

ABSTRACT
We have recently reported that treatment of disseminated pancreatic cancer with an attenuated Toxoplasma gondii uracil auxotroph vaccine promoted antitumor CD8+ T cell responses and long-term survival. Here, we optimized the treatment strategy for disseminated pancreatic cancer and show that attenuated Toxoplasma gondii therapy stimulated effective long-term immunity to pancreatic cancer through mechanisms involving CD4+ T cells and pancreatic tumor-specific IgG. Our results suggest that cell-mediated immunity in conjunction with humoral antibody immunity may offer greater resistance to recurrence of highly aggressive tumors. Cancer immunotherapeutic strategies using attenuated Toxoplasma gondii vaccines merit further investigation as a novel strategy to reawaken immunity to primary pancreatic carcinoma and to generate long-lasting immunity to pancreatic cancer recurrences.
KEYWORDS: Humoral immunity, immunotherapy, pancreatic cancer, T cell protection, Toxoplasma gondii, tumor-specific antibodies
Introduction
Despite advances made in the understanding of the development and tumor microenvironment and progress in the development of drugs and therapies, pancreatic ductal adenocarcinoma (PDAC) still remains one of the deadliest cancers with an average survival rate of only 4 to 6 mo following initial diagnosis.1 Thirty percent of patients present with locally advanced disease and 50% of patients present with metastatic disease upon diagnosis.2 Furthermore, metastases develop in ˜70% of patients that have received successful surgical resection of their primary tumor. For the 15 to 20% of patients eligible for and receiving surgery, the 5-year survival rate with additional therapeutic intervention remains around 20%.3-5 Although some progress has been made with regards to improving patient survival, there is still a strong need for the development of drugs and therapies to enhance the immune response to further combat advanced pancreatic cancer in conjunction with current therapies. In particular, innovative therapies that confer long-term protection against highly aggressive metastatic pancreatic cancer and tumor recurrence will be crucial to improve long-term patient survival.6
One recently reported effective immunotherapy for established solid tumors is immunotherapeutic treatment with the non-replicating attenuated Toxoplasma gondii uracil auxotroph CPS.7,8 CPS treatment of tumor-bearing mice promoted long-lasting tumor-free survival in highly aggressive murine solid tumor models for ovarian cancer, for melanoma, and for pancreatic cancer.9-11 This remarkably effective protection from the primary tumor is triggered and coordinated by CPS manipulation of tumor-associated myeloid cells, which leads to the elimination of tumor-associated immune suppression, thereby promoting the activation of significant antitumor immune responses.9-13 CPS therapy strongly induced the production of IL-12 and IFNγ, and stimulated tumor cell specific effector CD8+ T cell populations. Moreover, mice that survived B16 melanoma after CPS treatment exhibited increased survival against B16 melanoma re-challenge 40 d post-primary tumor challenge. However, this resistance to B16 tumor recurrence waned with time and only 15% of surviving mice re-challenged at 120 d post-primary challenge survived.9 We recently reported that CPS therapy of a highly aggressive model of disseminated pancreatic cancer promoted long-term tumor-free survival of mice (>1 year).11 Here, we report improved CPS therapy regimens and investigate CPS-triggered immunotherapeutic mechanisms that promote long-term protection against recurrence of pancreatic cancer.
