

ABSTRACT
The glucocorticoid (GC) steroid dexamethasone (Dex) is used as a supportive care co-medication for cancer patients undergoing standard care pemetrexed/platinum doublet chemotherapy. As trials for new cancer immunotherapy treatments increase in prevalence, it is important to track the immunological changes induced by co-medications commonly used in the clinic, but not specifically included in trial design or in pre-clinical models. Here, we document a number of Dex -induced immunological effects, including a large-scale lymphodepletive effect particularly affecting CD4+ T cells but also CD8+ T cells. The proportion of regulatory T cells within the CD4+ compartment did not change after Dex was administered, however a significant increase in proliferation and activation of regulatory T cells was observed. We also noted Dex -induced proportional changes in dendritic cell (DC) subtypes. We discuss these immunological effects in the context of chemoimmunotherapy strategies, and suggest a number of considerations to be taken into account when designing future studies where Dex and other GCs may be in use.
KEYWORDS: Immunotherapy, glucocorticoid, lymphodepletion, mesothelioma, dendritic cell, T cell, flow cytometry, treg
Introduction
The rapidly developing field of cancer immunotherapy aims to boost or direct a patient's immune system in a manner that can aid recognition of, and/or reactivity against, tumors. We and others have demonstrated that some chemotherapies can be immunogenic——inducing tumor cell death in such a way that tumor neoantigens are “unmasked” and become visible to the host immune system.1 “Chemoimmunotherapy” combines the properties of immunogenic chemotherapies with any one of a number of immunotherapeutic treatments currently under development. Immunotherapies are generally directed at specific rate-limiting steps in the development of an antitumor immune response——whether that be an enhancement of immune activation, or conversely the removal of immunological suppression.2 These finely tuned interventions undergo initial development in animal model systems; however it is commonplace for co-medications that accompany chemotherapy as part of standard clinical care to be omitted from pre-clinical models.
During retrospective analysis of data from two recent chemoimmunotherapy clinical trials in patients with malignant pleural mesothelioma (MPM), we observed large-scale lymphodepletion occurring between the first and second of two “immunological baseline” blood samples taken from each patient prior to receiving chemotherapy. This effect coincided with patients taking the GC steroid Dex. In standard care treatment of MPM (a highly aggressive, incurable, asbestos-induced cancer) Dex is given prior to and for three to five d after pemetrexed/platinum chemotherapy in order to control skin rash, emesis, and inflammatory side effects.3
Dex is routinely prescribed to patients with advanced cancer in a wide range of doses (0.5 mg up to 16 mg daily) for a variety of additional reasons: fatigue, stimulation of appetite, night sweats, and to combat the side effects of some chemotherapies (including platinum agents and taxanes) both as an antiemetic and to prevent hypersensitivity or allergic reactions.4 It is also used specifically to decrease oedema associated with primary and secondary tumors of the central nervous system, for brain metastases in advanced melanoma, or as a single agent therapy for leukemia, lymphoma, and multiple myeloma.
GCs induce immunosuppressive and anti-inflammatory effects predominantly through binding to the glucocorticoid receptor (GCR), of which there are several splice variants, and subsequent direct or indirect genomic interactions.5 In addition a number of non-genomic interactions have been described, including direct interaction with ion channels in the cell or mitochondrial membranes, and also the release of heat shock proteins and chaperonins.5 It has long been known that Dex and other GCs can modulate the immune system in a wide variety of ways, with varying mechanisms of action in different cell types (for recent comprehensive reviews, see refs. 6 and 7).6,7 However, the precise nature and scope of immunological changes induced in response to the particular regimen of Dex used in pemetrexed/platinum premedication has not been studied in detail––nor more generally in patients with cancer. We hypothesized that additional phenotypic and functional changes of importance to current chemoimmunotherapy strategies had occurred in response to Dex in this setting, and therefore further interrogated our data to highlight what these might be.
Understanding the immunological changes induced by Dex is important both in the context of combining immunotherapy with chemotherapy, and given the broadening indications and study settings for single agent or combination immunotherapy. The data presented here can inform future planning of combined chemoimmunotherapy modalities, timing of baseline immunological investigations, and suggest the importance of studying the effects of Dex in animal models of chemoimmunotherapy.
Results
We examined PBMC samples from 35 patients with MPM who had registered consent for either of two chemoimmunotherapy clinical trials. Criteria for acceptance onto either study were identical. Patients received three 4 mg doses of Dex in the 24 h prior to undergoing standard care pemetrexed + platinum (carboplatin or cisplatin) chemotherapy (Fig. 1A). Peripheral blood samples were collected on the day the patient consented to participation in the study, and again immediately prior to the first cycle of chemotherapy treatment––with a mean time between samples of 9.6 d (± 5.8 d). Immunological parameters were assessed by flow cytometry (see materials and methods). The patients were a typical cohort of advanced MPM, with median overall survival (OS) of 17.8 mo (Fig. 1B).
Figure 1.
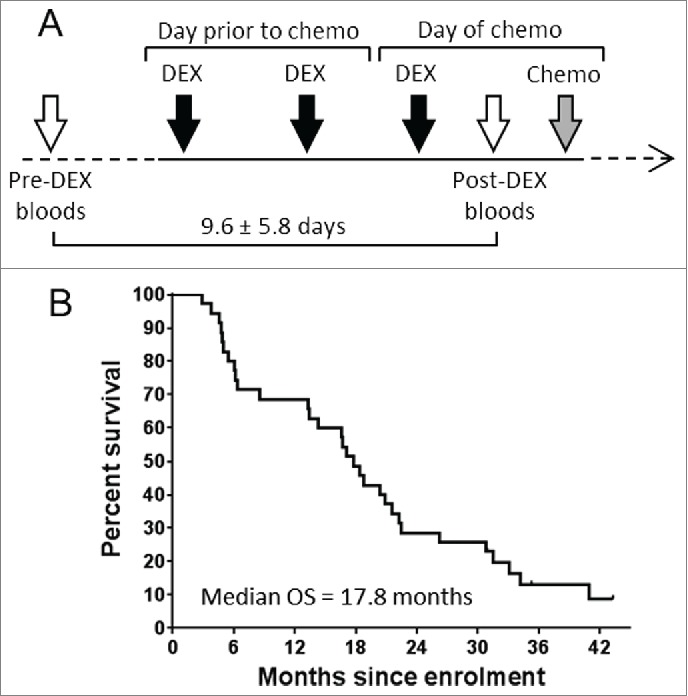
Study group treatment schedule and overall survival. Timeline of patient treatment schedule (A). Patient blood samples were drawn on the day of study enrolment, and again immediately prior to receiving chemotherapy. 3 × 4 mg doses of Dex were given in the lead up to chemotherapy as shown. Open arrows represent study blood collections, black arrows represent oral administration of 4 mg Dex, gray arrow represents infusion of chemotherapy. (B) Kaplan–Meier plot showing overall survival of all patients involved in this study.
The data for all parameters were collated, together with the percentage increase or decrease in that parameter between pre-Dex and post-Dex timepoints, and the corresponding p value resulting from paired t-test analysis (Table 1).
Table 1.
