

ABSTRACT
Therapeutic cancer vaccination is an attractive treatment modality for cancer, but with limitations using existing whole-cell, peptide, or protein vaccines. We propose that a cell-penetrating peptide (CPP)-based vaccine delivering multi-epitopic antigens into antigen-presenting cells (APCs) offers great potential to induce an integrated antitumor immune response and robust, sustained therapeutic effect.
KEYWORDS: Cell-penetrating peptide, CD4+ T cells, CD8+ T cells, therapeutic cancer vaccines
Abbreviations
- APC
antigen-presenting cell
- CPP
cell-penetrating peptide
- DC
dendritic cell.
Introduction
The historic success story of applied immunology is undoubtedly vaccination—capable of eradicating at least one of the major scourges of humanity, smallpox. However, achieving the dream of efficacious therapeutic cancer vaccination has remained elusive. Evidently, it is intrinsically simpler to amplify elements of our immune system that have principally evolved to combat pathogenic microbes. But putting aside the quantitative aspects of avidity of recognition of a cancer antigen versus a pathogen antigen, there are other aspects of antitumor immunity that need to be mastered. Solid cancers are composed of antigenically heterogenous malignant cells, with frequently hostile microenvironments, with variable accessibility for immune effector cells and molecules. Most cancer immunotherapists would agree that therapeutic vaccines should ideally target such tumors on multiple fronts. However, the current solutions proposed for advanced clinical development reflect the limited availability of safe and feasible vaccine formulations rather than optimal solutions. We therefore applaud the vision of individuals and companies in developing therapeutic vaccines, but we acknowledge that certain clinical disappointments are inevitable because of technological limitations.1 However, recent fundamental advances in vaccine vector design and application are highly promising for the next generation of therapeutic vaccines.
A simplified vision of the key requirements for therapeutic T-cell-mediated antitumor immunity highlights the induction of high numbers of CD4+and CD8+ T cells with specificity for multiple tumor antigens, with effector functions that include cytotoxicity and type I cytokine release, with capacity to reach, infiltrate and function at the tumor site, and with persistence or memory for long-term tumor surveillance. In short, we want it all—an integrated antitumor immune response, and to facilitate clinical development, vaccinologists need some sort of biological “Swiss Army knife” that can achieve all of this. The “all in one” cancer vaccine solution has been broadly developed in one of two directions. The first is to exploit the breadth of antigens expressed by each patient's tumor by using the autologous cancer cell or tissue itself as antigen source. Bypassing the necessity for antigen identification has enabled this approach to be rapidly tested clinically, but correlating occasional clinical responses with immune responses is clearly challenging.2 The alternative approach, which we consider more amenable to stepwise optimization, is to construct antigenically defined vaccines. This necessitates tumor antigen identification, but there are many advances in this field, from reverse immunology through to mining the mutanome and immunopeptidome, with an expanding list of candidate tumor rejection antigens.3 Nevertheless, there are still ongoing challenges in antigen discovery, particularly for MHC class II restricted epitopes. Indeed, a CD4+ T cell component of antitumor immunity is highly desirable, not only through intrinsic effector functions but also helper functions during CD8+ T cell priming, recruitment to the tumor site, and establishing memory. For some of these functions, antigens present in the tumor rather than “universal” but not tumor-expressed antigens are preferable.4 Therefore, the ideal antigenically defined therapeutic cancer vaccine will induce CD8+ and CD4+ T cells against several tumor antigens to cope with tumor heterogeneity and immune escape by antigen loss.
Once a tumor antigen panel has been selected, how can the vaccine be formulated? Perhaps, the simplest approach is a cocktail of synthetic peptides corresponding to the defined epitopes. There is extensive clinical experience of peptide-based vaccines,5 but overall limited clinical efficacy. Although short peptides are the most explored, there is now accumulating and compelling evidence that long peptides are advantageous, because they require processing, and because a given long peptide may potentially incorporate epitopes for both CD4+ and CD8+ T cells.6 A logical development of the long peptide-approach is to use recombinant proteins to incorporate even more of the desirable elements of the vaccine, particularly larger numbers of epitopes that are not only recognized by both CD4+ and CD8+ T cells, but which are also restricted by different HLA alleles. However, conventional recombinant protein vaccines have a major disadvantage: they are taken up by APCs and processed principally via the endosomal route, resulting in presentation on MHC class II molecules and stimulation of CD4+ T cells and a humoral response. These characteristics may be ideal for a prophylactic vaccine for HBV, but insufficient for an efficacious therapeutic cancer vaccine. This lack of an integrated CD4+ and CD8+ T cell response is likely to have contributed to recent disappointing results using recombinant MAGE-3 in clinical trials for melanoma and non-small-cell lung cancer (as discussed in6). We and others have explored a potential solution to benefit from the antigen-carrying capacity of a protein, while inducing an integrated immune response; this involves exploiting the properties of CPP that can transport cargoes across biological membranes.7
Studies of CPPs including those derived from viral proteins (such as TAT from HIV and ZEBRA from EBV) indicated that their cargo can include protein antigens that are in turn transported into APCs. We reported that a 42 residue fragment (Z12) of the ZEBRA CPP could transport multi-epitopic cargoes (Z12-Multi-E) into different processing compartments of DCs (Fig. 1A), resulting in presentation of the incorporated epitopes on different MHC class I and class II alleles, and subsequent stimulation of cognate CD4+ and CD8+ T cells (Fig. 1B-C).8 The efficiency and rapidity of protein transduction by Z12 was striking; cytosolic protein transduction was non-detectable in the absence of Z12, and far more efficient than achieved with the well-described CPP Tat57-49. Nevertheless, in vitro-proven antigen delivery does not necessarily equate with in vivo utility, in which a useful integrated antitumor response must have sufficient magnitude and functionality to impact on aggressive tumors. This was validated for Z12-based cancer vaccines (Fig. 1D), which had major therapeutic effect in three different mouse tumor models; remarkably, the most sustained therapeutic responses were with a challenging orthotopic brain tumor.