Results and discussion
CPS therapy confers protection against re-challenge with pancreatic tumor
CPS treatment of established Pan02 disseminated pancreatic cancer using a three-dose treatment schedule at 7, 19, and 31 d post-tumor challenge provided a 10 to 15% long-term survival rate.11 To determine whether additional CPS treatments would increase the proportion of mice protected against primary tumor, we tested a five-dose CPS treatment schedule at 7, 19, 31, 43, and 55 d post Pan02 tumor challenge (Fig. 1A). The five-dose CPS treatment schedule significantly increased long-term survival of pancreatic tumor-bearing mice, with ˜35% of mice surviving the primary pancreatic tumor (Fig. 1B). The CPS-treated mice that survived for >200 d were then re-challenged with Pan02 tumor cells to determine whether CPS treatment had generated detectable long-term protection against recurrence of pancreatic cancer. CPS-treated survivors or naive age-matched mice were re-challenged by intraperitoneal injection with 1.0 × 106 Pan02 cells. Following re-challenge, naive age-matched mice succumbed to Pan02 tumor within 35 d, a similar kinetic as seen in 8-week old mice bearing the same tumor type (Fig. 1C). In contrast, CPS-treated survivors re-challenged with pancreatic tumor did not succumb to tumor re-challenge. In fact, ˜80% percent of these mice survived Pan02 re-challenge (Fig. 1C). Thus, CPS treatment of the primary disseminated pancreatic tumor stimulated immune responses that strongly protected against disease recurrence after tumor re-challenge. To our knowledge, this is the first reported therapy against disseminated pancreatic cancer that confers long-lasting protection against tumor recurrence.
Figure 1.
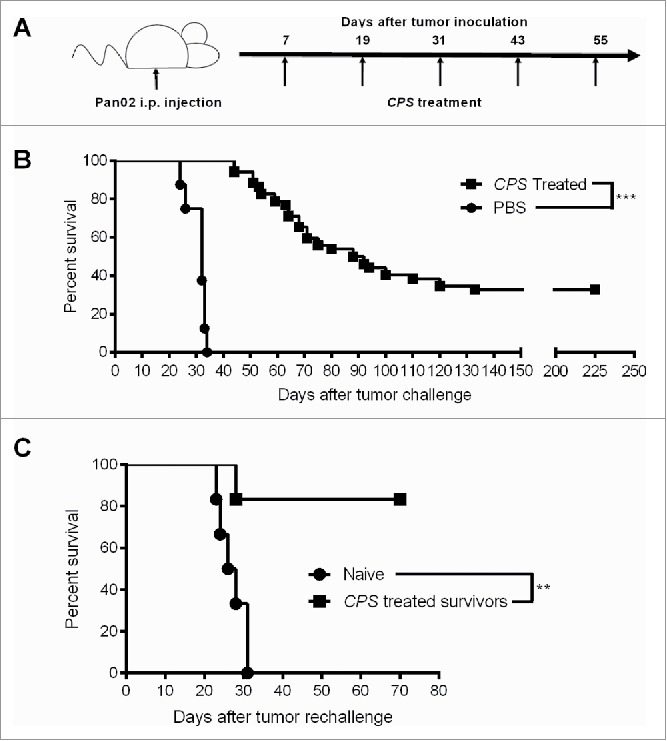
CPS treatment confers protection against pancreatic tumor re-challenge. (A) Treatment schematic outlining the treatment used for generating long-term survival. (B) One week after injection of 1.0 × 106 Pan02 cells i.p. mice were treated with CPS using a five-dose (n = 52) treatment schedule as outlined in Fig. 1A and survival was monitored. (C) 225 d after initial tumor inoculation, CPS-treated survivors (n = 5) or age-matched naive mice (n = 5) were re-challenged with 1.0 × 106 Pan02 cells and survival was monitored. **= p < 0.01, ***= p < 0.001.
CPS therapy increases pancreatic tumor-specific antibodies
Long-lasting protection against Pan02 re-challenge suggested the presence of immune memory to pancreatic tumor. Cancer patients often possess circulating tumor-specific antibody likely generated by the release of antigen during T cell lysis of tumor cells early during tumor progression.14 To detect the presence of a persistent humoral immune response following CPS therapy, we isolated serum from CPS-treated survivors and age-matched naive mice and measured bulk IgG antibody levels as well as the IgG populations able to recognize Pan02 cell lysate antigens. Serum obtained from CPS-treated survivors contained significantly higher levels of circulating IgG than serum from age-matched naive mice (˜326 µg/mL compared to ˜227 µg/mL) (Fig. 2A). Even more interesting was the presence of increased Pan02-specific IgG in CPS-treated survivors (Fig. 2B). While the concentration of circulating Pan02-specific antibody in untreated mice was relatively low (˜15 µg/mL), CPS-treated survivors expressed significantly more circulating Pan02-specific antibody (∼120 µg/mL) (Fig. 2B). The accumulation and persistence of pancreatic tumor-specific antibodies following CPS therapy highlights the ability of CPS to generate a broad antitumor immune response. Moreover, the generation of tumor-specific antibody responses to various solid tumors has been strongly linked as a positive prognostic factor for patient survival.14-17
Figure 2.
