to aortic rupture with evidence of aortic aneurysms and aortic tortuosity.11,12 Importantly, 80% of lysyl oxidase activity is lost with deletion of Lox, suggesting LOX encodes the majority of the lysyl oxidase in the aorta.14 Electron microscopic examination of Lox−/− mouse aortas shows decreased amount and fragmentation of the elastic laminae, along with marked disorganization of the elastin fibers, similar to what was observed in the aortic pathology associated with LOX mutations (Figure 2). Thus, other lysyl oxidase genes cannot compensate for the loss of Lox in aortas of mice to assure proper development of the aorta. In contrast, deletion of Loxl does not disrupt aortic development in mice, but instead leads to uterine and bowel prolapse and enlarged airspaces in the lung.15
Loss of lysyl oxidase activity due to the LOX mutations may predispose to thoracic aortic disease through aberrant development of the thoracic aorta due to fewer elastin cross links, leading to decreased and fragmented elastic fiber as observed in the Lox−/− mice. It is also established that irreversible inhibition of lysyl oxidase activity by administration of β-aminopropionitrile (BAPN) induces thoracic aortic aneurysms and dissections in both young turkeys and rats.16–21 The aortic pathology in BAPN-treated young rats shows features of medial degeneration, including elastin degradation, thickening of the medial layer, and SMC apoptosis.22 Thus, inhibition of lysyl oxidase activity postnatally also induces thoracic aortic disease. Since elastin layers are laid down during development, it raises the possibility that the stability of the aorta provided by lysyl oxidase activity may result from the cross linking of collagen, which has a relatively short half-life in the aorta in comparison to elastin.
Although LOX mutations have not been described in humans until this report, secondary lysyl oxidase deficiency can occur in individuals with Menkes syndrome and the less severe occipital horn syndrome (OHS). Mutations in ATP7A, a copper transporter necessary for copper absorption, cause both Menkes and OHS and loss of copper leads to reduced activities of copper-dependent enzymes, including all lysyl oxidases.23 Vascular abnormalities such as vascular tortuosity and arterial aneurysms have been observed in individuals with Menkes syndrome and OHS and are associated with fragmentation and disruption of the elastic lamellae. Furthermore, a mouse model of Menkes syndrome develops aneurysms in the ascending, descending thoracic, and abdominal aortas within six months of age.24 The overlap of the aortic disease in Menkes with the phenotype of FTAAD due to LOX mutations provides further evidence that lysyl oxidase activity is important for maintaining structural integrity of the aorta throughout a life time.
The rare variants in FTAAD that were classified as disease-causing fall in the catalytic domain, which is relatively invariant in the general population. Lysyl oxidase activity is predicted to be decreased by 50% with the decrease by p.Trp42* and p.Gly202* and we found the other rare variants, p.Thr248Ile, p.Ser280Arg and p.Ser348Arg, decrease activity from 50 – 92% of that of the wild type allele. Thus, with the addition of the wildtype allele, total LOX activity is predicted to be 75% to 96% that of normal, raising the question why the relatively modest decrease in enzymatic activity would cause thoracic aortic disease.-. One possibility is that the active mutant protein is less stable than the active wild type protein. We found that all the mutations lead to decrease active LOX protein levels when the proteins were overexpressed in HeLa cells, supporting this possibility. Additionally, although we have focused on disruption of the catalytic activity by the rare variants associated with TAAD, it is important to note that LOX has other functions in the cell. The LOX propeptide region targets LOX to elastin fibers in the extracellular matrix by mediating its interactions with tropoelastin.25 LOX is also reported to localize the nucleus in SMCs, where data indicate that it has a role in in chromatin structure regulation and gene transcription.26, 27 Thus, the LOX rare variants identified associated with FTAAD may disrupt other functions of the protein than just the catalytic activity. Finally, it is important to note that we were not able to confirm that the LOX variant that disrupted protein stability and activity minimally, p.Thr248Ile, segregated with disease in the family. Therefore, this variant may not be disease-causing.
In summary, accumulating evidence indicates that loss of lysyl oxidase activity due to mutations in LOX predisposes individuals to fusiform thoracic aortic aneurysms and ascending aortic dissections. The presence of bicuspid aortic valve in three of the 20 carriers of LOX mutations. Normal heart valves develop from cardiac cushions, which undergo extensive remodeling that involves cell differentiation, apoptosis, and remodeling of extracellular matrix. Thus, defects in LOX function has the potential to contribute to this congenital valve abnormality and thoracic aortic disease. Additional studies are needed to define the spectrum of mutations and further delineate the clinical phenotype.
Supplementary Material
Novelty and Significance.
What Is Known?
Approximately 25% of patients with thoracic aortic aneurysms that can lead to acute aortic dissections have an inherited predisposition for the condition and there is significant genetic heterogeneity in this inherited predisposition, with 11 genes identified to date.
The stability and integrity of elastin and collagen, two major matrix components of the aortic wall, are dependent on lysine-derived crosslinks formed by lysyl oxidases.
