

ABSTRACT
Despite identification of macrophages in tumors (tumor-associated macrophages, TAM) as potential targets for cancer therapy, the origin and function of TAM in the context of malignancy remain poorly characterized. Here, we show that microparticles (MPs), as a by-product, released by tumor cells act as a general mechanism to mediate M2 polarization of TAM. Taking up tumor MPs by macrophages is a very efficient process, which in turn results in the polarization of macrophages into M2 type, not only leading to promoting tumor growth and metastasis but also facilitating cancer stem cell development. Moreover, we demonstrate that the underlying mechanism involves the activation of the cGAS/STING/TBK1/STAT6 pathway by tumor MPs. Finally, in addition to murine tumor MPs, we show that human counterparts also possess consistent effect on human M2 polarization. These findings provide new insights into a critical role of tumor MPs in remodeling of tumor microenvironment and better understanding of the communications between tumors and macrophages.
KEYWORDS: cGAS/STING pathway, M2 tumor-associated macrophages, polarization, tumor cell-derived microparticles, tumor progression
Introduction
M2-type TAM as a hallmark of cancers not only promote tumor immunosuppression, angiogenesis and metastasis, but also mediate chemotherapeutic drug resistance and cancer cell survival as well as facilitate the formation of cancer stem cells.1-5 Notwithstanding these tumor-promoting consequences, potential therapeutic strategies against cancers are conceived by targeting M2 TAMs.6-8 Indeed, there are clinical trial designs in which tumor-infiltrating macrophages are being targeted.8 As opposed to M2 TAMs, tumor-infiltrating macrophages, when expressing M1 phenotype, can on the other hand play a positive role through producing substantial nitric oxide and other mediators favoring antitumor immunity.9,10 Therefore, better understanding the molecular pathways through which macrophages are polarized is necessary for optimal M2 TAM targeting. How macrophages are polarized in tumor microenvironment seems to be very complex and remains poorly understood. Various factors such as IL-4, IL-13, PGE2, M-CSF and VEGF are capable of inducing M2 macrophage development.11 However, individual tumors may not express some or even any of these factors under certain conditions. In addition, high levels of lactic acid were recently found to be able to induce M2 macrophages.12 Again, peripheral tumor tissue may not include enough lactic acid due to high blood supply. Whether tumor cells use a common pathway to educate tumor-infiltrating macrophages into M2 phenotype remains elusive.
Normal cells as well as tumor cells are capable of releasing different types of vesicles for intercellular communication. In response to stimulation or apoptotic signals, cells may change their cytoskeletal structure, leading to plasma membranes encapsulating cytosolic elements. The latter is then expelled or downloaded into the extracellular space. These specialized subcellular vesicles are called MPs with sizes of 100 to 1000 nm in diameter. Functionally, MPs may act as vectors to deliver various messenger molecules such as enzymes, RNAs and even DNAs between cells.13-15 Regardless of their rapid proliferation, abundant tumor cells still undergo apoptosis in vivo, leading to the generation of tumor MPs. Moreover, a variety of signal molecules in tumor microenvironment can also stimulate tumor cells to produce MPs.16 In practice, both chemotherapy and radiotherapy can kill plenty of tumor cells, leading to the production of MPs. Therefore, the generation of tumor cell-derived MPs (T-MP) might be a very common phenomenon in tumor microenvironment. To date, how T-MPs mediate the communication between tumor cells and immune cells remains unclear. Previously, we found that T-MPs contain abundant tumor information including membrane molecules, nuclear histones, caspases, microRNAs and DNA; these T-MPs can be taken up by phagocytes such as macrophages.17 Given the presence of a large number of macrophages in tumor microenvironment, we wonder whether T-MPs can educate tumor-infiltrating macrophages once they are taken up by macrophages. In this study, we provide evidence that T-MPs may function as a common pathway to induce M2-type TAMs through cGAS/STING/TBK1/STAT6 pathway, promoting tumor growth, metastasis and cancer stem cell development.
Results
T-MPs induce the differentiation of macrophages toward M2 phenotype
Radiation is a conventional clinical means to induce tumor cell apoptosis, leading to the generation of T-MPs. In this study, we used UV radiation to treat H22 (hepatocarcinoma), B16 (melanoma) and CT-26 (colon cancer) murine tumor cell lines, respectively, to generate T-MPs. Consistent with previous reports,17 these T-MPs could be readily taken up by tumor cells. Interestingly, however, we found that macrophages had much higher efficiency to take up T-MPs, and such high efficiency were observed with a variety of macrophage subtypes, including primary bone-marrow-derived macrophages (M0), LPS-activated macrophages (M1), IL-4-activated macrophages (M2), Raw264.7 cell line as well as macrophages cultured under hypoxic condition (Fig. S1). As a result, macrophages' phenotypes were markedly influenced by taking up T-MPs. As shown in Fig. 1A and Fig. S2A, the expression of arginase 1, a typical marker for M2 phenotype, was significantly upregulated by T-MPs under both normoxic and hypoxic conditions in monocyte precursors, M0, M1 and M2 macrophages and primary peritoneal macrophages as well as Raw264.7 cell line. Meanwhile, other M2 markers, including CD206, IL-10 and CD301 were also upregulated in M0 and M2 macrophages by H22-MPs (Fig. 1B and Fig. S2B). By contrast, the expression of M1-related genes such as NOS2, TNF-α, IL12p35 and IL12p40 was found to be downregulated under either normoxic or hypoxic condition (Fig. 1C and Fig. S2C). The inhibition of TNF-α, IL-12 and iNOS expression at the protein levels were also confirmed by flow cytometric analysis (Fig. 1D and Fig. S2D) and Western blot (Fig. 1E). In addition to the above T-MPs from H22, MPs generated from irradiated LLC lung cancer, CT26 colon cancer and B16 melanoma tumor cells consistently showed their ability to induce M2 type macrophages (Fig. S3).
Figure 1.
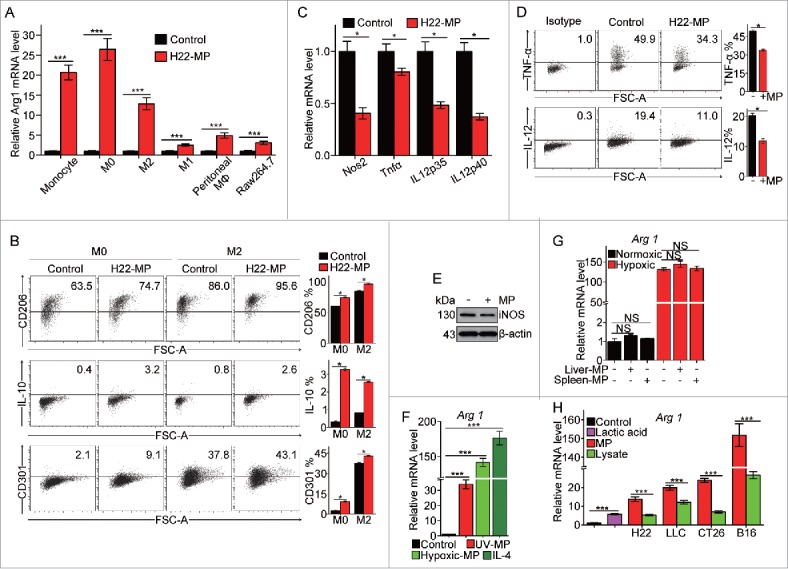
T-MPs polarize macrophages toward M2 phenotype under normoxia. (A) Real-time PCR analysis of arginase1 mRNA expression in monocytes, bone marrow-derived M0, M2 and M1 macrophages, primary peritoneal macrophages and Raw264.7 cell line treated with H22-MPs for 24 h under normoxic condition. (B) The expression of CD206, IL-10 and CD301 in M0 and M2 macrophages treated with H22-MPs for 24 h under normoxic condition was analyzed by flow cytometry. (C) The expression of NOS2, TNF-α, IL12p35 and IL12p40 in M1 macrophages treated with H22-MPs for 24 h under normoxic condition was detected by real-time PCR. (D) Flow cytometry analysis the expression of TNF-α and IL-12 in M1 macrophages treated with H22-MPs. (E) The expression of iNOS in M1 macrophages treated with H22-MPs for 24 h under normoxic condition was analyzed by protein gel blot. (F) M0 macrophages were treated with T-MPs from H22 tumor cells under UV irradiation or hypoxic condition. IL-4 was used as a positive control. (G) The arginase1 expression in M0 macrophages treated with MPs generated from liver and spleen cells under normoxic and hypoxic condition. MPs generated from liver and spleen cells were added to M0 macrophages for 24 h, and then the expression of arginase1 was analyzed by real-time PCR. (H) MPs or lysate derived from H22, LLC, CT26, B16 and 5 mM lactic acid were added to M0 macrophages for 24 h, the expression of arginase1 was detected by real-time PCR. All experiments were performed at least twice. The histogram bars represent the expression level of three biological replicates, displayed as means±s.e.m. *p <0.05, ***p <0.001. NS, not statistically significant.