Results of flow cytometry analysis.
| Pre-Dex |
Post-Dex |
|||||||
|---|---|---|---|---|---|---|---|---|
| Parameter measured | Mean | StDev | n = | Mean | StDev | n = | % change | p value |
| CD3 T cells per μL | 848 | 401 | 30 | 370 | 207 | 32 | −56.3 | <0.001 |
| CD4 T cells per μL | 551 | 199 | 31 | 236 | 149 | 33 | −57.2 | <0.001 |
| CD4 % of CD3 T cells | 65.4 | 10.8 | 32 | 57.6 | 12.8 | 35 | −11.9 | <0.001 |
| CD8 T cells per μL | 246 | 212 | 30 | 124 | 103 | 31 | −49.8 | <0.001 |
| CD8 % of CD3 T cells | 27.1 | 10.3 | 32 | 33.7 | 13.4 | 34 | 24.4 | <0.001 |
| CD4 to CD8 T cell ratio | 3.0 | 1.9 | 32 | 2.3 | 1.7 | 34 | −25.3 | <0.001 |
| Tregs per μL | 24.0 | 11.9 | 31 | 9.7 | 6.5 | 33 | −59.3 | <0.001 |
| Treg % of CD4+ T cells | 4.2 | 1.3 | 35 | 4.2 | 1.4 | 35 | −1.2 | 0.755 |
| Ki67+ % of Tregs | 21.3 | 6.9 | 35 | 38.3 | 13.0 | 35 | 79.5 | <0.001 |
| Ki67+ % of non-Treg CD4 T cells | 3.1 | 1.4 | 35 | 4.8 | 2.4 | 35 | 53.1 | <0.001 |
| ICOS+ % of Tregs | 35.2 | 15.3 | 35 | 46.7 | 15.7 | 35 | 32.4 | <0.001 |
| ICOS+ % of non-Treg CD4 T cells | 6.5 | 5.0 | 35 | 7.0 | 5.2 | 35 | 8.4 | 0.121 |
| effector (CD38hiHLA-DRhi) CD8 T cells per μL | 4.4 | 3.9 | 30 | 2.3 | 1.3 | 31 | −46.5 | 0.003 |
| effector % of CD8 T cells | 1.9 | 1.5 | 35 | 2.5 | 1.9 | 35 | 33.4 | <0.001 |
| Bcl2lo % of effector CD8 T cells | 66.5 | 17.0 | 35 | 64.1 | 18.1 | 35 | −3.6 | 0.293 |
| Ki67+ % of effector CD8 T cells | 63.3 | 17.2 | 35 | 69.7 | 17.1 | 35 | 10.0 | 0.009 |
| ICOS+ % of effector CD8 T cells | 27.9 | 14.2 | 35 | 28.8 | 13.7 | 35 | 3.3 | 0.608 |
| Bcl2lo % of CD8 T cells | 3.8 | 2.2 | 35 | 3.6 | 2.6 | 35 | −5.9 | 0.527 |
| Ki67+ % of CD8 T cells | 2.0 | 1.5 | 35 | 2.9 | 2.0 | 35 | 45.7 | 0.001 |
| ICOS+ % of CD8 T cells | 2.1 | 3.8 | 35 | 1.7 | 1.9 | 35 | −20.7 | 0.353 |
| CD8 T cells to Treg ratio | 10.7 | 6.3 | 30 | 17.0 | 13.5 | 31 | 59.2 | <0.001 |
| DC (HLA-DR+ Lin-) % of PBMC | 0.4 | 0.2 | 15 | 0.3 | 0.2 | 15 | −31.4 | 0.035 |
| HLA-DR+ lin- BDCA1+ % of total PBMC | 0.159 | 0.120 | 15 | 0.085 | 0.102 | 15 | −46.3 | 0.024 |
| HLA-DR+ lin- BDCA2+ % of total PBMC | 0.147 | 0.095 | 15 | 0.048 | 0.053 | 15 | −67.2 | 0.001 |
| HLA-DR+ lin- BDCA3+ % of total PBMC | 0.028 | 0.019 | 15 | 0.022 | 0.015 | 15 | −21.9 | 0.279 |
| BDCA1+ % of DC | 35.6 | 19.8 | 15 | 21.8 | 18.8 | 15 | −38.8 | 0.023 |
| BDCA2+ % of DC | 32.6 | 13.7 | 15 | 17.2 | 14.4 | 15 | −47.1 | 0.008 |
| BDCA3+ % of DC | 5.6 | 2.4 | 15 | 12.0 | 8.4 | 15 | 116.2 | 0.003 |
| BDCA1+ Dc to BDCA2+ DC ratio | 2.1 | 2.6 | 15 | 2.2 | 2.3 | 15 | 5.3 | 0.884 |
| BDCA1+ DC CD40 MFI | 43.0 | 32.3 | 15 | 75.6 | 58.3 | 15 | 75.7 | 0.005 |
| BDCA1+ DC CD83 MFI | 78.7 | 34.1 | 15 | 64.8 | 27.8 | 15 | −17.7 | 0.188 |
| BDCA1+ DC CD80 MFI | 110 | 42 | 15 | 135 | 69 | 15 | 22.7 | 0.036 |
| BDCA2+ DC CD40 MFI | 27.0 | 24.1 | 15 | 17.0 | 19.3 | 15 | −37.0 | 0.138 |
| BDCA2+ DC CD83 MFI | 34.7 | 13.8 | 15 | 35.8 | 12.7 | 15 | 2.9 | 0.831 |
| BDCA2+ DC CD80 MFI | 129 | 25 | 15 | 241 | 440 | 15 | 86.4 | 0.344 |
| BDCA3+ DC CD40 MFI | 29.5 | 70.9 | 15 | 25.2 | 84.3 | 15 | −14.5 | 0.744 |
| BDCA3+ DC CD83 MFI | 46.6 | 58.2 | 15 | 37.5 | 30.2 | 15 | −19.5 | 0.408 |
| BDCA3+ DC CD80 MFI | 198 | 45 | 15 | 229 | 96 | 15 | 15.8 | 0.118 |
List of leukocyte parameters retrospectively examined for changes in response to Dex. For timing of blood sample withdrawal and Dex administration, see Figure 1A. “% change” refers to difference in mean values between “pre-Dex” and “post-Dex” timepoints. P values were calculated using the paired t-test.
We assessed changes in concentrations of total CD4+ and CD8+ T cells per μL of peripheral whole blood, by appropriate gating (Fig. 2A). Both CD4+ and CD8+ cell numbers approximately halved in response to Dex treatment (Fig. 2B and C). This lymphodepletive effect was significantly more pronounced in CD4+ T cells, as shown by the CD4:CD8 ratio (Fig. 2D).
Figure 2.

Dex treatment reduces the number circulating of CD4+ and CD8+ T cells. (A) Representative flow cytometry data showing gating strategy on whole blood samples used to obtain absolute volumetric cell count data. Lymphocytes were identified on the basis of forward scatter (FSC) vs. side scatter (SSC), with CD4+ or CD8 T cells were subsequently identified as CD3+CD4+ and CD3+CD8+ respectively. (B–D) Analysis of T cell subsets in peripheral blood samples collected before (pre-Dex) and after (post-Dex) administration of dexamethasone. (B) Concentration of CD3+CD4+CD8− lymphocytes per μL of peripheral whole blood. (C) Concentration of CD3+CD4−CD8+ lymphocytes per μL of whole blood. (D) The CD4:CD8 ratio, calculated by dividing the number of CD4+ T cells per μL by the number of CD8+ T cells per μL. Each dot represents an individual patient; significant difference between pre-Dex and post-Dex values: ***p < 0.0001, paired students t-test.