Mammalian genomes have 5 lysyl oxidase genes, including the prototypic LOX gene and LOX-like genes 1 through 4, but no genetic mutations predisposing to thoracic aortic disease have been identified to date.
What New Information Does This Article Contribute?
Mutations in LOX were identified in families with an inherited predisposition for thoracic aortic disease.
These LOX variants are loss-of-function mutations, leading either to haploinsufficiency or disruption of the catalytic domain and decreased enzymatic activity.
Individuals with LOX mutations had fusiform enlargement of the root and ascending thoracic aorta, and in the absence of surgical repair of the aneurysms, this leads to acute type A (ascending) aortic dissections.
Identification of the genes predisposing to thoracic aortic aneurysms and acute aortic dissections is important to identify at-risk individuals so that medical and surgical management can be initiated to prevent premature death. However, no mutations causing thoracic aortic disease have been identified in any of the five genes for human lysyl oxidases. Here we report loss-of-function mutations in LOX that predispose to thoracic aortic disease. This finding adds to the list of genes predisposing to thoracic aortic disease and confirms the role of lysyl oxidase in regulating the stability and integrity of elastin and collagen in the aortic wall.
Acknowledgments
The authors are extremely grateful to the individuals and families who participated in this study and the physicians and genetic counselors who aided in the collection of clinical data from the families.
SOURCES OF FUNDING
The following sources provided funding for these studies: RO1 HL62594, P01HL110869-01, UL1 RR024148, John Ritter Foundation, Vivian L. Smith Foundation, and Richard T. Pisani Funds (all to D.M.M.) and GIS-Maladies rares (C.B.), PHRC AOM09093 (G.J.), and ANR 2010 BLAN 1129 from the French National Research Agency (G.J.). Sequencing was provided by the University of Washington Center for Mendelian Genomics (UW CMG) and was funded by the National Human Genome Research Institute and the National Heart, Lung and Blood Institute grant 1U54HG006493 to D.A.N, J.S. and M.J.B.
Nonstandard Abbreviations and Acronyms
- BAPN
β-aminopropionitrile
- ECM
extracellular matrix
- ESP
the NHLBI Exome Sequencing Project
- ExAC
Exome Aggregation Consortium
- FTAAD
familial thoracic aortic aneurysms and dissections
- HA
hemagglutinin
- MAF
minor allele frequency
- OHS
occipital horn syndrome
- SMC
smooth muscle cell
- TAAD
thoracic aortic aneurysms and dissections
- WT
wild type
Footnotes
Subject Terms:
Aneurysm, Aortic Dissection, Genetics, Vascular Biology
DISCLOSURES
None.
References
- 1.Biddinger A, Rocklin M, Coselli J, Milewicz DM. Familial thoracic aortic dilatations and dissections: a case control study. J Vasc Surg. 1997;25:506–511. doi: 10.1016/s0741-5214(97)70261-1. [DOI] [PubMed] [Google Scholar]
- 2.Albornoz G, Coady MA, Roberts M, Davies RR, Tranquilli M, Rizzo JA, Elefteriades JA. Familial thoracic aortic aneurysms and dissections–incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg. 2006;82:1400–1405. doi: 10.1016/j.athoracsur.2006.04.098. [DOI] [PubMed] [Google Scholar]
- 3.Mizuguchi T, Collod-Beroud G, Akiyama T, et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004;36:855–860. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pannu H, Fadulu V, Chang J, Lafont A, Hasham SN, Sparks E, Giampietro PF, Zaleski C, Estrera AL, Safi HJ, Shete S, Willing MC, Raman CS, Milewicz DM. Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005;112:513–520. doi: 10.1161/CIRCULATIONAHA.105.537340. [DOI] [PubMed] [Google Scholar]
- 5.Zhu L, Vranckx R, Khau Van KP, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc JM, Brunotte F, Bruneval P, Wolf JE, Michel JB, Jeunemaitre X. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38:343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 6.Guo DC, Pannu H, Papke CL, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 7.Wang L, Guo DC, Cao J, et al. Mutations in Myosin light chain kinase cause familial aortic dissections. Am J Hum Genet. 2010;87:701–707. doi: 10.1016/j.ajhg.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Regalado ES, Guo DC, Villamizar C, et al. Exome Sequencing Identifies SMAD3 Mutations as a Cause of Familial Thoracic Aortic Aneurysm and Dissection With Intracranial and Other Arterial Aneurysms. Circ Res. 2011;109:680–686. doi: 10.1161/CIRCRESAHA.111.248161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boileau C, Guo DC, Hanna N, et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet. 2012;44:916–921. doi: 10.1038/ng.