Hypoxia generally exists in solid tumors and is a common pathway to induce tumor cell death, leading to the release of T-MPs. Therefore, we also prepared T-MPs from hypoxic tumor cells. Like UV-MPs, hypoxic-MPs were also capable of upregulating the expression of arginase 1 in M0 macrophages (Fig. 1F). As a comparison, in this study we also generated MPs from spleen or hepatocytes. Intriguingly, neither spleen-MPs nor hepatocyte-MPs could induce M2 macrophage development (Fig. 1G), suggesting that T-MP-mediated induction of M2 macrophage seems to be tumor-specific. To further characterize the effect of T-MPs on M2 macrophages, we compared the effect on M2 polarization between T-MPs and tumor lysates or lactic acid, two known factors inducing M2 macrophage polarization.12,18 Notably, although the addition of lysates from H22 (liver), LLC (lung), CT26 (colon) and B16 (melanoma) tumor cells or lactic acid resulted in the upregulation of arginase 1 expression in M0 macrophages, the corresponding T-MPs caused even higher expression of arginase 1 (Fig. 1H). Taken together, these data suggest that T-MPs may function as a common pathway to induce the development of macrophages toward M2 type.
T-MPs promote M2 macrophage proliferation but induce M1 apoptosis
Next, we explored the effect of T-MPs on the behaviors of macrophages such as proliferation and apoptosis. Although activated macrophages are considered as mature cells, they seem to still have the ability to proliferate.19,20 When T-MPs were incubated with IL-4-induced M2 macrophages, we found that the number of M2 macrophages was significantly increased after the addition of T-MPs (Fig. 2A). In line with this result, the number of immature M0 macrophages was also higher in the T-MP group, compared to the control group (Fig. 2A). The increase in cell number could not be explained by the less death of macrophages, since T-MPs treatment did not alter the apoptosis of either M0 or M2 macrophages (Fig. 2B). On the other hand, when we detected the cell cycle by flow cytometry, we found that T-MPs significantly increased the ratio of S phase and G1/G0 phase of those macrophages (Fig. 2C). In addition, similar effects of T-MPs on proliferation and apoptosis of M2 or M0 macrophages were also observed under hypoxic condition (Fig. S4A and S4B), suggesting that T-MPs are capable of promoting the proliferation of M0 and M2 macrophages. Unexpectedly, we found that T-MPs did not promote the proliferation of M1 macrophages (Fig. 2D and Fig. S5). By contrast, the addition of T-MPs significantly induced the apoptosis of M1 macrophages (Fig. 2E). Taken together, these data suggest that T-MPs promote M2 macrophage proliferation but induce M1 apoptosis.
Figure 2.

H22-MPs promote M2 macrophage proliferation but induce M1apoptosis. (A) 5 × 105 M0 and M2 macrophages were treated with H22-MPs and cell number was analyzed at 24 h and 48 h, respectively. (B, C) M0 and M2 macrophages were treated with H22-MPs for 24 h, and then cells were strained with FITC-Annexin V and PI for apoptosis analysis (B) or labeled with FITC-conjugated anti-BrdU antibody for cell cycle analysis (C). (D, E) 5 × 105 M1 macrophages were treated with H22-MPs and cell number was analyzed (D). Meanwhile, cells were strained with FITC-Annexin V and PtdIns for flow cytometric analysis of apoptosis (E). Data shown are representative of three reproducible experiments expressed as means±s.e.m. *p < 0.05, **p < 0.01, NS, not statistically significant.
T-MP-induced M2 macrophages promote tumor growth and metastasis
Next, we tested whether T-MP-induced M2 macrophages exerted the tumor-promoting effect. To this end, we first adapted an intramuscular tumor model to dissect the potential effect of T-MPs. 5 × 104 H22 tumor cells were injected to the right thigh muscle of mice. Six days later, 1 × 107 T-MPs were injected to the tumors at the peripheral and central sites, respectively, once per day for three times. On day 15, mice were sacrificed and the tumor weight was measured. We found that only peripheral but not central injection of T-MPs resulted in the promotion of tumor growth (Fig. 3A). Such different consequences could be reconciled by the finding that in this intramuscular tumor model, macrophages preferred to reside in the periphery rather than the center of tumor mass (Fig. S6); and this tumor-promoting effect was blunted by the depletion of macrophages by clodronate (Fig. 3B), suggesting that T-MPs may educate macrophages leading to tumor promotion and progression. To further confirm the tumor-promoting effect of T-MP-induced M2 macrophages, we incubated M0, M2 and Raw264.7 macrophages with T-MPs for 24 h, respectively. Thereafter, 2 × 105 H22 plus 6 × 104 treated or untreated macrophages were subcutaneously injected into BALB/c mice for tumor growth. As expected, either M0, M2 or Raw264.7 macrophages after education by T-MPs acquired the ability to promote tumor growth, compared to the control macrophages (Fig. 3C). In line with these in vivo data, M0 and M2 macrophages pre-treated with T-MPs significantly promoted H22 tumor cell growth in vitro (Fig. 3D).
Figure 3.
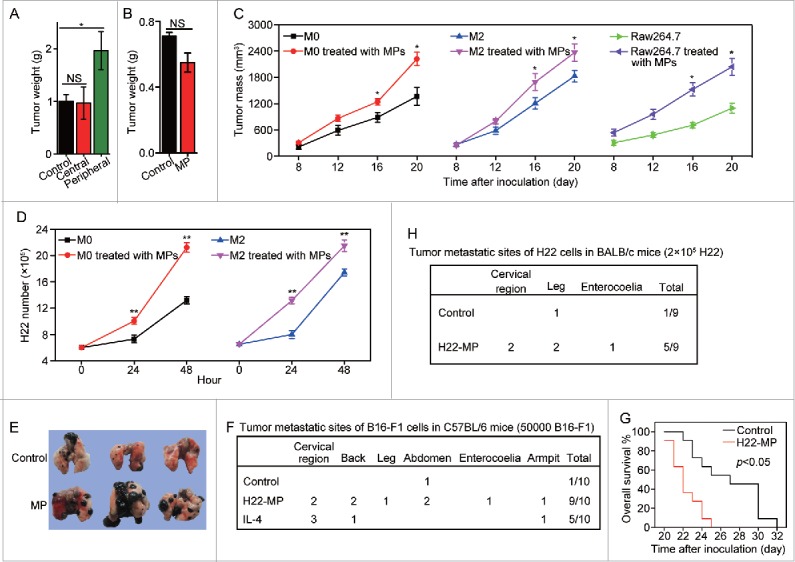
T-MP-educated macrophages promote tumor growth and metastasis. (A) 5 × 104 H22 tumor cells were injected to the right thigh muscle of mice. Six days later, 1×107 H22-MPs were injected to either peripheral or central site of tumors once per day for three times. On day 15, mice were sacrificed and the tumor weight was measured. (B) 5 × 104 H22 tumor cells were injected to the right thigh muscle of BALB/c mice on day 1. Clodronate (FormuMax Scientific Inc., CA) was i.p. injected into mice on day 5 (200 μL) and day 8 (100 μL), respectively. 1 × 107 H22-MPs were injected to peripheral site of tumors on day 7. On day 14, mice were sacrificed and the tumor weight was measured. (C) T-MP-educated macrophages promote tumor growth in vivo. M0, M2 and Raw264.7 macrophages were incubated with H22-MPs for 24 h, respectively. Thereafter, 2 × 105 H22 plus 6 × 104 treated or untreated macrophages were subcutaneously injected into BALB/c mice for tumor growth. The growth of tumor was monitored (n = 6, each group). (D) H22-MP-educated macrophages promote H22 tumor cell growth in vitro. M0 and M2 macrophages were incubated with H22-MPs for 24 h, and then H22 cells were added to the above group, H22 tumor cell number was analyzed at different time points. (E–G) H22-MP-educated macrophages promote melanoma metastasis. 5 × 104 B16 tumor cells plus 1 × 104 H22-MP-treated or untreated M0 macrophages were intravenously injected into C57BL/6 mice. Three weeks later, the formed tumor nodules in the lungs were examined (E), and apart from lung metastasis, metastatic tumors in various sites were analyzed (F). Meanwhile, the long-term survival of mice was analyzed by Kaplan–Meier analysis (G). (H) H22-MP-educated macrophages promote H22 tumor metastasis in BALB/c mice. 2 × 105 H22 tumor cells plus 6 × 104 H22-MP-treated M0 macrophages were intravenously injected into BALB/c mice. Metastatic tumors in various sites were analyzed. Data shown are representative of three reproducible experiments expressed as means±s.e.m. *p < 0.05, **p < 0.01, NS, not statistically significant.