Within the overall CD4+ T cell subset, we focused on regulatory T cells (Treg). The absolute concentration of CD4+CD25+CD127loFoxp3+ Treg cells (see Fig. 3A for a description of gating strategy) in peripheral blood was also seen to decrease in response to Dex, with the overall Treg proportion of total CD4+ T cells remaining constant (Fig. 3B). However, the proliferating proportion of Tregs (determined by intracellular staining for Ki67, a nuclear protein expressed in dividing and recently-divided cells, but not in resting or naïve lymphocytes)8 rose by around 80% in comparison to the approximately 50% increase seen in the non-Treg CD4+ population (Fig. 3C and D). The inducible co-stimulator molecule (ICOS), a member of the CD28 family of co-stimulatory molecules, is expressed on activated T cells, and has been described as an indicator of antigen-specific activation.9,10 An increasing proportion of Tregs were seen to express ICOS in response to Dex, whereas the ICOS-expressing proportion of non-Treg CD4+ T cells remained low (Fig. 3E and F).
Figure 3.

Dex treatment increases the proliferation and activation state of Tregs. (A) Representative flow cytometry data, demonstrating the gating strategy used for Treg identification and analysis. Forward scatter (FSC) area vs. FSC-height was used for doublet discrimination, and lymphocytes subsequently selected by FSC vs. side scatter. A “dump” channel was used to gate out dead cells (LIVE/DEAD fixable aqua viability stain), CD14+, CD56+, and CD19+ cells. CD4+ T cells were subsequently selected on the basis of CD4 vs. CD3 staining, followed by the identification of Tregs as CD25hi, CD127lo, and Foxp3+. Tregs, or non-Treg CD4+ T cells, were further gated for expression of Ki67 and ICOS. (B–F) Analysis of Tregs in patient PBMC samples collected before (pre-Dex) and after (post-Dex) administration of Dex. (B) Percentage of Tregs (CD25+CD127loFoxp3+) as a proportion of total CD4+ T cells in PBMC samples. (C–D) Proportion of Tregs (C) and non-Treg CD4+ lymphocytes (D) expressing the proliferation marker Ki67. (E–F) Changes in proportional expression of the activation marker ICOS, in Treg (E) and non-Treg CD4+ lymphocytes (F). Each dot represents an individual patient; significant difference between pre-Dex and post-Dex values: ***p < 0.0001, paired students t-test.
We examined the CD8+ T cell compartment in more detail, focusing on the “effector” CD38hiHLA-DRhi population (see Fig. 4A for gating strategy). The number of effector cells (CD38hiHLA-DRhi cells as a proportion of CD3+CD8+ cells) underwent a small but significant increase in response to Dex (Fig. 4B). This population consists of predominantly proliferating (Ki67+) cells with low expression of Bcl-2 (a constitutively expressed anti-apoptotic protein that becomes downregulated in T cells upon their activation in response to antigen).11,12 The proportion of Ki67-expressing cells within this effector population increased by ˜10% in response to Dex; however expression of ICOS did not change. Although the overall proportion of proliferating (Ki67+) CD8 T cells was small––as expected––there was a significant increase in response to Dex (Fig. 4C). In terms of activation, however, the cell-surface expression of ICOS remained constant (Fig. 4D).
Figure 4.

Dex treatment increase the proportion of CD8+ T cells displaying an effector phenotype. (A) Representative flow cytometry data demonstrating the gating strategy used for CD8+ T cells. Forward scatter (FSC) area vs. FSC-height was used for doublet discrimination, and lymphocytes were selected by FSC vs. side scatter. A “dump” channel was then used to gate out dead cells (LIVE/DEAD fixable aqua viability stain), monocytes/macrophages (CD14), NK cells (CD56) and B cells CD19). CD8+ T cells were subsequently selected on the basis of CD8 vs. CD3 staining, followed by the identification of “effector CD8” cells as HLA-DRhiCD38hi. This population was further gated for expression of Ki67, Bcl-2 and ICOS, using HLA-DR−CD38− CD8 T cells from the same sample as gating controls (filled histogram peaks, “negative” in respect to Ki67 and ICOS, and “positive” regarding Bcl-2). (B–E) Analysis of CD8+ T cells in patient PBMC samples collected before (pre-Dex) and after (post-Dex) administration of dexamethasone. (B) HLA-DRhiCD38hi “effector” lymphocytes as a percentage of total CD8+ T cells. (C–D) Proportion of CD8+ T cells expressing the proliferation marker Ki67+ (C) and activation marker ICOS (D). (E) CD8+ T cell to Treg ratio, calculated by dividing the CD8+ percentage of total T cells by the Treg percentage of total T cells. Each dot represents an individual patient; significant difference between pre-Dex and post-Dex values: ***p < 0.0001, paired students t-test.
All T cell subsets underwent substantial depletion from blood, however we wanted to examine whether the balance between CD8+ T cells and Tregs had changed as this might alter the stoichiometric potential between immune suppression versus an antitumor immune response. The ratio of Tregs to CD8+ T cells was calculated here as follows: total CD4+ and CD8+ T cell concentrations were obtained from whole blood cell count experiments; Treg concentrations were subsequently calculated using the CD25hiCD127loFoxp3+ percentage of total CD4+ T cells from PBMC staining as described above. The number of CD8+ T cells per Treg showed approximately a 65% increase in favor of CD8+ T cells (Fig. 4E).
The effect of Dex on DC subpopulations of patient PBMC was also examined. PBMC from patients recruited to only one of the two clinical trials were analyzed for DC markers by flow cytometry, in line with the outcomes targeted in that particular study (n = 15). The gating strategy identified DCs——classically defined as HLA-DR+lin−—— with subsets identified as “myeloid” or “plasmacytoid” through use of BDCA antibodies (Fig. 5A; for recent reviews on DC nomenclature and subtypes, see refs. 38 and 39).13,14 These subsets are often reported as a percentage of total PBMC (Fig. 5B). However, due to the substantial depletive effects of Dex on T cells, the proportion of PBMCs identified as DCs would change whether or not DCs were actually affected by Dex. Therefore, we also assessed DC subsets as a proportion of the HLA-DR+lin− DC population (Fig. 5C). Notably, the BDCA-1+ (CD1c+, major myeloid) and BDCA-2+ (CD303+, plasmacytoid) proportion of both total PBMC and HLA-DR+lin− cells reduced significantly in response to Dex. The BDCA-3+ (CD141+ myeloid) DC proportion of total PBMC did not change, and indeed was seen to increase as a proportion of HLA-DR+lin− cells. Relative expression of the CD80 (B7.1) co-stimulatory molecule, and the maturational markers CD40 and CD83, were analyzed by mean fluorescence intensity (MFI) on all three of the DC subsets mentioned above. No significant changes were observed in response to Dex treatment with the exception of significant (but highly heterogeneous) increases in the MFI of CD40 and, to a lesser extent, CD80 on the BDCA1+ DC subset.
Figure 5.