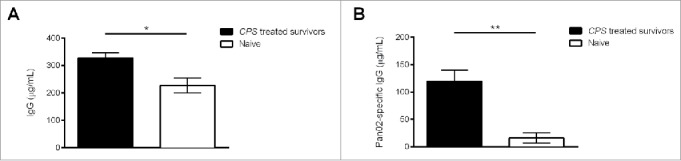
CPS therapy increases tumor-specific antibodies in pancreatic tumor-surviving mice. About 200 d after initial Pan02 tumor inoculation, blood serum was isolated from CPS-treated survivors or age-matched naive mice. Detection of circulating IgG (A) or Pan02-specific IgG (B) was conducted by ELISA coated with IgG detecting antibodies or with Pan02 cell lysates. *= p < 0.05, **= p < 0.01.
Role of T cell responses in protection from pancreatic cancer recurrence
The presence of circulating CD8+ T cells is a positive prognostic in pancreatic cancer.18 In mice bearing disseminated Pan02 tumors, CPS treatment significantly increased activated CD8+ T cell infiltration into the tumor microenvironment, and also increased the number of circulating pancreatic tumor-specific T cells.11 Elimination of CD8+ T cells abrogated the immune protection conferred by CPS treatment of mice bearing disseminated Pan02 tumors. To determine whether the CD8+ T cell population was required for long-term protection against pancreatic tumor in CPS-treated survivors, we depleted CD8+ T cell populations prior to and after Pan02 tumor re-challenge (Fig. 3A). Following the depletion of CD8+ T cells, age-matched naive mice succumbed to pancreatic cancer with similar kinetics as 8-week old mice (Fig. 3B). Unexpectedly, antibody depletion of CD8+ T cells in CPS-treated survivors did not abolish immunity to Pan02 tumor re-challenge compared to CPS-treated survivors receiving control isotype antibody (Fig. 3B).
Figure 3.
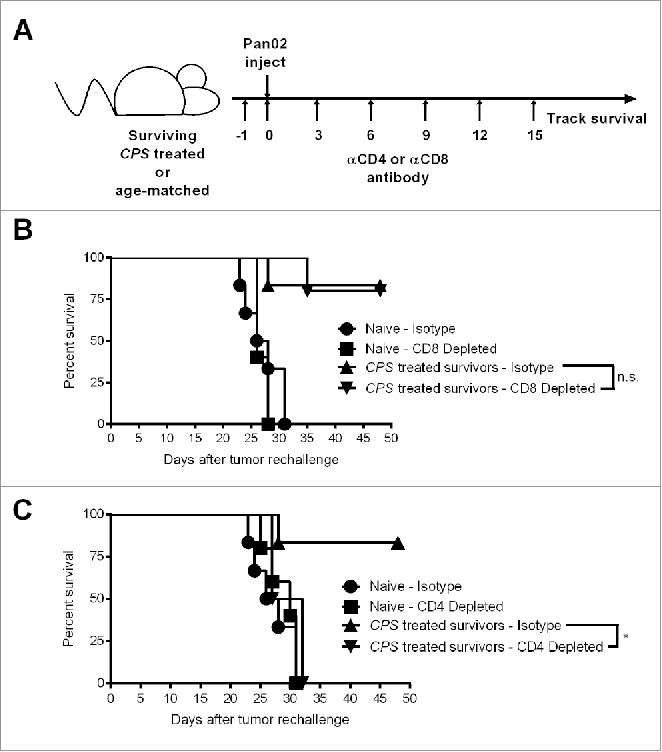
Protection against Pan02 re-challenge depends on CD4+ T cells. (A) CPS-treated survivors or age-matched naive mice were re-challenged by i.p. injection with 1.0 × 106 Pan02 cells at 225 d after initial tumor inoculation and survival was monitored. (B) αCD8 antibody was injected i.p. on days indicated in the schedule shown in panel (A) (n = 5 per group). Depletion of CD8+ T cells was verified to be >99% by flow cytometry 16 d after tumor re-challenge. (C) αCD4 antibody was injected i.p. on days indicated in the schedule shown in panel A (n = 5 for CD4+-depleted age-matched naive mice and n = 4 for CD4+-depleted CPS-treated survivors). Depletion of CD4+ T cells was verified to be >99% by flow cytometry 16 d after tumor re-challenge. ns = not significant, *= p < 0.05.