In addition to tumor growth, M2 macrophages are also capable of promoting tumor metastasis. In this regard, we first used B16 melanoma lung metastasis as a model to test this possibility. 5 × 104 B16 tumor cells plus 1 × 104 T-MPs-treated or untreated M0 macrophages were intravenously injected into C57BL/6 mice. Three weeks later, much more and larger lung tumor nodules in T-MP group were observed, compared to the control group (Fig. 3E). More significantly, besides lung metastasis, tumors were also formed in other tissues or organs, including cervical region, back, leg, abdomen, enterocoelia and armpit (Fig. 3F). Consistently, the mice in T-MP group showed much shortened survival (Fig. 3G). In addition to B16 tumor cells, T-MP-induced macrophages also promoted H22 hepatocarcinoma tumor cell metastasis. 2 × 105 H22 tumor cells plus 6 × 104 T-MP-treated M0 macrophages were intravenously injected into BALB/c mice. Metastatic tumors in various sites such as cervix, thigh and peritoneal cavity were found (Fig. 3H). In line with these in vivo data, M0 macrophages pre-treated with T-MPs significantly promoted the growth and migration of B16 cells in vitro (Fig. S7A and S7B). Taken together, these data suggest that T-MPs-educated M2 macrophages promote tumor growth and metastasis.
T-MP-induced M2 macrophages promote tumor-repopulating cells for tumor growth and metastasis
Stem cell-like cancer cells (SCLCCs) are essential for tumor formation and metastasis.21 Recently, we developed a mechanical method to select and grow SCLCCs from the bulk population of tumor cells by culturing single tumor cells in 3D soft fibrin gels, and found that as few as 10 selected cells are sufficient to grow tumors in immunocompetent mice.22 We thus functionally define these soft fibrin gel-selected cells as tumor-repopulating cells (TRC).23 Using this method, we here further tested whether T-MP-induced M2 macrophages could promote TRC growth, thus facilitating tumor development and metastasis. To this end, M0 macrophages were treated with or without T-MPs for 24 h and the supernatants were used to culture H22 tumor cells in 3D fibrin gels for TRC growth. We found that T-MP-supernatants significantly increased the size and number of H22 TRC colonies (Fig. 4A–C). Consistently, the TRC expression of stemness-related genes, such as Bmi1, CD44, Hif1α, and c-myc was significantly upregulated in the T-MP group (Fig. 4D). To further confirm the promoting effect of T-MP-induced M2 macrophages on TRCs, we additionally tested B16 tumor cells. Consistently, T-MP-supernatant treatment also significantly increased the size and number of B16 TRC colonies (Fig. S8A–S8C), as well as the expression of SOX2, a crucial stemness gene of B16 tumor cells (Fig. S8D).24 To further dissect the effect of T-MP-induced M2 macrophages on TRCs, we also compared IL-4-educated macrophages, since IL-4 is a prototypic inducer of M2 macrophages. Intriguingly, the supernatants of IL-4-induced M2 macrophages showed little promoting effect on TRCs (Fig. 4A–C), suggesting that T-MP-induced macrophages release different factor(s) for TRC growth. Indeed, we found that T-MP-induced macrophages release milk-fat globule-epidermal growth factor (MFG-E8) and TGF-β1 (Fig. 4E), two cytokines that have the promoting effect on cancer stem cells.4,25 If we used siRNAs to knock down MFG-E8 or TGF-β1 (Fig. S9A and S9B), the above TRC-promoting effect was obliterated (Fig. 4F–H), suggesting that T-MPs educate macrophages to release MFGE8 and TGF-β1 for TRC growth. To validate the above in vitro data, H22 and B16 tumor cells, pre-treated with the supernatants of T-MP-induced macrophages, were subcutaneously injected to BALB/c and C57BL/6 mice, respectively. As a result, H22 and B16 tumor cells from pretreatment group showed much higher tumorigenic capability, respectively, compared with the control tumor cells (Fig. S10).
Figure 4.
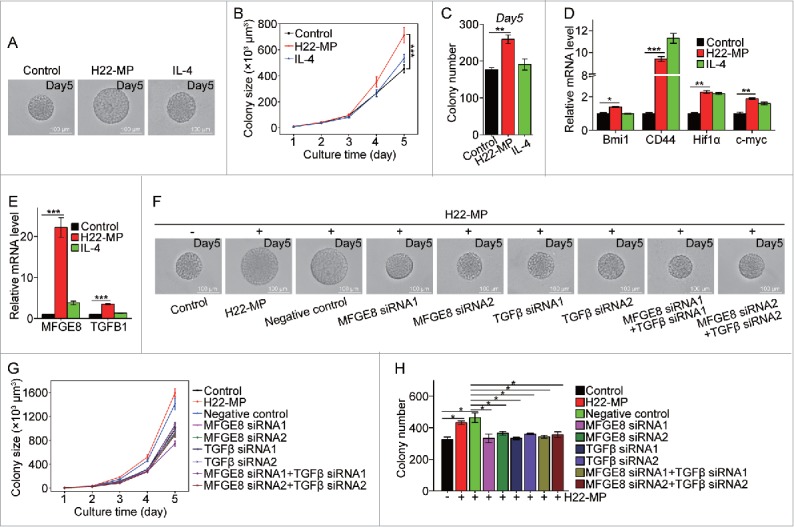
T-MP-educated macrophages promote TRCs growth via producing MFGE8 and TGF-β1. (A–C) H22-MPs-educated macrophages increased colony size and number of H22 TRCs. M0 macrophages were treated with or without H22-MPs for 24 h and the supernatants were used to culture H22 tumor cells in 3D fibrin gels for TRC growth. On day 5, the colony size of H22 TRCs was visualized by microscope (A) and analyzed by Image J software (B). Meanwhile, the colony number of H22 TRCs was analyzed by microscope (C). (D) The expression of Bmi1, CD44, Hif1α and c-myc in H22 TRCs was detected by real-time PCR. (E) The expression of MFGE8 and TGFB1 in H22-MPs-treated or untreated macrophages was analyzed by real-time PCR. (F–H) Downregulating the expression of MFGE8 and TGFB1 in H22-MPs-educated macrophage inhibits H22 TRCs colony size and number. The scale bar represents 100 μm. Data shown are representative of three reproducible experiments expressed as means±s.e.m. *p < 0.05, **p < 0.01, *** p < 0.001.