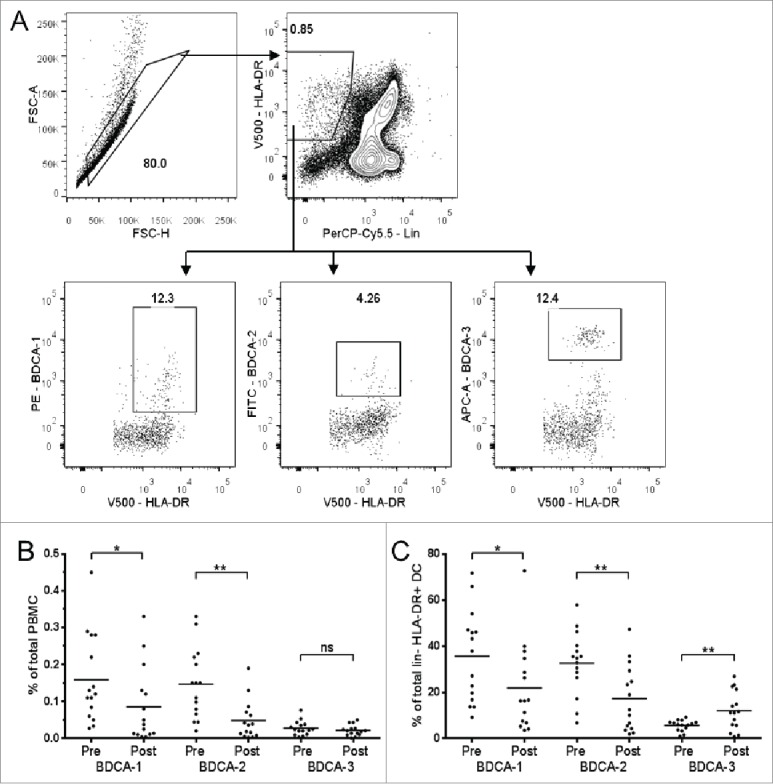
Effect of Dex treatment on peripheral blood DC subsets. (A) Representative flow cytometry data demonstrating the gating strategy used for dendritic cells. Forward scatter (FSC) area vs. FSC-height was used for doublet discrimination. A “dump” channel was used to gate out dead cells (LIVE/DEAD fixable red viability stain) plus those staining positively with a CD3/CD14/CD16/CD19/CD56 “lin” cocktail. DCs were identified as lin− HLA-DR+ cells, and respective DC subpopulations identified by BDCA-1, BDCA-2 or BDCA-3. (B) Compiled flow cytometric data from PBMC analysis, depicting individual patients, showing values before and after administration of dexamethasone. (B–C) DC subsets as a percentage of total PBMCs (B) and lin−HLA-DR+ cells (C) in patient samples collected before (pre-Dex) and after (post-Dex) administration of dexamethasone. Each dot represents an individual patient; significant difference between pre-Dex and post-Dex values: *p < 0.05, **p < 0.001, paired students t-test.
Discussion
Understanding the immunological changes induced by Dex is likely to be an important consideration in the context of combined immunotherapy and chemotherapy, and also given the broadening indications and study settings for single agent or combination immunotherapy. While most current clinical trials of anti-CTLA4 and anti-PD-pathway “checkpoint blockade” agents exclude patients requiring Dex treatment, or limit the dose of Dex, this is empirical and there is little data upon which to base a recommendation. Furthermore, some studies are now enrolling participants who may be Dex-dependent, such as those with intracranial primary or metastatic disease, without a clear understanding of how this may affect treatment efficacy.
Here, we describe a large-scale lymphodepletive effect in response to Dex administration as a pre-chemotherapy agent, with a particular bias toward CD4+ T cell loss. A similar outcome has been observed with the GC methylprednisolone in patients with multiple sclerosis, with increased numbers of apoptotic cells observed in the CD4+ over the CD8+ T cell populations following treatment.15 GC-induced apoptosis of T cells has been shown in part to be mediated by GC-GCR complex binding to the mitochondrial membrane.16
Regulatory T cells (Tregs) were of particular interest to us. These cells have the capacity to suppress an antitumor immune response,17 and are typically found to be increased in the peripheral blood of patients with a number of cancers including MPM.18-26 Infiltration of Treg into tumor tissue has been associated with a poor prognosis in several cancers including NSCLC,27,28 ovarian cancer,29 breast cancer,30 gastric cancer,31 pancreatic cancer 32 and hepatocellular carcinoma.33,34 In particular, the intratumoral balance between CD8+ tumor-infiltrating lymphocytes (TILs) and Tregs has been shown to be predictive in a number of malignancies, with a low CD8/Treg TIL ratio correlating with reduced survival in cervical and ovarian cancers for example. 35-37 GCs are known to have selective effects on Tregs over conventional T cells, although the nature of these effects remains controversial. GCs have been previously described as causing apoptosis in pro-inflammatory T cells, while aiding survival of Tregs.7,38-40 Conversely, a recent in vitro study by Pandolfi and colleagues using human PBMC report significantly increased apoptosis in Tregs, and a relative increase in effector T cell frequency in response to Dex treatment——however, the authors also report that these effects can be modulated by the addition of IL-2.41 Our findings show the proportion of Tregs within the overall CD4+ T cell compartment does not change, indicating that Treg survival was not selectively affected in response to the brief but high dose exposure to Dex in our patient group. We therefore think that the in vivo concentration of IL-2 is likely to be sufficient to provide Tregs with a pro-survival signal in these patients. GCs have also been described to increase the proportion of CD4+CD25hi “Tregs” in patients with systemic lupus erythematosus.42
All T cells subsets examined here were seen to proliferate in the post Dex environment; however, Tregs were more likely to be cycling and to show an activated phenotype. The increase in the turnover rate of Tregs compared to non-Treg T cells after Dex-mediated lymphodepletion is consistent with previous findings from models of irradiation and cytotoxic lymphodepletion by cyclophosphamide.43 This higher proliferation of Tregs in the lymphopenic environment may likely be due to a greater affinity for IL-2, although TCR-mediated activation of CD4+ T cells in the presence of Dex has been reported to increase the proportion of IL-10-producing cells, highlighting a possible additional mechanism whereby Tregs may be induced.44 This view is supported by our observation that the frequency of ICOS-expressing Tregs is significantly increased following Dex treatment, suggesting antigen-specific activation of these cells. In contrast, non-Treg CD4+ and the general CD8+ T cell populations did not show any increase in ICOS-positive cells, indicating that these cells were proliferating in an antigen-independent, homeostatic manner.
Previous studies have shown that high intratumoral CD8/Treg ratios are associated with better survival in a number of cancers including cervical and ovarian.35,45 Similar observations have also been reported in peripheral blood, from dogs with osteosarcoma.46 We were interested to see whether, in the PBMC samples we had available, the Dex-mediated depletion of T cells had the effect of altering the CD8/Treg ratio. Indeed, we observed a significant relative increase in CD8 T cells compared to Tregs. It would seem that this change simply reflects the higher susceptibility of CD4+ T cells over CD8+ T cells to Dex-mediated depletion, since the overall Treg proportion of total CD4+ T cells showed no significant change.
HLA-DR and CD38 are markers of antigen-specific T cell activation, previously documented through their expression during chronic viral infection.12,47-49 This CD8+ effector population is increased in MPM and non-small cell lung cancer (NSCLC) compared with healthy controls.18 At this stage, it is not clear whether the change in the balance of suppressive Tregs vs. CD8 potential effector cells has any meaningful repercussions as far as the antitumor response is concerned; our study was not designed to, or able to test this.
Although we did not specifically examine B cells and NK cells, it has been previously published that pro-apoptotic effects on these cells are not observed in response to GC treatment.15
GCs have a wide range of effects on DCs, with the potential to suppress their maturation, disrupt their migration, and induce tolerogenic DC phenotypes.6,50 GC treatment can prevent DCs from upregulating cell surface expression of MHC class II and the co-stimulatory molecules CD86 (B7.2), CD80 (B7.1), CD83 and CD40, in response to activating stimuli.51-53 Decreased expression of mRNA for pro-inflammatory cytokines IL-1, IL-6, and IL-12 has also been reported.54,55 In addition, GCs are known as potent inducers of apoptosis in immature DCs.54 Although we were not able to unequivocally determine whether loss of DC subsets in response to Dex resulted from cell death or from migration of DCs out of peripheral blood and into tissue, the evidence from the literature above suggests that increased maturation and relocation of DCs was not the cause of observed changes in the frequency of myeloid (BDCA-1+) and plasmacytoid (BDCA-2+) DC subsets following Dex. Indeed, we saw no change in expression of maturational markers such as MHC class II, CD80, CD86, and CD40. However, peripheral blood DC in general express low levels of these markers, with terminal differentiation occurring after migration has occurred, so at this stage a firm conclusion cannot be drawn.56-58 The observed increase of BDCA-3+ DCs most likely results from a proportional decrease in the more prevalent BDCA-1+ and BDCA-2+ populations.