The absence of a strict dependence on CD8+ T cells for immune protection led us to investigate the potential importance of CD4+ T cells in mediating protection against tumor recurrence after Pan02 re-challenge. Following the depletion of CD4+ T cells, age-matched naive mice succumbed to pancreatic cancer with similar kinetics as 8-week old mice (Fig. 3C). Surprisingly, CPS-treated survivors depleted of CD4+ T cells were not protected against pancreatic tumor re-challenge (Fig. 3C). Thus, while CD4+ T cells were not required for the treatment efficacy of the primary tumor, the CD4+ T cell population was required for protection against tumor re-challenge. The critical role of CD4+ T cells in re-challenge against Pan02 tumor, in addition to the diminished role of CD8+ T cells in re-challenge, reinforces the evidence that CPS therapy of the primary pancreatic tumor induced a robust and dynamic antitumor response to promote long-term survival.
In the B16 melanoma model, immunity to tumor recurrence lasted less than ∼120 d in CPS-treated survivors.9 Here, we find that long-term immunity to pancreatic cancer recurrence lasted more than 200 d in mice that survived the primary tumor after treatment with CPS. CPS therapy triggered a diverse cell-mediated (CD4+ and CD8+ T cells) and a significant humoral antibody response against pancreatic cancer. While CD8+ T cells were essential for survival against the primary Pan02 tumor, immune protection against tumor re-challenge did not rely on the CD8+ T cell population. On the other hand, while CD4+ T cells were not essential for survival against the primary Pan02 tumor, immune protection against tumor re-challenge was dependent on the CD4+ T cell population. In view that the CD4+ T cell population was associated with immune protection, our results do not yet rule out a supporting role for CD8+ T cells in the immunity to tumor recurrence.
CD4+ T cells were shown to be required for development of optimal effector CD8+ T cell populations following vaccination with the CPS vaccine strain.19 Moreover, the relationship between CD4+ T cells and B cells plays a critical role in protection against viral infections and in the generation of protection against tumor growth.20,21 Vaccination of µMT mice (deficient in B cells) with CPS failed to provide protective immunity to re-challenge with virulent T. gondii infection.22 While significant levels of antibody are not detected within 10 d after CPS therapy of primary Pan02 tumors (data not shown), antibody may play a central role in early identification of recurrent tumor. The persistence of circulating antibody raises interesting questions regarding the role these antibodies and circulating CD4+ T cells play during re-challenge. These data suggest the CD8+ effector T cells recruited during primary treatment with CPS may release high levels of pancreatic tumor antigen that stimulate circulating pancreatic tumor-specific IgG as well as potential CD4+ T cell memory.