Development of M2 type macrophages by T-MPs is mediated through cGAS/STING pathway
Next, we investigated the pathway through which T-MPs educated macrophages toward M2 type. Cytokines IL-4 and IL-13 are known as the prototypical inducer of M2 macrophages through STAT6 signaling pathway.26 Here, we found that the addition of T-MPs induced the phosphorylation of STAT6 in M0 macrophages, in parallel with the upregulation of the expression of arginase 1 (Fig. 5A). On the other hand, the knockdown of STAT6 by siRNAs decreased the expression of arginase 1 (Fig. 5B). Besides STAT6, STAT3 is another member of STATs that is capable of inducing M2 macrophages.26 Consistently, we found that phosphorylation of STAT3 was enhanced by T-MP treatment (Fig. 5A). Recently, we identified that T-MPs included mitochondrial and nuclear DNA fragments that activated the cGAS/STING signaling pathway in dendritic cells.27 We thus examined the involvement of TBK1, because activated STING can recruit TBK1 and TBK1 has been reported as one of the upstream signal molecules of STAT6.28 As expected, the phosphorylation of TBK1 was enhanced in T-MP-treated M0 macrophages, and more importantly, knocking down TBK1 resulted in decreases in the phosphorylation of STAT6 and the expression of arginase 1 (Fig. 5C–D). In parallel, using STING or cGAS siRNAs consistently resulted in the decreased expression of arginase 1 (Fig. 5E–F). Taken together, these data suggest that activation of cGAS/STING/TBK1/STAT6 pathway is involved in T-MP-induced M2 macrophage development.
Figure 5.

T-MPs polarize M2 macrophages through the cGAS/STING pathway. (A) The expression of arginase1 and phosphorylation of TBK1, STAT6 and STAT3 in M0 macrophages treated with H22-MPs were detected by Western blot. (B) The expression of STAT6 and arginase1 in M0 macrophages transfected with STAT6 siRNAs was detected by real-time PCR. (C, D) The expression of TBK1, arginase1 or STAT6 in M0 macrophages transfected with TBK1 siRNAs was analyzed by real-time PCR (C) and Western blot (D). (E) The expression of STING and arginase1 in M0 macrophages transfected with STING siRNAs was analyzed by real-time PCR. (F) The expression of cGAS and arginase1 in M0 macrophages transfected with cGAS siRNA was analyzed by real-time PCR. Data shown are representative of three independent experiments expressed as means ± s.e.m. *p < 0.05, ***p < 0.001.
Human tumor cell-derived microparticles (hT-MPs) induce the differentiation of PMA-stimulated THP-1 cells toward M2 phenotype
Finally, we investigated whether hT-MPs could induce macrophage to differentiate into M2 phenotype. PMA-stimulated THP-1 cells were used as a macrophage model, since THP-1 is a human monocytic leukemia cell line that can be induced to differentiate to macrophage-like cells by PMA stimulation.29 The expression of arginase 1 in PMA-treated THP-1 cells was strikingly upregulated by hT-MPs from A2780 ovarian, A549 lung or SMMC7721 liver cancer cell line (Fig. 6A). Consistent with THP-1 cell line, human primary monocyte-derived macrophages also highly upregulated the expression of arginase 1 upon hT-MP stimulation (Fig. S11). Notably, these hT-MPs seemed to possess stronger ability to stimulate the expression of arginase 1, compared to tumor lysates or lactic acid (Fig. 6B). To verify the effect of hT-MPs on TRCs, the supernatants from PMA-stimulated THP-1 cells, treated with SMMC7721-MPs or human IL-4, were used to culture SMMC7721 tumor cells in 3D fibrin gels for TRC growth. We found that hT-MP-supernatants but not IL-4-supernatants significantly increased the size of SMMC7721 TRC colonies (Fig. 6C-D). Moreover, 1 × 106 SMMC7721 tumor cells plus 3 × 105 SMMC7721-MP-treated PMA-stimulated THP-1 cells were subcutaneously injected to nude mice (n = 10 per group). On day 35, the SMMC7721-MP group formed more subcutaneous tumors compared with control (Fig. 6E).
Figure 6.
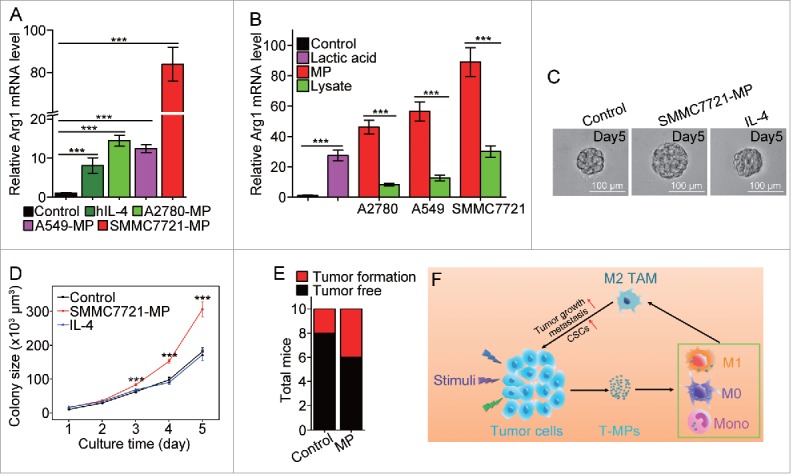
SMMC7721-MPs induce the differentiation of PMA-stimulated THP-1 cells toward M2 phenotype. (A) THP-1 cells treated with 100 ng/mL PMA for 48 h were incubated with hIL-4, A2780-MPs, A549-MPs, SMMC7721-MPs for 24 h and the expression of arginase1 was detected with real-time PCR. (B) The expression of arginase 1 in PMA-stimulated THP-1 cells treated with A2780-MPs, A2780-lysate, A549-MPs, A549-lysate, SMMC7721-MPs, SMMC7721-lysate and 5 mM lactic acid was analyzed with real-time PCR. (C, D) SMMC7721-MPs-educated PMA-stimulated THP-1 cells promote SMMC7721 TRCs colony size. PMA-stimulated THP-1 cells were treated with or without SMMC7721-MPs for 24 h and the supernatants were used to culture SMMC7721 tumor cells in 3D fibrin gels for TRC growth. On day 5, the colony size of SMMC7721 TRCs was visualized under microscope (C) and analyzed by Image J software (D). (E) 1 × 106 SMMC7721 tumor cells plus 3 × 105 SMMC7721-MPs-treated or untreated PMA-stimulated THP-1 cells were subcutaneously injected to nude mice (n = 10 per group). On day 35, the tumor formation was obtained. (F) T-MPs act as a general mechanism to polarize macrophages into M2 type tumor-associated macrophages. In tumor microenvironment, a variety of stimuli (apoptotic signals, live stimulatory signals, chemo- and radio-therapy) induce tumor cells to release T-MPs, which are then taken up by monocytes, M0 or even M1 macrophages. In turn, the entered T-MPs educate monocytes, M0 or M1 macrophages into M2 like TAMs, leading to tumor growth, metastasis as well as cancer stem cell development. Data shown are representative of three independent experiments expressed as means±s.e.m. ***p < 0.001.
Discussion
Tumor-infiltrating macrophages in solid tumors are widely educated into M2-type TAMs so to promote tumor immune escape, angiogenesis and metastasis as well as cancer stem cell development.1-5 However, the mechanism through which macrophages are educated into M2 TAMs remains largely incompletely understood. The present study showed that tumor cell-derived MPs function as a common pathway to generate M2 TAMs through cGAS/STING/TBK1/STAT6 pathway.