Figure 1.
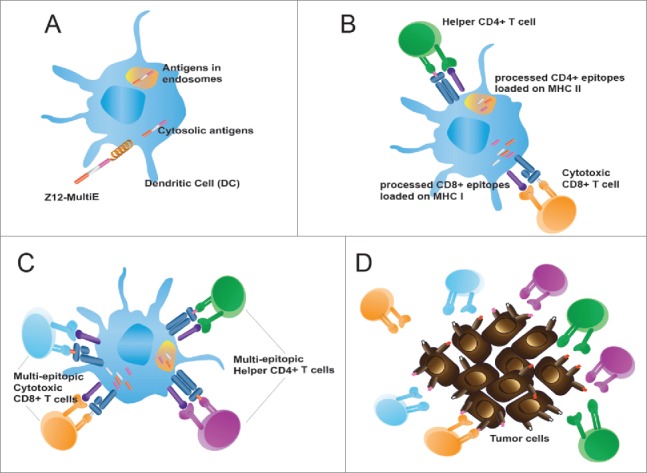
Cell-penetrating peptides (CPPs) transport antigenic cargoes into dendritic cells (DCs) and induce robust CD4+ and CD8+ T-cell mediated antitumor immunity. (A) The Z12-MultiE CPP transports antigenic cargoes into both endosomal and cytosolic compartments. (B) Processing and loading of Z12-MultiE antigens on MHC class I and MHC class II. (C) Stimulation of multiepitopic CD4+ and CD8+ T cells. (D) CPP-based vaccine induction of an integrated effector T cell response directed against tumor with heterogeneous antigen expression.
The clinical context and biological complexity of human malignancy means that there are multiple optimizations that may need to be tested to achieve major clinical impact, especially for application to different tumor types. These will undoubtedly include the selection of antigens, but must also incorporate continuing advances in adjuvants and potential ameliorations such as DC targeting. In this regard, the potential benefits of an “all in one” CPP-protein-based platform are considerable. At least for the Z12 CPP we have tested, cargo capacity is substantial, and greater than currently reported for viral-vector based approaches. Therefore, this allows great flexibility in selection of antigens. Moreover, adding targeting or adjuvanting moieties will apply to all constituent epitopes within the protein rather than to individual antigens, as would be the case with short or long peptide-based vaccines. The current opportunities and tools in cancer immunotherapy offer many opportunities for future cancer treatment, with checkpoint blockade attracting much deserved attention. But a single immunotherapy modality is clearly insufficient for most patients; the potential of vaccination should therefore be fully exploited using the opportunities to further explore, develop, and combine tools such as CPP-based vectors.
Disclosure of potential conflicts of interest
Madiha Derouazi has ownership interest (including patents) in Amal Therapeutics. Paul Walker and Pierre-Yves Dietrich have ownership interest in patents related to Z12 (University of Geneva) and are consultant/advisory board member for Amal Therapeutics.
References
- 1.Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I et al.. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol 2014; 11:509-24; PMID:25001465; http://dx.doi.org/ 10.1038/nrclinonc.2014.111 [DOI] [PubMed] [Google Scholar]
- 2.Butterfield LH. Cancer vaccines. BMJ 2015; 350:h988; PMID:25904595; http://dx.doi.org/ 10.1136/bmj.h988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 2014; 14:135-46; PMID:24457417; http://dx.doi.org/ 10.1038/nrc3670 [DOI] [PubMed] [Google Scholar]
- 4.Hoepner S, Walker PR. Getting by with a little help from the right CD4+ T cells. Oncoimmunology 2013; 2:e25772; PMID:24244904; http://dx.doi.org/ 10.4161/onci.25772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pol J, Bloy N, Buque A, Eggermont A, Cremer I, Sautes-Fridman C, Galon J, Tartour E, Zitvogel L, Kroemer G et al.. Trial Watch: Peptide-based anticancer vaccines. Oncoimmunology 2015; 4:e974411; PMID:26137405; http://dx.doi.org/ 10.4161/2162402X.2014.974411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melief CJ, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. J Clin Invest 2015; 125:3401-12; PMID:26214521; http://dx.doi.org/ 10.1172/JCI80009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milletti F. Cell-penetrating peptides: classes, origin, and current landscape. Drug Discov Today 2012; 17(15-16):850-60; PMID:22465171; http://dx.doi.org/ 10.1016/j.drudis.2012.03.002 [DOI] [PubMed] [Google Scholar]
- 8.Derouazi M, Di Berardino-Besson W, Belnoue E, Hoepner S, Walther R, Benkhoucha M, Teta P, Dufour Y, Yacoub Maroun C, Salazar AM et al.. Novel Cell-Penetrating Peptide-Based Vaccine Induces Robust CD4+ and CD8+ T Cell-Mediated Antitumor Immunity. Cancer Res 2015; 75:3020-31; PMID:26116496; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-3017 [DOI] [PubMed] [Google Scholar]