In conclusion, our data show that CPS treatment of mice bearing primary Pan02 tumors generates long-lasting antitumor immunity to disseminated pancreatic cancer. These results reveal early modifications made to the tumor microenvironment mediated by CPS invasion of tumor-associated myeloid cells leads to recognition of Pan02 tumor and generates immune memory against future recurrence.11 Further work is necessary to examine the basis for protective immunity to pancreatic cancer to understand the relationship between memory CD4+ T cells, potential memory B cells, and circulating persistent antibody specific to pancreatic tumor cells. Our results present compelling evidence of the potency of CPS therapy for treatment of primary disseminated pancreatic cancer. Mice surviving the primary pancreatic tumor after CPS treatment exhibited potent immune protection against the recurrence of pancreatic cancer. Our results suggest that diverse cell mediated and humoral antibody mediated antitumor immune responses generated by primary immunotherapeutic treatment may be critical to the generation of long-lasting protective immunity to highly aggressive cancers such as PDAC.
Materials and methods
Mice and cell lines
Six–eight week old female C57BL/6 (000664) were purchased from Jackson Laboratory. All animal work was performed at the Dartmouth Hitchcock Medical Center animal facility with Dartmouth IACUC approval. The murine pancreatic adenocarcinoma Pan02 cell line, also known as Panc02, was acquired from the Division of Cancer Treatment Tumor Repository (NCI).23 Pan02 cells were maintained in high glucose Roswell Park Memorial Institute (RPMI) 1640 media. Cell culture media was supplemented with 10% FBS, L-glutamine, and penicillin/streptomycin.
Parasites and tachyzoite isolation
CPS tachyzoites were grown in HFF cells supplemented with 300 μM of uracil.7,8 Tachyzoites were purified through a 3.0 μM nuclepore membrane and washed twice with phosphate buffer saline (PBS) prior to treatment of tumor-bearing mice.
Tumor inoculation and treatment
106 Pan02 cells were injected intraperitoneally (i.p.) in 200 µL of PBS. All CPS treatments used 2.0 × 106 tachyzoites injected i.p. For survival studies, mice were treated with CPS using a five-dose schedule (7, 19, 31, 43, and 55 d). Surviving mice were re-challenged 225 d after primary tumor inoculation.
Circulating IgG and Pan02-specific antibody ELISA
Blood sera were collected via cheek bleeds from animals ˜200 d after primary tumor inoculation. Blood was allowed to clot for one hour at room temperature and spun at 1500 rcf for 10 min without break. Collected sera were stored at −80°C until needed.
Pan02 cell lysates were generated by isolating cells from confluent cell cultures. Cells were incubated with protease inhibitor and RIPA buffer (Sigma). Detached cells were isolated utilizing the protocol provided by Sigma. Protein levels were measured utilizing BCA Assay (Life Technologies). For IgG ELISA, purified mouse IgG was diluted in coating buffer (BD Bioscience) at 500 ng/mL. Following overnight incubation, cells were blocked with animal serum in a buffered solution with ProClin®-150 (assay diluent) (BD Bioscience), followed by three washes with 1X wash buffer (concentrated detergent solution with ProClin®-150 as a preservative). Blood sera were serially diluted in assay diluent and incubated for 2 h at room temperature. Bound IgG was detected by incubating goat anti-mouse IgG (Life Technologies) for 1 h at room temperature. Following incubation, samples were incubated with HRP-streptavidin (Biolegend). Samples were then incubated in substrate solution (BD Bioscience) and the reaction was stopped with Stop Solution (BD Bioscience). For Pan02-specific ELISA, Pan02 lysates (0.2 mg/mL) were incubated in coating buffer. Subsequent steps were conducted as stated above.
CD4+ and CD8+ T cell depletions
Purified anti-CD4 (GK1.5), anti-CD8 (2.43), and isotype control (rat IgG2a) antibodies were purchased from BioXCell. 500 μg of antibody was administered i.p. by the schedule outlined in Fig. 3A. Target cell populations were depleted by greater than 99%.