MPs can be generated under both pathological and physiological conditions. For instance, MPs released by neutrophils or platelets commonly exist in the peripheral blood.30-32 However, MPs are also released from damaged or inflamed tissues into the blood, providing potential strategies on disease diagnosis.33,34 Cytoskeletal alteration is a key event involved in the generation of MPs through cellular membrane bubbling and shedding. Therefore, signals that cause the cytoskeletal change might be potential to produce MPs by cells. For instance, the activated TLR4 signaling can be transduced to cellular cytoskeletons, leading to tumor cell releasing MPs.16 In fact, various damage-associated molecular patterns always commonly exist in the tumor microenvironment that recognize and activate tumor TLRs.35 In addition, hypoxia, nutrient deficiency, and apoptotic signals may induce tumor cells to produce MPs in tumor microenvironment. In practice, both chemotherapy and radiotherapy cause abundant tumor cell death, leading to their releasing MPs. Therefore, the existence of T-MPs in tumor microenvironment might be fundamental in tumor biology.
In this study, we found that T-MPs not only promoted M0 macrophages but also monocytes to differentiate into M2 phenotype. Notably, although tumor lysates and lactic acid are capable of inducing M2 macrophages, this capability appeared not to be as strong as that of T-MPs. Moreover, the M2 macrophage-promoting effect of T-MPs can be further amplified, as T-MPs facilitate the proliferation of M0 and M2 macrophages but inducing the apoptosis of M1 macrophages. Although we did not dissect the molecular mechanism underlying this unusual phenomenon in the present study, we speculate that lysosomes might be involved in this process. Lysosomes are conventionally thought as waste bags, but recent studies reveal that lysosomes also play critical roles in regulating cellular homeostasis, including cell growth, metabolism, drug resistance and apoptosis,36-40 and all these regulatory processes seem to be associated with the pH value of lysosomes.41,42 Intriguingly, when we measured the lysosomal pH value in another study, we found that M2 macrophages had much lower pH value, compared to M1 macrophages. On the other hand, when we used T-MPs to treat macrophages, we found that T-MPs were taken up by macrophages through phagocytosis pathway and entered the lysosomes, where T-MPs decreased the lysosomal pH value of both M1 and M2 macrophages. Given the different lysosomal pH values between M1 and M2 macrophages, we propose that in response to T-MP stimulation M2 macrophages might be suitable to the lower lysosomal pH value for degrading wastes; however, M1 macrophages are not suitable to such decreased lysosomal pH value and enter apoptotic pathway. This might partially explain why T-MPs might be a common pathway to induce M2 type TAMs but negatively regulate M1 macrophages in tumor microenvironment. Notably, tumor-infiltrating macrophages as the major immune cell type in tumor microenvironment may have multiple cellular sources. Monocytes might be a major contributor, which can readily differentiate into macrophages.43,44 However, myeloid-derived suppressor cells (MDSC) and local resident macrophages may also be the cellular source of tumor-infiltrating macrophages.45,46 Here, we should clarify that T-MP-educated M2-type macrophages are different from MDSCs. Although macrophages and MDSCs are derived from the common myeloid precursor cells and both cell types can express F4/80, Gr-1, arginase 1 and others, T-MP-educated macrophages still show some difference from MDSCs. First, our macrophages adhere on the plate; however, MDSCs grow in a suspension manner; secondly, the density of MDSCs are lower compared to macrophages. When the centrifugation on a Percoll density gradient was performed, different cell bands were formed between 40% and 50% as fraction 1, between 50% and 60% as fraction 2, and between 60% and 70% as fraction 3. We found that MDSCs were at the fraction 2 and T-MP-educated macrophages were at the fraction 3, which was consistent with the previous study.47 Despite the differences between MDSCs and M2 macrophages, whether T-MPs are involved in the differentiation of MDSCs toward M2 macrophages remains unclear, but is worthy of further investigation, considering the pivotal role of MDSCs in tumor progression.
The molecular mechanism through which T-MPs induce the differentiation of macrophages/monocytes toward M2 macrophages is a key point of this work. Recent studies highlight the cGAS/STING pathway as a DNA sensor to induce the type I interferon production.48-50 Upon binding DNA sequence, the conformation of cGAS is changed and its enzymatic activity is subsequently activated, leading to catalyzing ATP and GTP to cyclic dinucleotide cGAMP. The latter then binds to and activates ER membrane-localized STING for the recruitment of downstream signal molecules.48,49,51 Our previous study showed that T-MPs contain both nuclear and mitochondrial DNA fragments and effectively activate dendritic cells through cGAS/STING pathway.27 In the present study, we further demonstrated that T-MPs activate the cGAS/STING pathway in macrophages. The key signal transducer downstream of STING is TBK1, a central IKK family member of serine/threonine kinase essential for IFN induction. Upon engagement with STING on ER membrane, TBK1 undergoes dimerization and autophosphorylation leading to the acquisition of kinase activity which is responsible for the phosphorylation of IRF3 and IRF7, the key transcriptional activators driving type I IFN expression. It has been reported that under certain conditions, activated TBK1 results in the phosphorylation of Stat6, leading to homodimerization and nuclear translocation of Stat6.28 However, how to regulate these activities remains unclear and requires further investigation.
In addition to STAT6, we also found STAT3 was phosphorylated in this study, which might be due to the autocrine or paracrine effect of IL-10, released from T-MP-induced M2 macrophages. Given that STAT6 and STAT3 are the two prototypical transcription factors to induce M2 macrophages,26 this study thus clearly identifies the pathway through which T-MPs induce the development of M2 type TAMs. Nevertheless, several issues still remain to be addressed regarding the activation of cGAS/STING pathway. One is whether T-MP-contained DNAs are processed in endosome/lysosome system and released to the cytosol for cGAS recognition. Another is whether cGAS also exists in endosome/lysosome system, where it directly catalyzes the synthesis of cGAMP and the latter is then released to the cytosol. Related to these issues, this study found that the induction of M2 macrophages by MPs seems to be tumor specific. When we used the same protocol to prepare normal splenocyte- or liver-cell-derived MPs, we found that these normal cell-derived MPs did not cause the M2 macrophage polarization. This intriguing phenomenon might be probably due to the different modification of DNAs between normal and cancerous cells, that is, only the tumor-modified but not the normal cell DNA sequence can be effectively recognized by cGAS. This hypothesis is attracting and further experiments are needed to establish such connection. Regarding to the cGAS/STING pathway, another issue is whether this pathway may regulate the expression of MFGE8 and TGF-β1, since T-MP-educated macrophages upregulate the expression of these two cytokines. In this study, we did not address this question; however, the underlying mechanism is worthy of investigation. Although in this study we found that T-MP-educated macrophages may use MFGE8 and TGF-β1 to mediate the development of TRCs, whether such educated macrophages also use other means to activate stemness remains unclear. Previous study showed that human ovarian cancer MDSCs may activate cancer stemness such as SOX2 activation,52 Given the similarities such as intracellular signal molecules IDO, arginase 1 and STAT3 shared by MDSCs and M2 macrophages, T-MP-educated macrophages probably are similar to MDSCs to induce cancer stemness and metastasis. In addition, both MDSCs and M2 macrophages express M-CSFR (CD115), which mediates the important functions of both cell types.47,53 Together, T-MP-educated macrophages might use various means to promote cancer stemness and metastasis.
The highly heterogenic and plastic features of M2 macrophages implicate that the combination of phenotypic markers and functional analysis might be a better way to define M2 macrophages.54,55 In this study, we found that T-MP-induced M2 macrophages effectively promote tumor cell growth, migration, metastasis, as well as cancer stem cell development. These findings might be very significant in explaining issues on pre-metastatic niches and targeting therapies. It has been hypothesized that primary tumor has the capability to alter the tissue microenvironment of target organs such as the lungs before primary tumor cells arrive at the metastatic site.56 To date, amounting evidence has been provided to support this hypothesis, however the underlying mechanism remain elusive. On the basis of our present findings, we speculate that T-MPs might operate to remodel the pre-metastatic niches through continuous steps: primary tumor releases T-MPs; T-MPs cross the blood vessel and circulate to the pre-metastatic site; local macrophages then take up the circulated T-MPs and are educated into M2 macrophages; and M2 macrophages remodel the pre-metastatic niche by releasing various factors that inhibit antitumor immunity but promote cancer stem cell development. In addition to pre-metastatic niches, our present findings also provide a new perspective on current TKI targeting therapies. Blockade of EGFR or HER-2 may generate an ideal treatment result at the beginning; however, once drug resistance is generated or the recurrence occurs, an accelerated development of the disease are usually observed in treated cancer patients.57 Similarly, we speculate that tumor cell apoptosis triggered by target drugs results in the release of T-MPs, concomitant with the attraction of macrophages. As a result, macrophages take up T-MPs but are educated into M2-type TAMs so to favor the remnant tumor cells growth and metastasis. The above two issues are intriguing and require future investigation.