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo DC, Regalado E, Casteel DE, et al. Recurrent Gain-of-Function Mutation in PRKG1 Causes Thoracic Aortic Aneurysms and Acute Aortic Dissections. Am J Hum Genet. 2013;93:398–404. doi: 10.1016/j.ajhg.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arribas SM, Hinek A, Gonzalez MC. Elastic fibres and vascular structure in hypertension. Pharmacol Ther. 2006;111:771–791. doi: 10.1016/j.pharmthera.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Maki JM, Rasanen J, Tikkanen H, Sormunen R, Makikallio K, Kivirikko KI, Soininen R. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation. 2002;106:2503–2509. doi: 10.1161/01.cir.0000038109.84500.1e. [DOI] [PubMed] [Google Scholar]
- 13.Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De BJ, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P, De Paepe AM. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476–485. doi: 10.1136/jmg.2009.072785. [DOI] [PubMed] [Google Scholar]
- 14.Maki JM, Sormunen R, Lippo S, Kaarteenaho-Wiik R, Soininen R, Myllyharju J. Lysyl oxidase is essential for normal development and function of the respiratory system and for the integrity of elastic and collagen fibers in various tissues. Am J Pathol. 2005;167:927–936. doi: 10.1016/S0002-9440(10)61183-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X, Zhao Y, Gao J, Pawlyk B, Starcher B, Spencer JA, Yanagisawa H, Zuo J, Li T. Elastic fiber homeostasis requires lysyl oxidase-like 1 protein. Nat Genet. 2004;36:178–182. doi: 10.1038/ng1297. [DOI] [PubMed] [Google Scholar]
- 16.BARNETT BD, BIRD HR, LALICH JJ, STRONG FM. Toxicity of beta-amino-propionitrile for turkey poults. Proc Soc Exp Biol Med. 1957;94:67–70. [PubMed] [Google Scholar]
- 17.Simpson CF, Kling JM, Palmer RF. The use of propranolol for the protection of turkeys from the development of beta-aminopropionitrile-induced aortic ruptures. Angiology. 1968;19:414–418. doi: 10.1177/000331976801900705. [DOI] [PubMed] [Google Scholar]
- 18.Nakashima Y, Sueishi K. Alteration of elastic architecture in the lathyritic rat aorta implies the pathogenesis of aortic dissecting aneurysm. Am J Pathol. 1992;140:959–969. [PMC free article] [PubMed] [Google Scholar]
- 19.Kurihara T, Shimizu-Hirota R, Shimoda M, Adachi T, Shimizu H, Weiss SJ, Itoh H, Hori S, Aikawa N, Okada Y. Neutrophil-derived matrix metalloproteinase 9 triggers acute aortic dissection. Circulation. 2012;126:3070–3080. doi: 10.1161/CIRCULATIONAHA.112.097097. [DOI] [PubMed] [Google Scholar]
- 20.Anzai A, Shimoda M, Endo J, et al. Adventitial CXCL1/G-CSF expression in response to acute aortic dissection triggers local neutrophil recruitment and activation leading to aortic rupture. Circ Res. 2015;116:612–623. doi: 10.1161/CIRCRESAHA.116.304918. [DOI] [PubMed] [Google Scholar]
- 21.Li JS, Li HY, Wang L, Zhang L, Jing ZP. Comparison of beta-aminopropionitrile-induced aortic dissection model in rats by different administration and dosage. Vascular. 2013 doi: 10.1177/1708538113478741. [DOI] [PubMed] [Google Scholar]
- 22.Jia LX, Zhang WM, Zhang HJ, Li TT, Wang YL, Qin YW, Gu H, Du J. Mechanical stretch-induced endoplasmic reticulum stress, apoptosis and inflammation contribute to thoracic aortic aneurysm and dissection. J Pathol. 2015;236:373–383. doi: 10.1002/path.4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaler SG. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat Rev Neurol. 2011;7:15–29. doi: 10.1038/nrneurol.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brophy CM, Tilson JE, Braverman IM, Tilson MD. Age of onset, pattern of distribution, and histology of aneurysm development in a genetically predisposed mouse model. J Vasc Surg. 1988;8:45–48. [PubMed] [Google Scholar]
- 25.Thomassin L, Werneck CC, Broekelmann TJ, Gleyzal C, Hornstra IK, Mecham RP, Sommer P. The Pro-regions of lysyl oxidase and lysyl oxidase-like 1 are required for deposition onto elastic fibers. J Biol Chem. 2005;280:42848–42855. doi: 10.1074/jbc.M506832200. [DOI] [PubMed] [Google Scholar]
- 26.Li W, Nellaiappan K, Strassmaier T, Graham L, Thomas KM, Kagan HM. Localization and activity of lysyl oxidase within nuclei of fibrogenic cells. Proc Natl Acad Sci U S A. 1997;94:12817–12822. doi: 10.1073/pnas.94.24.12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mello ML, Alvarenga EM, Vidal BC, Di DA. Chromatin supraorganization, mitotic abnormalities and proliferation in cells with increased or down-regulated lox expression: Indirect evidence of a LOX-histone H1 interaction in vivo. Micron. 2011;42:8–16. doi: 10.1016/j.micron.2010.09.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.