Statistical analysis
Statistical analysis was performed using Graphpad Prism 5 software. The log-rank Mantel–Cox test was used for survival analysis. Bar graph samples were compared using the unpaired student t-test. Error bars show the SEM. p values of less than 0.05, 0.01, or 0.001 are indicated by *, **, ***, respectively, and non-significant differences are indicated by “n.s.”.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank DartLab for flow cytometry instrument resources. The authors would also like to thank Charles Sentman and Edward Usherwood for advice.
Funding
This work was supported by NIH grant AI041930 to DJB.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics , 2014. CA: a cancer journal for clinicians 2014; 64:9-29; PMID:24399786; http://dx.doi.org/ 10.3322/caac.21208 [DOI] [PubMed] [Google Scholar]
- 2.Malik NK, May KS, Chandrasekhar R, Wee W, Flaherty L, Iyer R, Gibbs J, Kuvshinoff B, Wilding G, Warren G et al.. Treatment of locally advanced unresectable pancreatic cancer: a 10-year experience. J Gastrointestinal Oncol 2012; 3:326-34; PMID:23205309; http://dx.doi.org/ 10.3978/j.issn.2078-6891.2012.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mayo SC, Nathan H, Cameron JL, Olino K, Edil BH, Herman JM, Hirose K, Schulick RD, Choti MA, Wolfgang CL et al.. Conditional survival in patients with pancreatic ductal adenocarcinoma resected with curative intent. Cancer 2012; 118:2674-81; PMID:21935914; http://dx.doi.org/ 10.1002/cncr.26553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hartwig W, Hackert T, Hinz U, Gluth A, Bergmann F, Strobel O, Büchler MW, Werner J. Pancreatic cancer surgery in the new millennium: better prediction of outcome. Annals of Surg 2011; 254:311-9; PMID:21606835; http://dx.doi.org/ 10.1097/SLA.0b013e31821fd334 [DOI] [PubMed] [Google Scholar]
- 5.Werner J, Combs SE, Springfeld C, Hartwig W, Hackert T, Buchler MW. Advanced-stage pancreatic cancer: therapy options. Nat Rev Clin Oncology 2013; 10:323-33; PMID:23629472; http://dx.doi.org/ 10.1038/nrclinonc.2013.66 [DOI] [PubMed] [Google Scholar]
- 6.Tuveson DA, Neoptolemos JP. Understanding metastasis in pancreatic cancer: a call for new clinical approaches. Cell 2012; 148:21-3; PMID:22265397; http://dx.doi.org/ 10.1016/j.cell.2011.12.021 [DOI] [PubMed] [Google Scholar]
- 7.Fox BA, Bzik DJ. De novo pyrimidine biosynthesis is required for virulence of Toxoplasma gondii. Nature 2002; 415:926-9; PMID:11859373; http://dx.doi.org/ 10.1038/415926a [DOI] [PubMed] [Google Scholar]
- 8.Fox BA, Bzik DJ. Avirulent uracil auxotrophs based on disruption of orotidine-5'-monophosphate decarboxylase elicit protective immunity to Toxoplasma gondii. Infect Immun 2010; 78:3744-52; PMID:20605980; http://dx.doi.org/ 10.1128/IAI.00287-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baird JR, Byrne KT, Lizotte PH, Toraya-Brown S, Scarlett UK, Alexander MP, Sheen MR, Fox BA, Bzik DJ, Bosenberg M et al.. Immune-mediated regression of established B16F10 melanoma by intratumoral injection of attenuated Toxoplasma gondii protects against rechallenge. J Immunol 2013; 190:469-78; PMID:23225891; http://dx.doi.org/ 10.4049/jimmunol.1201209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baird JR, Fox BA, Sanders KL, Lizotte PH, Cubillos-Ruiz JR, Scarlett UK, Rutkowski MR, Conejo-Garcia JR, Fiering S, Bzik DJ. Avirulent Toxoplasma gondii generates therapeutic antitumor immunity by reversing immunosuppression in the ovarian cancer microenvironment. Cancer Res 2013; 