In summary, the data in this study clearly show that T-MPs by virtue of their intrinsic biological information, can act as a general mechanism to educate macrophages into M2-type TAM, leading to tumor growth, metastasis as well as cancer stem cell development (Fig. 6F). This study provides new insight into crucial roles of T-MPs in remodeling of tumor microenvironment.
Materials and methods
Mice and cell lines
Female BALB/c, C57BL/6 and nude mice, 6- to 8-week-old, were purchased from the Center of Medical Experimental Animals of Hubei Province (Wuhan, China) for studies approved by the Animal Care and Use Committee of Tongji Medical College. Murine cell lines H22 (hepatocarcinoma), B16 (melanoma), LLC (Lewis lung carcinoma), CT-26 (colon cancer), murine macrophage cell line Raw264.7 and human monocytic leukemia cell line THP-1 were purchased from the China Center for Type Culture Collection (Wuhan, China) and cultured according to the guidelines given.
Preparation of T-MPs
Tumor cells were exposed to ultraviolet radiation (300 J/m2) for 1 h, and 12 h later, supernatants were used for UV-induced MP isolation as described previously.17 For hypoxia-induced MPs, tumor cells were cultured under 0.1% oxygen hypoxic incubator for 24 h, and supernatants were used for MP isolation. Briefly, supernatants were centrifuged at 1,300 rpm for 10 min to remove whole cells and then centrifuged at 5,000 g for 10 min and 14,000 g for 2 min to remove debris. The supernatants were further centrifuged at 14,000 g for 60 min to pellet MPs. The MPs were washed three times and suspended in culture medium for the following experiments.
Polarization of mouse bone marrow-derived macrophages
Bone marrow cells isolated from femurs of mice were cultured for 5 d in the presence of 20 ng/mL recombinant mouse macrophage colony-stimulating factor (M-CSF, PeproTech) in complete RPMI-1640 medium containing 10% fetal bovine serum (FBS), 10 mM glucose, 2 mM L-glutamine, 100 U/mL penicillin-streptomycin. On day 6, M0 macrophages were harvested and then were stimulated for 24 h with 20 ng/mL IL-4 (PeproTech) or 100 ng/mL LPS (Sigma) plus 10 ng/mL IFNγ (PeproTech) for the generation of M2 or M1 macrophages.
Preparation of primary peritoneal macrophages
Mouse peritoneal macrophages were harvested by peritoneal lavage. Cold PBS was injected into the peritoneal cavity and extracted after gentle agitation. The peritoneal cell suspension was centrifuged at 1300 rpm, and the cell pellet was mixed with 2 mL Red blood cell lysis Buffer for 5 min at room temperature. After washing, the cells were cultured on six-well plate for 2 h. The adhesion cells were collected as peritoneal macrophages.
Isolation of mouse monocytes
Mice peripheral blood cells were obtained from anesthetizing mice heart. Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll Paque density gradient separation solution (GE healthecare). Monocytes were purified from PBMCs with mouse CD14 MicroBeads (MACS).
Isolation of human monocytes
Human PBMCs were isolated from human peripheral blood using density gradient separation. Monocytes were purified by human CD14 MicroBeads (MACS) and then were cultured in complete RPMI 1640 medium containing 20 ng/mL recombinant human M-CSF (PeproTech) for the induction of macrophages. 7 d later, human macrophages were harvested and stimulated with T-MPs or 20ng/mL human IL-4 for M2 differentiation.
Three-dimensional fibrin gel culture of tumor cells
Salmon fibrinogen and thrombin were purchased from Searun Holdings. Detailed methods are described as previously.22 Tumor cells were detached from the standard culture conditions and suspended in MEM (10% FBS) and cell density was adjusted to 104 cells/mL. Fibrinogen was diluted into 2 mg/mL with T7 buffer (pH 7.4, 50 mM Tris, 150 mM NaCl). 1:1 fibrinogen and cell solution mixture was made, resulting in 1 mg/mL fibrinogen and 5000 cells/mL in the mixture. 250 µL cell/fibrinogen mixtures were seeded into each well of 24-well plate and mixed well with pre-added 5 µL thrombin (0.1 U/µL) for culture under 37°C condition.
Gene silencing experiments
siRNAs targeting mouse cGAS (siRNA: GGATTGAGCTACAAGAATA), STING (siRNA1#: GGAGCCGAAGACTGTACAT; siRNA2#: CCACAGACGGAAACAGTTT), TBK1 (siRNA1#: GGAAGTGTCCAAGTATCAA; siRNA2#: GCAGAACGCAGACTAGCTT), STAT6 (siRNA1 #:GCTGATCATTGGCTTTATT; siRNA2#: CCTGCAACCATCTCCTTAT), MFGE8 (siRNA1 #:CGGAGTACCTGAAGACCTT; siRNA2#: GTATATGAGGAGCAAGGAA), TGFB1 (siRN A1#: CCGCAACAACGCCATCTAT; siRNA2#: CCAGAAATATAGCAACAAT) and negative control siRNAs were purchased from RiboBio (Guangzhou, China). siRNA (50 nM) was transfected into macrophages using lipofectamine RNAiMax (Invitrogen) according to the manufacturer's instruction.
Real-time quantitative PCR
Total RNA extracted from cells with TRIzol reagent (Invitrogen) was used for real-time quantitative PCR. Total RNA (2μg) was reverse-transcribed into cDNA using the ReverTra Ace qPCR RT Kit (Toyobo). Real-time PCR was performed with the THUNDERBIRD SYBR qPCR Mix (Toyobo) on a Bio-Rad CFX Connect. The mRNA levels were normalized to β-actin. The primer sequences are shown in Table S1.
Western blot
Cell lysates and prestained molecular weight marker were separated by SDS-PAGE, followed by transfer onto nitrocellulose membranes. The membranes were blocked in TBST (Tris-buffered saline with 0.5% TritonX-100) containing 5% bull serum albumin (BSA) and probed with specific anti-Arginase-1 (1:1000 dilution, Cell Signaling Technology), anti-iNOS (1:1000 dilution, Abcam), anti-Phospho-TBK (Ser172) (1:1000 dilution, Cell Signaling Technology), anti-TBK (1:1000 dilution, Cell Signaling Technology), anti-Phospho-STAT6 (Tyr641) (1:500 dilution, Abcam), anti-STAT6 (1:1000 dilution, Abcam), anti-Phospho-STAT3 (Tyr705) (1:1000 dilution, Cell Signaling Technology), anti-STAT3 (1:1000 dilution, Cell Signaling Technology) or anti-β-actin (1:3000 dilution, Sigma) overnight at 4°C. The membranes were washed three times and incubated with horseradish peroxidase-conjugated secondary antibodies. The immunoreactivity was visualized by enhanced chemiluminescence according to the manufacturer's protocol (ECL Kit, Pierce).
Flow cytometry
Cells were kept at 4°C and nonspecific binding was blocked with anti-CD16/32 (eBiosciences) before cell surfaces were stained with APC-conjugated anti-CD206 (Biolegend) or PE-conjugated anti-CD301 (Biolegend). For intracellular staining, live cells were first fixed with the fixation buffer, then treated with Permeabilization buffer and stained with PE-conjugated anti-IL-10 (eBiosciences), APC-conjugated anti-TNF-α (eBiosciences) and PE-conjugated anti-IL-12 (eBiosciences). For cell cycle analysis, cells were labeled with FITC-conjugated anti-BrdU antibody according to the manufacturer's instruction (FITC BrdU Flow Kit, BD Biosciences). Data were acquired on an Accuri C6 (BD Biosciences) and analyzed with FlowJo software.