In the context of the two chemoimmunotherapy phase I clinical trials from which the data in this paper were taken, there are effects of Dex that could potentially affect the activity of the study drug combination. Low-dose cyclophosphamide has been shown to preferentially deplete Tregs in a number of animal tumor models, with greatest efficacy at low doses,59-65 and also in patients with metastatic melanoma and other advanced cancers.66-68 Dex affects Treg such that the population remains proportionally constant but undergoes increased proliferation and activation. This would be counter to the desired outcome of Treg depletion by cyclophosphamide. Indeed, Dex-treated murine DC cell lines have been used to induce formation of Tregs,55 and to subsequently protect against autoimmunity or a graft-vs.-host response.69 The anti-CD40 study was a clinical translation of a successful pre-clinical treatment in a mouse mesothelioma model.70 The postulated mechanism of action of this treatment is to activate DCs without the specific requirement for CD4+ T cell help, thereby tipping the balance of a possible antigen-specific CD8+ T cell response away from tolerogenic and toward antitumor cytotoxicity (for a recent review, see ref. 71)71. However, due to the modulatory effects on DCs of the particular dose and timing combination of Dex, it may be that any beneficial effects of DC activation are lessened. The potential for DC loss, and/or the prevention of increased CD40 expression on these cells, may have a negative impact on the intended target (and downstream efficacy) of the treatment. However, it should also be noted that terminally differentiated DCs are resistant to the effects of GC and continue to express these maturational markers72 Finally, GC treatment has been reported to inhibit TCR signaling itself——providing yet another method to subvert a potentially successful antitumor cellular immune response. 73,74 The primary aim of both of these studies was to assess safety and not efficacy, hence we are not powered to comment on survival. Since neither trial set out to look at the effects of Dex, which was administered to all patients as part of standard care chemotherapy, there are no “Dex-free” control arms with which to compare its effect on patient outcomes in the context of either study.
The data presented here can inform future planning of combined chemoimmunotherapy modalities. It may be that further work will highlight an optimal treatment schedule or dosage of Dex whereby any potentially negative impacts on the mechanisms of chemoimmunotherapy are minimised——enabling current or future treatments to realize their full potential. We recommend further in-depth analysis of the immunological effects of different dosage and timing of Dex, and to examine the duration of the effects observed here. It may be that any negative impacts of Dex on immunotherapy modalities can be minimized by adjusting dose and/or timing of GC administration. We would also recommend consideration of including Dex (or any other appropriate concomitant medications) in future pre-clinical development of chemoimmunotherapy strategies.
Patients/materials and methods
Eligibility criteria
Participants were enrolled on either one of two phase Ib chemoimmunotherapy clinical trials at Sir Charles Gairdner Hospital (Perth, WA, Australia). The first study involved patients that subsequently received treatment with metronomic low dose cyclophosphamide in combination with standard care pemetrexed/platinum doublet therapy. In the second trial, an anti-CD40 agonistic antibody was given in combination with chemotherapy. Eligible patients had a histologically or cytologically confirmed diagnosis of MPM, Eastern Co-operative Oncology Group (ECOG) performance status (PS) of 0–1, and were planned for first-line treatment with platinum and pemetrexed. All participants had measureable disease on thoracic CT scan as defined by the modified RECIST criteria.75 All had adequate haematological parameters, renal function, and hepatic function. Patients were ineligible if they had previous therapy for MPM (including immunotherapy or investigational agents), radiotherapy to all measurable lesions, symptomatic central nervous system involvement, or a second primary malignancy within the past 10 y. Pregnant or lactating women and patients with other serious medical disorders were also ineligible. The protocols were approved by the Institutional Human Research Ethics Committee and all patients provided written informed consent. Study drug and partial funding to conduct the clinical trials were provided by Pfizer Oncology Australia, the National Health and Medical Research Council Australia, and the Cancer Council Western Australia. Clinical trial registration numbers on the Australia New Zealand Clinical Trials Registry were ACTRN12609000294257 and ACTRN12609000260224.
Treatment administration
Dex (Aspen Pharmacare) was given as standard prophylactic medication for chemotherapy. 2 × 4 mg oral doses were given the day before, and 1 × 4 mg oral dose given the day of (but prior to), drawing of the second study blood sample and receiving chemotherapy.
Assessment of immunological parameters
“Pre-Dex” peripheral blood samples were collected at enrolment, followed within 14 d by collection of the “Post-Dex” blood sample prior to chemotherapy administration. Blood was collected into BD K2EDTA Vacutainers (BD Diagnostics). Whole blood was analyzed by flow cytometry on the day of collection to obtain absolute volumetric cell counts (cells per μL) of CD3+CD8+ and CD3+CD4+ T cells. Blood samples were stained using the “panel 1” antibodies described in Table 2, fixation and red blood cell lysis was performed using BD FACS lysing buffer, and data collected by three-color analysis using a Millipore Guava and Guava ExpressPro Software.
Table 2.
List of antibodies.
| Antigen | Fluor | Clone | Isotype | Supplier | Catalog # | Antibody registry # | Panel | Dilution |
|---|---|---|---|---|---|---|---|---|
| CD4 | AF488 | RPA-T4 | ms IgG1 | BD PharMingen | 557695 | 396804 | 1 | 1/20 |
| CD3 | PE | SK7 | ms IgG1 | BD PharMingen | 347347 | 400287 | 1 | 1/50 |
| CD8 | PECy7 | RPA-T8 | ms IgG1 | BD PharMingen | 555368 | 395771 | 1 | 1/50 |
| CD4 | APC-H7 | RPA-T4 | ms IgG1 | BD PharMingen | 560158 | 1645478 | 2 | 1/40 |
| Foxp3 | PE | PCH101 | rt IgG2a | eBioscience | 12-4776-42 | 1518782 | 2 | 1/20 |
| CD25 | APC | M-A251 | ms IgG1 | BD PharMingen | 555434 | 398598 | 2 | 1/5 |
| CD127 | PECy7 | eBioRDR5 | ms IgG1 | eBioscience | 25-1278-42 | 1659672 | 2 | 1/100 |
| Ki67 | FITC | B56 | ms IgG1 | BD PharMingen | 556026 | 396302 | 2,3 | 1/10 |
| ICOS | PerCP-Cy5.5 | C398.4A | ha IgG | Biolegend | 313518 | 10641280 | 2,3 | 1/80 |
| CD14 | V500 | M5E2 | ms IgG2a | BD PharMingen | 561391 | 10611856 | 2,3 | 1/80 |
| CD19 | V500 | HIB19 | ms IgG1 | BD PharMingen | 561121 | 10562391 | 2,3 | 1/80 |
| CD3 | V450 | UCHT1 | ms IgG1 | BD PharMingen | 560365 | 1645570 | 2,3 | 1/40 |
| CD8 | APC-H7 | SK1 | ms IgG1 | BD PharMingen | 560179 | 1645481 | 3 | 1/40 |
| CD38 | AF647 | HIT2 | ms IgG1 | Biolegend | 303514 | 493090 | 3 | 1/40 |
| HLA-DR | PECy7 | L243 | ms IgG2a | BD PharMingen | 335795 | 399973 | 3 | 1/80 |
| Bcl-2 | PE | Bcl-2/100 | ms IgG1 | BD PharMingen | 556535 | 396455 | 3 | 1/10 |
| BDCA-1(CD1c) | PE | AD5-8E7 | ms IgG1 | Miltenyi | 130-090-508 | 244316 | 4 | 1/10 |
| BDCA-2(CD303) | FITC | AC144 | ms IgG1 | Miltenyi | 130-090-510 | 244167 | 4 | 1/10 |
| BDCA-3(CD141) | APC | AD5-14H12 | ms IgG1 | Miltenyi | 130-090-907 | 244170 | 4 | 1/10 |
| HLA-DR | V500 | G46.6 | ms IgG2a | BD PharMingen | 561224 | 10563765 | 4 | 1/100 |
| CD3 | PerCP-Cy5.5 | SK7 | ms IgG1 | Biolegend | 344808 | 10640736 | 4 | 1/100 |
| CD14 | PerCP-Cy5.5 | HCD14 | ms IgG1 | Biolegend | 325622 | 893250 | 4 | 1/100 |
| CD16 | PerCP-Cy5.5 | 3G8 | ms IgG1 | Biolegend | 302028 | 893262 | 4 | 1/100 |
| CD19 | PerCP-Cy5.5 | HIB19 | ms IgG1 | Biolegend | 2072925 | 115534 | 4 | 1/100 |
| CD56 | PerCP-Cy5.5 | HCD56 | ms IgG1 | Biolegend | 318322 | 893389 | 4 | 1/100 |
| CD83 | PECy7 | HB15e | ms IgG1 | BD PharMingen | 561132 | 10562565 | 4 | 1/20 |
| CD80 | V450 | L307.4 | ms IgG1 | BD PharMingen | 560442 | 1645583 | 4 | 1/20 |
| CD40 | APC-H7 | 5C3 | ms IgG1 | BD PharMingen | 561211 | 10584325 | 4 | 1/10 |
List of monoclonal antibodies used for flow cytometric staining. Panels were used for absolute cell counts of whole blood (panel 1), Treg staining of PBMC (panel 2), CD8 T cell staining of PBMC (panel 3) or dendritic cell staining of PBMC (panel 4). Abbreviations: AF = AlexaFluor, ms = mouse, rt = rat, ha = hamster.
Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll-Paque™ density gradient centrifugation following the manufacturer's instructions, then cryopreserved in 1mL aliquots of RPMI (Invitrogen, cat# 11875-119) supplemented with 10% FCS, 20mM HEPES and 10% DMSO, at 2 × 106 cells/mL. Cells were placed in a “Mr Frosty” container and frozen at –80°C for 24–48 h before transferring to storage in liquid nitrogen. PBMC from any one patient were analyzed simultaneously by flow cytometry once samples from all time points were available. Prior to analysis, PBMCs were thawed for 1 min in a 37°C water bath and washed once in RPMI, followed by two washes in PBS. Dead cells were identified using LIVE/DEAD Fixable Dead Cell Stain Kit (Molecular Probes, cat. # L34957 (Aqua) or L23102 (Red)). Antibodies were used in three further staining panels, as described in Table 2. Data was collected on a BD FACScanto II, using BD FACSDiva software (BD Biosciences), and analyzed using FlowJo software (Tree Star Inc.).
Statistical considerations
Sample sizes were calculated for each of the studies independently and the patient numbers were derived from those available; a formal power calculation was not performed for this sub-study. Results describing the mean of data at pre-Dex and post-Dex timepoints are reported ± standard deviation. P values reporting statistical significance were calculated using the paired t-test in Prism 6 (GraphPad Software). p values on figures are represented as: *<0.05, **<0.01, ***<0.001, ns>0.05 (not significant).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors acknowledge the facilities, and the scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments.
References
- 1.Nowak AK, Robinson BWS, Lake RA. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res 2003; 63:4490-6; PMID:12907622 [PubMed] [Google Scholar]
- 2.McDonnell AM, Nowak AK, Lake RA. Contribution of the immune system to the chemotherapeutic response. Semin Immunopathol 2011; 33:353-67; PMID:21274535; http://dx.doi.org/ 10.1007/s00281-011-0246-z [DOI] [PubMed] [Google Scholar]
- 3.Vogelzang NJ, Rusthoven JJ, Symanowski J, Denham C, Kaukel E, Ruffie P, Gatzemeier U, Boyer M, Emri S, Manegold C et al.. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol 2003; 21:2636-44; PMID:12860938; http://dx.doi.org/ 10.1200/JCO.2003.11.136 [DOI] [PubMed] [Google Scholar]
- 4.Shih A, Jackson KC. Role of corticosteroids in palliative care. J Pain Palliat Care Pharmacother 2007; 21:69-76; PMID:18032321; http://dx.doi.org/ 10.1080/J354v21n04_14 [DOI] [PubMed] [Google Scholar]
- 5.Stahn C, Buttgereit F. Genomic and nongenomic effects of glucocorticoids. Nat Clin Pract Rheumatol 2008; 4:525-33; PMID:18762788; http://dx.doi.org/ 10.1038/ncprheum0898 [DOI] [PubMed] [Google Scholar]
- 6.Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci 2013; 34:518-30; PMID:23953592; http://dx.doi.org/ 10.1016/j.tips.2013.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zen M, Canova M, Campana C, Bettio S, Nalotto L, Rampudda M, Ramonda R, Iaccarino L, Doria A. The kaleidoscope of glucorticoid effects on immune system. Autoimmun Rev 2011; 10:305-10; PMID:21224015; http://dx.doi.org/ 10.1016/j.autrev.2010.11.009 [DOI] [PubMed] [Google Scholar]
- 8.Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol 1984; 133:1710-5; PMID:6206131 [PubMed] [Google Scholar]
- 9.Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, Kroczek RA. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 1999; 397:263-6; PMID:9930702; http://dx.doi.org/ 10.1038/16717 [DOI] [PubMed] [Google Scholar]
- 10.Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol 2003; 3:939-51; PMID:14647476; http://dx.doi.org/ 10.1038/nri1248 [DOI] [PubMed] [Google Scholar]
- 11.Hildeman DA, Zhu Y, Mitchell TC, Bouillet P, Strasser A, Kappler J, Marrack P. Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity 2002; 16:759-67; PMID:12121658; http://dx.doi.org/ 10.1016/S1074-7613(02)00322-9 [DOI] [PubMed] [Google Scholar]
- 12.Miller JD, van der Most RG, Akondy RS, Glidewell JT, Albott S, Masopust D, Murali-Krishna K, Mahar PL, Edupuganti S, Lalor S et al.. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity 2008; 28:710-22; PMID:18468462; http://dx.doi.org/ 10.1016/j.immuni.2008.02.020 [DOI] [PubMed] [Google Scholar]
- 13.Boltjes A, Van Wijk F. Human dendritic cell functional specialization in steady-state and inflammation. Front Immunol 2014; 5:131; PMID:24744755; http://dx.doi.org/ 10.3389/fimmu.2014.00131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collin M, McGovern N, Haniffa M. Human dendritic cell subsets. Immunology 2013; 140:22-30; PMID:23621371; http://dx.doi.org/ 10.1111/imm.12117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leussink VI, Jung S, Merschdorf U, Toyka KV, Gold R. High-dose methylprednisolone therapy in multiple sclerosis induces apoptosis in peripheral blood leukocytes. Arch Neurol 2001; 58:91-7; PMID:11176941; http://dx.doi.org/ 10.1001/archneur.58.1.91 [DOI] [PubMed] [Google Scholar]
- 16.Sionov RV, Cohen O, Kfir S, Zilberman Y, Yefenof E. Role of mitochondrial glucocorticoid receptor in glucocorticoid-induced apoptosis. J Exp Med 2006; 203:189-201; PMID:16390935; http://dx.doi.org/ 10.1084/jem.20050433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 2006; 6:295-307; PMID:16557261; http://dx.doi.org/ 10.1038/nri1806 [DOI] [PubMed] [Google Scholar]
- 18.McCoy MJ, Nowak AK, van der Most RG, Dick IM, Lake RA. Peripheral CD8(+) T cell proliferation is prognostic for patients with advanced thoracic malignancies. Cancer Immunol Immunother 2012; 62:529-39; PMID:23069871; http://dx.doi.org/ 10.1007/s00262-012-1360-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res 2003; 9:606-12; PMID:12576425 [PubMed] [Google Scholar]
- 20.Ichihara F, Kono K, Takahashi A, Kawaida H, Sugai H, Fujii H. Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin Cancer Res 2003; 9:4404-8; PMID:14555512 [PubMed] [Google Scholar]