73:3842-51; PMID:23704211; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanders KL, Fox BA, Bzik DJ. Attenuated toxoplasma gondii stimulates immunity to pancreatic cancer by manipulation of myeloid cell populations. Cancer Immunol Res 2015; 3:891-901; PMID:25804437; http://dx.doi.org/ 10.1158/2326-6066.CIR-14-0235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fox BA, Sanders KL, Bzik DJ. Non-replicating Toxoplasma gondii reverses tumor-associated immunosuppression. Oncoimmunology 2013; 2:e26296; PMID:24353916; http://dx.doi.org/ 10.4161/onci.26296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fox BA, Sanders KL, Chen S, Bzik DJ. Targeting tumors with nonreplicating Toxoplasma gondii uracil auxotroph vaccines. Trends Parasitol 2013; 29:431-7; PMID:23928100; http://dx.doi.org/ 10.1016/j.pt.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahn SS, Irie RF, Weisenburger TH, Jones PC, Juillard G, Roe DJ, Morton DL. Humoral immune response to intralymphatic immunotherapy for disseminated melanoma: correlation with clinical response. Surgery 1982; 92:362-7; PMID:7101129 [PubMed] [Google Scholar]
- 15.GuhaThakurta D, Sheikh NA, Fan LQ, Kandadi H, Meagher T, Hall SJ, Kantoff PW, Higano CS, Small EJ, Gardner TA et al.. Humoral immune response against non-targeted tumor antigens after treatment with sipuleucel-T and its association with improved clinical outcome. Clin Cancer Res 2015; 21:3619-30; PMID:25649018; http://dx.doi.org/9738558 10.1158/1078-0432.CCR-14-2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsueh EC, Gupta RK, Qi K, Morton DL. Correlation of specific immune responses with survival in melanoma patients with distant metastases receiving polyvalent melanoma cell vaccine. J Clin Oncol 1998; 16:2913-20; PMID:9738558 [DOI] [PubMed] [Google Scholar]
- 17.Qin L, Smith BD, Tsai HL, Yaghi NK, Neela PH, Moake M, Fu J, Kasamon YL, Prince GT, Goswami M et al.. Induction of high-titer IgG antibodies against multiple leukemia-associated antigens in CML patients with clinical responses to K562/GVAX immunotherapy. Blood Cancer J 2013; 3:e145; PMID:24013666; http://dx.doi.org/ 10.1038/bcj.2013.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, Hiraoka N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer 2013; 108:914-23; PMID:23385730; http://dx.doi.org/ 10.1038/bjc.2013.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jordan KA, Wilson EH, Tait ED, Fox BA, Roos DS, Bzik DJ, Dzierszinski F, Hunter CA. Kinetics and phenotype of vaccine-induced CD8+ T-cell responses to Toxoplasma gondii. Infect Immun 2009; 77:3894-901; PMID:19528214; http://dx.doi.org/ 10.1128/IAI.00024-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013; 39:782-95; PMID:24138885; http://dx.doi.org/ 10.1016/j.immuni.2013.10.003 [DOI] [PubMed] [Google Scholar]
- 21.Tsai LM, Yu D. Follicular helper T-cell memory: establishing new frontiers during antibody response. Immunol Cell Biol 2014; 92:57-63; PMID:24189164; http://dx.doi.org/ 10.1038/icb.2013.68 [DOI] [PubMed] [Google Scholar]
- 22.Gigley JP, Fox BA, Bzik DJ. Cell-mediated immunity to Toxoplasma gondii develops primarily by local Th1 host immune responses in the absence of parasite replication. J Immunol 2009; 182:1069-78; PMID:19124750; http://dx.doi.org/ 10.4049/jimmunol.182.2.1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corbett TH, Roberts BJ, Leopold WR, Peckham JC, Wilkoff LJ, Griswold DP Jr, Schabel FM Jr. Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res 1984; 44:717-26; PMID:6692374 [PubMed] [Google Scholar]