Trans-well migration assay
1 × 105 B16-F1 cells were loaded in the upper chambers (8 μm pore size, Costar) of six-well transewell plates, and the lower chambers were seeded with M0 macrophages treated or untreated with H22-MPs. Migration was allowed for 6 h at 37°C, and cells were fixed with methanol and stained with 1% crystal violet. Cells remaining on the upper chamber side of the filter were removed with cotton swabs. The number of migrated cells was determined by counting stained cell from multiple randomly selected microscopic visual fields.
Animal experiments
2 × 105 H22 plus 6 × 104 macrophages treated or untreated with T-MPs were subcutaneously injected into the right flank of BALB/c to generate subcutaneous tumor model. Subcutaneous tumor growth was monitored by measuring the length (L) and width (W) of tumors using vernier calipers, and the volume (V) of the tumor was calculated by formula V=(L × W2)/2. In tumor metastasis model, 5 × 104 B16 tumor cells plus 1 × 104 T-MP-treated or untreated M0 macrophages were intravenously injected into C57BL/6 mice. Or 2 × 105 H22 tumor cells plus 6 × 104 T-MP-treated M0 macrophages were intravenously injected into BALB/c mice. In tumorigenic model, 5 × 103 B16 or 1 × 104 H22 or 1 × 106 SMMC7721 and corresponding macrophages were subcutaneously injected into BALB/c or C57BL/6 or nude mice. The tumor growth of injected mice was carefully monitored every day.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
RM, TJ, DC, WD, HZ, XY, JM, XL, YZ and YL designed and performed the experiments and analyzed the data. TJ, GS and XQ analyzed the data. RM and BH designed the experiments, interpreted the results and wrote the manuscript.
Funding
This work was supported by National Basic Research Program of China (2014CB542100), National Science Fund for Distinguished Young Scholars of China (81225021), National Natural Science Foundation of China (81502415, 81472653), Special Fund of Health Public Welfare Profession of China (201302018), and Soundny (Sheng-Qi-An) Biotech (Wuhan, China).
References
- 1.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 2004; 4:71-8; PMID:14708027; http://dx.doi.org/ 10.1038/nrc1256 [DOI] [PubMed] [Google Scholar]
- 2.Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015; 27:462-72; PMID:25858805; http://dx.doi.org/ 10.1016/j.ccell.2015.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013; 23:277-86; PMID:23518347; http://dx.doi.org/ 10.1016/j.ccr.2013.02.013 [DOI] [PubMed] [Google Scholar]
- 4.Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H, Yagita H, Takaoka A, Tahara H. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci U S A 2011; 108:12425-30; PMID:21746895; http://dx.doi.org/ 10.1073/pnas.1106645108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okuda H, Kobayashi A, Xia B, Watabe M, Pai SK, Hirota S, Xing F, Liu W, Pandey PR, Fukuda K et al.. Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer Res 2012; 72:537-47; PMID:22113945; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, Balkwill FR. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med 2008; 205:1261-8; PMID:18490490; http://dx.doi.org/ 10.1084/jem.20080108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang W, Zhu XD, Sun HC, Xiong YQ, Zhuang PY, Xu HX, Kong LQ, Wang L, Wu WZ, Tang ZY. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res 2010; 16:3420-30; PMID:20570927; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-2904 [DOI] [PubMed] [Google Scholar]
- 8.Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, Erba E, Uboldi S, Zucchetti M, Pasqualini F et al.. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013; 23:249-62; PMID:23410977; http://dx.doi.org/ 10.1016/j.ccr.2013.01.008 [DOI] [PubMed] [Google Scholar]
- 9.Ong SM, Tan YC, Beretta O, Jiang D, Yeap WH, Tai JJ, Wong WC, Yang H, Schwarz H, Lim KH et al.. Macrophages in human colorectal cancer are pro-inflammatory and prime T cells towards an anti-tumour type-1 inflammatory response. Eur J Immunol 2012; 42:89-100; PMID:22009685; http://dx.doi.org/ 10.1002/eji.201141825 [DOI] [PubMed] [Google Scholar]
- 10.Qin Z, Blankenstein T. CD4+ T cell–mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity 2000; 12:677-86; PMID:10894167; http://dx.doi.org/ 10.1016/S1074-7613(00)80218-6 [DOI] [PubMed] [Google Scholar]
- 11.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012; 122:787-95; PMID:22378047; http://dx.doi.org/ 10.1172/JCI59643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM et al.. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014; 513:559-63; PMID:25043024; http://dx.doi.org/ 10.1038/nature13490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mause SF, Weber C. Microparticles: protagonists of a novel communication network for intercellular information exchange. Circ Res 2010; 107:1047-57; PMID:21030722; http://dx.doi.org/ 10.1161/CIRCRESAHA.110.226456 [DOI] [PubMed] [Google Scholar]
- 14.Tual-Chalot S, Leonetti D, Andriantsitohaina R, Martinez MC. Microvesicles: intercellular vectors of biological messages. Mol Interv 2011; 11:88-94; PMID:21540467; http://dx.doi.org/ 10.1124/mi.11.2.5 [DOI] [PubMed] [Google Scholar]
- 15.Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 2009; 9:581-93; PMID:19498381; http://dx.doi.org/ 10.1038/nri2567 [DOI] [PubMed] [Google Scholar]
- 16.Li D, Jia H, Zhang H, Lv M, Liu J, Zhang Y, Huang T, Huang B. TLR4 signaling induces the release of microparticles by tumor cells that regulate inflammatory cytokine IL-6 of macrophages via microRNA let-7b. Oncoimmunology 2012; 1:687-93; PMID:22934260; http://dx.doi.org/ 10.4161/onci.19854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang K, Zhang Y, Zhang H, Xu P, Liu J, Ma J, Lv M, Li D, Katirai F, Shen GX et al.. Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat Commun 2012; 3:1282; PMID:23250412; http://dx.doi.org/ 10.1038/ncomms2282 [DOI] [PubMed] [Google Scholar]
- 18.Hiwatashi K, Tamiya T, Hasegawa E, Fukaya T, Hashimoto M, Kakoi K, Kashiwagi I, Kimura A, Inoue N, Morita R et al.. Suppression of SOCS3 in macrophages prevents cancer metastasis by modifying macrophage phase and MCP2/CCL8 induction. Cancer Lett 2011; 308:172-80; PMID:21624767; http://dx.doi.org/ 10.1016/j.canlet.2011.04.024 [DOI] [PubMed] [Google Scholar]
- 19.Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, MacDonald AS, Allen JE. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 2011; 332:1284-8; PMID:21566158; http://dx.doi.org/ 10.1126/science.1204351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science 2013; 342:1242974; PMID:24264994; http://dx.doi.org/ 10.1126/science.1242974 [DOI] [PubMed] [Google Scholar]
- 21.Medema JP. Cancer stem cells: the challenges ahead. Nat Cell Biol 2013; 15:338-44; PMID:23548926; http://dx.doi.org/ 10.1038/ncb2717 [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Tan Y, Zhang H, Zhang Y, Xu P, Chen J, Poh YC, Tang K, Wang N, Huang B. Soft fibrin gels promote selection and growth of tumorigenic cells. Nat Mater 2012; 11:734-41; PMID:22751180; http://dx.doi.org/ 10.1038/nmat3361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan Y, Tajik A, Chen J, Jia Q, Chowdhury F, Wang L, Chen J, Zhang S, Hong Y, Yi H et al.. Matrix softness regulates plasticity of tumour-repopulating cells via H3K9 demethylation and Sox2 expression. Nat Commun 2014; 5:4619; PMID:25099074; http://dx.doi.org/ 10.1038/ncomms5619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santini R, Pietrobono S, Pandolfi S, Montagnani V, D'Amico M, Penachioni JY, Vinci MC, Borgognoni L, Stecca B. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene 2014; 33:4697-708; PMID:24681955; http://dx.doi.org/ 10.1038/onc.2014.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan QM, Jing YY, Yu GF, Kou XR, Ye F, Gao L, Li R, Zhao QD, Yang Y, Lu ZH et al.. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett 2014; 352:160-8; PMID:24892648; http://dx.doi.org/ 10.1016/j.canlet.2014.05.008 [DOI] [PubMed] [Google Scholar]