- 21.Liu L, Wu G, Yao J, Ding Q, Huang S. CD4+CD25high regulatory cells in peripheral blood of cancer patients. Neuro Endocrinol Lett 2008; 29:240-5; PMID:18404145 [PubMed] [Google Scholar]
- 22.Liyanage UK, Moore TT, Joo H, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ, Goedegebuure PS et al.. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol 2002; 169:2756-61; PMID:12193750; http://dx.doi.org/ 10.4049/jimmunol.169.5.2756 [DOI] [PubMed] [Google Scholar]
- 23.Okita R, Saeki T, Takashima S, Yamaguchi Y, Toge T. CD4+CD25+ regulatory T cells in the peripheral blood of patients with breast cancer and non-small cell lung cancer. Oncol Rep 2005; 14:1269-73; PMID:16211295 [PubMed] [Google Scholar]
- 24.Ormandy LA, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res 2005; 65:2457-64; PMID:15781662; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-3232 [DOI] [PubMed] [Google Scholar]
- 25.Meloni F, Morosini M, Solari N, Bini F, Vitulo P, Arbustini E, Pellegrini C, Fietta AM. Peripheral CD4+ CD25+ Treg cell expansion in lung transplant recipients is not affected by calcineurin inhibitors. Int Immunopharmacol 2006; 6:2002-10; PMID:17161354; http://dx.doi.org/ 10.1016/j.intimp.2006.07.019 [DOI] [PubMed] [Google Scholar]
- 26.Miller AM, Lundberg K, Ozenci V, Banham AH, Hellström M, Egevad L, Pisa P. CD4+CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J Immunol 2006; 177:7398-405; PMID:17082659; http://dx.doi.org/ 10.4049/jimmunol.177.10.7398 [DOI] [PubMed] [Google Scholar]
- 27.Petersen RP, Campa MJ, Sperlazza J, Conlon D, Joshi M, Harpole DH, Patz EF. Tumor infiltrating Foxp3+ regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients. Cancer 2006; 107:2866-72; PMID:17099880; http://dx.doi.org/ 10.1002/cncr.22282 [DOI] [PubMed] [Google Scholar]
- 28.Shimizu K, Nakata M, Hirami Y, Yukawa T, Maeda A, Tanemoto K. Tumor-infiltrating Foxp3+ regulatory T cells are correlated with cyclooxygenase-2 expression and are associated with recurrence in resected non-small cell lung cancer. J Thorac Oncol 2010; 5:585-90; PMID:20234320 [DOI] [PubMed] [Google Scholar]
- 29.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M et al.. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004; 10:942-9; PMID:15322536; http://dx.doi.org/ 10.1038/nm1093 [DOI] [PubMed] [Google Scholar]
- 30.Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, Banham AH. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol 2006; 24:5373-80; PMID:17135638; http://dx.doi.org/ 10.1200/JCO.2006.05.9584 [DOI] [PubMed] [Google Scholar]
- 31.Perrone G, Ruffini PA, Catalano V, Spino C, Santini D, Pietro M, Spoto C, Zingaretti C, Sisti V, Alessandroni P et al.. Intratumoural FOXP3-positive regulatory T cells are associated with adverse prognosis in radically resected gastric cancer. Eur J Cancer 2008; 44:1875-82; PMID:18617393; http://dx.doi.org/ 10.1016/j.ejca.2008.05.017 [DOI] [PubMed] [Google Scholar]
- 32.Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res 2006; 12:5423-34; PMID:17000676; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-0369 [DOI] [PubMed] [Google Scholar]
- 33.Gao Q, Qiu S, Fan J, Zhou J, Wang X, Xiao Y, Xu Y, Li Y, Tang Z. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol 2007; 25:2586-93; PMID:17577038; http://dx.doi.org/ 10.1200/JCO.2006.09.4565 [DOI] [PubMed] [Google Scholar]
- 34.Zhou J, Ding T, Pan W, Zhu L, Li L, Zheng L. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. Int J Cancer 2009; 125:1640-8; PMID:19569243; http://dx.doi.org/ 10.1002/ijc.24556 [DOI] [PubMed] [Google Scholar]
- 35.Jordanova ES, Gorter A, Ayachi O, Prins F, Durrant LG, Kenter GG, van der Burg SH, Fleuren GJ. Human leukocyte antigen class I, MHC class I chain-related molecule A, and CD8+/regulatory T-cell ratio: which variable determines survival of cervical cancer patients? Clin Cancer Res 2008; 14:2028-2035; PMID:18381941; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-4554 [DOI] [PubMed] [Google Scholar]
- 36.Koyama K, Kagamu H, Miura S, Hiura T, Miyabayashi T, Itoh R, Kuriyama H, Tanaka H, Tanaka J, Yoshizawa H et al.. Reciprocal CD4+ T-cell balance of effector CD62Llow CD4+ and CD62LhighCD25+ CD4+ regulatory T cells in small cell lung cancer reflects disease stage. Clin Cancer Res 2008; 14:6770-9; PMID:18980970; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-1156 [DOI] [PubMed] [Google Scholar]
- 37.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C et al.. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A 2005; 102:18538-43; PMID:16344461; http://dx.doi.org/ 10.1073/pnas.0509182102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen X, Murakami T, Oppenheim JJ, Howard OMZ. Differential response of murine CD4+CD25+ and CD4+CD25- T cells to dexamethasone-induced cell death. Eur J Immunol 2004; 34:859-69; PMID:14991616; http://dx.doi.org/ 10.1002/eji.200324506 [DOI] [PubMed] [Google Scholar]
- 39.Baschant U, Tuckermann J. The role of the glucocorticoid receptor in inflammation and immunity. J Steroid Biochem Mol Biol 2010; 120:69-75; PMID:20346397; http://dx.doi.org/ 10.1016/j.jsbmb.2010.03.058 [DOI] [PubMed] [Google Scholar]
- 40.Kang Y, Xu L, Bin W, Chen A, Zheng G. Cutting edge: immunosuppressant as adjuvant for tolerogenic immunization. J Immunol 2008; 180:5172-6; PMID:18390698; http://dx.doi.org/ 10.4049/jimmunol.180.8.5172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pandolfi J, Baz P, Fernández P, Lupi AD, Payaslián F, Billordo LA, Fainboim L, Arruvito L. Regulatory and effector T-cells are differentially modulated by Dexamethasone. Clin Immunol 2013; 149:400-10; PMID:24211714; http://dx.doi.org/ 10.1016/j.clim.2013.09.008 [DOI] [PubMed] [Google Scholar]
- 42.Suárez A, López P, Gómez J, Gutiérrez C. Enrichment of CD4+ CD25high T cell population in patients with systemic lupus erythematosus treated with glucocorticoids. Ann Rheum Dis 2006; 65:1512-7; http://dx.doi.org/ 10.1136/ard.2005.049924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baba J, Watanabe S, Saida Y, Tanaka T, Miyabayashi T, Koshio J, Ichikawa K, Nozaki K, Koya T, Deguchi K et al.. Depletion of radio-resistant regulatory T cells enhances antitumor immunity during recovery from lymphopenia. Blood 2012; 120:2417-27; PMID:22806892; http://dx.doi.org/ 10.1182/blood-2012-02-411124 [DOI] [PubMed] [Google Scholar]