- 26.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest 2007; 117:1155-66; PMID:17476345; http://dx.doi.org/ 10.1172/JCI31422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H, Tang K, Zhang Y, Ma R, Ma J, Li Y, Luo S, Liang X, Ji T, Gu Z et al.. Cell-free tumor microparticle vaccines stimulate dendritic cells via cGAS/STING signaling. Cancer Immunol Res 2015; 3:196-205; PMID:25477253; http://dx.doi.org/ 10.1158/2326-6066.CIR-14-0177 [DOI] [PubMed] [Google Scholar]
- 28.Chen H, Sun H, You F, Sun W, Zhou X, Chen L, Yang J, Wang Y, Tang H, Guan Y et al.. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell 2011; 147:436-46; PMID:22000020; http://dx.doi.org/ 10.1016/j.cell.2011.09.022 [DOI] [PubMed] [Google Scholar]
- 29.Qin Z. The use of THP-1 cells as a model for mimicking the function and regulation of monocytes and macrophages in the vasculature. Atherosclerosis 2012; 221:2-11; PMID:21978918; http://dx.doi.org/ 10.1016/j.atherosclerosis.2011.09.003 [DOI] [PubMed] [Google Scholar]
- 30.Daniel L, Fakhouri F, Joly D, Mouthon L, Nusbaum P, Grunfeld JP, Schifferli J, Guillevin L, Lesavre P, Halbwachs-Mecarelli L. Increase of circulating neutrophil and platelet microparticles during acute vasculitis and hemodialysis. Kidney Int 2006; 69:1416-23; PMID:16531979; http://dx.doi.org/ 10.1038/sj.ki.5000306 [DOI] [PubMed] [Google Scholar]
- 31.Pitanga TN, de Aragao Franca L, Rocha VC, Meirelles T, Borges VM, Goncalves MS, Pontes-de-Carvalho LC, Noronha-Dutra AA, dos-Santos WL. Neutrophil-derived microparticles induce myeloperoxidase-mediated damage of vascular endothelial cells. BMC Cell Biol 2014; 15:21; PMID:24915973; http://dx.doi.org/ 10.1186/1471-2121-15-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siljander P, Carpen O, Lassila R. Platelet-derived microparticles associate with fibrin during thrombosis. Blood 1996; 87:4651-63; PMID:8639834 [PubMed] [Google Scholar]
- 33.Kanazawa S, Nomura S, Kuwana M, Muramatsu M, Yamaguchi K, Fukuhara S. Monocyte-derived microparticles may be a sign of vascular complication in patients with lung cancer. Lung Cancer 2003; 39:145-9; PMID:12581566; http://dx.doi.org/ 10.1016/S0169-5002(02)00441-5 [DOI] [PubMed] [Google Scholar]
- 34.Kim HK, Song KS, Park YS, Kang YH, Lee YJ, Lee KR, Kim HK, Ryu KW, Bae JM, Kim S. Elevated levels of circulating platelet microparticles, VEGF, IL-6 and RANTES in patients with gastric cancer: possible role of a metastasis predictor. Eur J Cancer 2003; 39:184-91; PMID:12509950; http://dx.doi.org/ 10.1016/S0959-8049(02)00596-8 [DOI] [PubMed] [Google Scholar]
- 35.Huang B, Zhao J, Unkeless JC, Feng ZH, Xiong H. TLR signaling by tumor and immune cells: a double-edged sword. Oncogene 2008; 27:218-24; PMID:18176603; http://dx.doi.org/ 10.1038/sj.onc.1210904 [DOI] [PubMed] [Google Scholar]
- 36.Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 2013; 14:283-96; PMID:23609508; http://dx.doi.org/ 10.1038/nrm3565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Settembre C, Ballabio A. Lysosome: regulator of lipid degradation pathways. Trends Cell Biol 2014; 24:743-50; PMID:25061009; http://dx.doi.org/ 10.1016/j.tcb.2014.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhitomirsky B, Assaraf YG. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget 2015; 6:1143-56; PMID:25544758; http://dx.doi.org/ 10.18632/oncotarget.2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Groth-Pedersen L, Jaattela M. Combating apoptosis and multidrug resistant cancers by targeting lysosomes. Cancer Lett 2013; 332:265-74; PMID:20598437; http://dx.doi.org/ 10.1016/j.canlet.2010.05.021 [DOI] [PubMed] [Google Scholar]
- 40.Winchester B. Lysosomal metabolism of glycoproteins. Glycobiology 2005; 15:1R-15R; PMID:15647514; http://dx.doi.org/ 10.1093/glycob/cwi041 [DOI] [PubMed] [Google Scholar]
- 41.Schneider DL. ATP-dependent acidification of intact and disrupted lysosomes. Evidence for an ATP-driven proton pump. J Biol Chem 1981; 256:3858-64; PMID:7217060 [PubMed] [Google Scholar]
- 42.Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol 2012; 74:69-86; PMID:22335796; http://dx.doi.org/ 10.1146/annurev-physiol-012110-142317 [DOI] [PubMed] [Google Scholar]
- 43.van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. J Exp Med 1968; 128:415-35; PMID:5666958; http://dx.doi.org/ 10.1084/jem.128.3.415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van oud Alblas AB, van Furth R. Origin, Kinetics, and characteristics of pulmonary macrophages in the normal steady state. J Exp Med 1979; 149:1504-18; PMID:448291; http://dx.doi.org/ 10.1084/jem.149.6.1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, Ouyang GF, Okada M, Balazs M, Adany R et al.. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol 2008; 83:1136-44; PMID:18285406; http://dx.doi.org/ 10.1189/jlb.0907611 [DOI] [PubMed] [Google Scholar]
- 46.Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004; 104:2224-34; PMID:15231578; http://dx.doi.org/ 10.1182/blood-2004-03-1109 [DOI] [PubMed] [Google Scholar]
- 47.Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res 2006; 66:1123-31; PMID:16424049; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-1299 [DOI] [PubMed] [Google Scholar]
- 48.Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, Zuo X, Kao CC, Herr AB, Li P. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 2013; 39:1019-31; PMID:24332030; http://dx.doi.org/ 10.1016/j.immuni.2013.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhat N, Fitzgerald KA. Recognition of cytosolic DNA by cGAS and other STING-dependent sensors. Eur J Immunol 2014; 44:634-40; PMID:24356864; http://dx.doi.org/ 10.1002/eji.201344127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lam E, Stein S, Falck-Pedersen E. Adenovirus detection by the cGAS/STING/TBK1 DNA sensing cascade. J Virol 2014; 88:974-81; PMID:24198409; http://dx.doi.org/ 10.1128/JVI.02702-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell 2013; 51:226-35; PMID:23747010; http://dx.doi.org/ 10.1016/j.molcel.2013.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, Vatan L, Szeliga W, Mao Y, Thomas DG et al.. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 2013; 39:611-21; PMID:24012420; http://dx.doi.org/ 10.1016/j.immuni.2013.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laoui D, Van Overmeire E, De Baetselier P, Van Ginderachter JA, Raes G. Functional Relationship between Tumor-Associated Macrophages and Macrophage Colony-Stimulating Factor as Contributors to Cancer Progression. Front Immunol 2014; 5:489; PMID:25339957; http://dx.doi.org/ 10.3389/fimmu.2014.00489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008; 8:958-69; PMID:19029990; http://dx.doi.org/ 10.1038/nri2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol 2013; 229:176-85; PMID:23096265; http://dx.doi.org/ 10.1002/path.4133 [DOI] [PubMed] [Google Scholar]
- 56.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA et al.. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005; 438:820-7; PMID:16341007; http://dx.doi.org/ 10.1038/nature04186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, Kong X, Bosenberg MC, Wiesner T, Rosen N et al.. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015; 520:368-72; PMID:25807485; http://dx.doi.org/ 10.1038/nature14336 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.