- 44.Barrat FJ, Cua DJ, Boonstra A, Richards DF, Crain C, Savelkoul HF, de Waal-Malefyt R, Coffman RL, Hawrylowicz CM, O'Garra A. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J Exp Med 2002; 195:603-16; PMID:11877483; http://dx.doi.org/ 10.1084/jem.20011629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sato M, Liu M, Anraku M, Ogura T, D'Cruz G, Alman BA, Waddell TK, Kim E, Zhang L, Keshavjee S. Allograft airway fibrosis in the pulmonary milieu: a disorder of tissue remodeling. Am J Transpl: Off J Am Soc Transpl Am Soc Transpl Surg 2008; 8:517-28; PMID:18294148; http://dx.doi.org/ 10.1111/j.1600-6143.2007.02106.x [DOI] [PubMed] [Google Scholar]
- 46.Biller BJ, Guth A, Burton JH, Dow SW. Decreased ratio of CD8+ T cells to regulatory T cells associated with decreased survival in dogs with osteosarcoma. J Vet Intern Med 2010; 24:1118-23; PMID:20666983; http://dx.doi.org/ 10.1111/j.1939-1676.2010.0557.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lechner F, Wong DK, Dunbar PR, Chapman R, Chung RT, Dohrenwend P, Robbins G, Phillips R, Klenerman P, Walker BD. Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med 2000; 191:1499-512; PMID:10790425; http://dx.doi.org/ 10.1084/jem.191.9.1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Callan MF, Tan L, Annels N, Ogg GS, Wilson JD, O'Callaghan CA, Steven N, McMichael AJ, Rickinson AB. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus In vivo. J Exp Med 1998; 187:1395-402; PMID:9565632; http://dx.doi.org/ 10.1084/jem.187.9.1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GMA, Papagno L, Ogg GS, King A, Lechner F, Spina CA et al.. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med 2002; 8:379-85; PMID:11927944; http://dx.doi.org/ 10.1038/nm0402-379 [DOI] [PubMed] [Google Scholar]
- 50.Cumberbatch M, Dearman RJ, Kimber I. Inhibition by dexamethasone of Langerhans cell migration: influence of epidermal cytokine signals. Immunopharmacology 1999; 41:235-43; PMID:10428652; http://dx.doi.org/ 10.1016/S0162-3109(99)00037-5 [DOI] [PubMed] [Google Scholar]
- 51.Piemonti L, Monti P, Allavena P, Sironi M, Soldini L, Leone BE, Socci C, Di Carlo V. Glucocorticoids affect human dendritic cell differentiation and maturation. J Immunol 1999; 162:6473-81; PMID:10352262 [PubMed] [Google Scholar]
- 52.Vanderheyde N, Verhasselt V, Goldman M, Willems F. Inhibition of human dendritic cell functions by methylprednisolone. Transplantation 1999; 67:1342-7; PMID:10360588; http://dx.doi.org/ 10.1097/00007890-199905270-00009 [DOI] [PubMed] [Google Scholar]
- 53.Kitajima T, Ariizumi K, Bergstresser PR, Takashima A. A novel mechanism of glucocorticoid-induced immune suppression: the inhibiton of T cell-mediated terminal maturation of a murine dendritic cell line. J Clin Invest 1996; 98:142-7; PMID:8690786; http://dx.doi.org/ 10.1172/JCI118759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moser M, De Smedt T, Sornasse T, Tielemans F, Chentoufi AA, Muraille E, Van Mechelen M, Urbain J, Leo O. Glucocorticoids down-regulate dendritic cell function in vitro and in vivo. Eur J Immunol 1995; 25:2818–24; PMID:7589077; http://dx.doi.org/ 10.1002/eji.1830251016 [DOI] [PubMed] [Google Scholar]
- 55.Bros M, Jährling F, Renzing A, Wiechmann N, Dang N, Sutter A, Ross R, Knop J, Sudowe S, Reske-Kunz AB. A newly established murine immature dendritic cell line can be differentiated into a mature state, but exerts tolerogenic function upon maturation in the presence of glucocorticoid. Blood 2007; 109:3820-9; PMID:17209058; http://dx.doi.org/ 10.1182/blood-2006-07-035576 [DOI] [PubMed] [Google Scholar]
- 56.Zaba LC, Fuentes-Duculan J, Steinman RM, Krueger JG, Lowes MA. Normal human dermis contains distinct populations of CD11c+BDCA-1+ dendritic cells and CD163+FXIIIA+ macrophages. J Clin Invest 2007; 117:2517-25; PMID:17786242; http://dx.doi.org/ 10.1172/JCI32282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Angel CE, George E, Brooks AES, Ostrovsky LL, La H Brown T, Dunbar PR. Cutting edge: CD1a+ antigen-presenting cells in human dermis respond rapidly to CCR7 ligands. J Immunol 2006; 176:5730-34; PMID:16670277; http://dx.doi.org/ 10.4049/jimmunol.176.10.5730 [DOI] [PubMed] [Google Scholar]
- 58.McLellan AD, Heiser A, Sorg RV, Fearnley DB, Hart DN. Dermal dendritic cells associated with T lymphocytes in normal human skin display an activated phenotype. J Invest Dermatol 1998; 111:841-9; PMID:9804348; http://dx.doi.org/ 10.1046/j.1523-1747.1998.00375.x [DOI] [PubMed] [Google Scholar]
- 59.Röllinghoff M, Starzinski-Powitz A, Pfizenmaier K, Wagner H. Cyclophosphamide-sensitive T lymphocytes suppress the in vivo generation of antigen-specific cytotoxic T lymphocytes. J Exp Med 1977; 145:455-9; PMID:299883; http://dx.doi.org/ 10.1084/jem.145.2.455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Glaser M. Augmentation of specific immune response against a syngeneic SV40-induced sarcoma in mice by depletion of suppressor T cells with cyclophosphamide. Cell Immunol 1979; 48:339-45; PMID:228870; http://dx.doi.org/ 10.1016/0008-8749(79)90128-X [DOI] [PubMed] [Google Scholar]
- 61.Yoshida S, Nomoto K, Himeno K, Takeya K. Immune response to syngeneic or autologous testicular cells in mice. I. Augmented delayed footpad reaction in cyclophosphamide-treated mice. Clin Exp Immunol 1979; 38:211-7; PMID:527259 [PMC free article] [PubMed] [Google Scholar]
- 62.Man S, Bocci G, Francia G, Green SK, Jothy S, Hanahan D, Bohlen P, Hicklin DJ, Bergers G, Kerbel RS. Antitumor effects in mice of low-dose (metronomic) cyclophosphamide administered continuously through the drinking water. Cancer Res 2002; 62:2731-5; PMID:12019144 [PubMed] [Google Scholar]
- 63.Hanahan D, Bergers G, Bergsland E. Less is more, regularly: metronomic dosing of cytotoxic drugs can target tumor angiogenesis in mice. J Clin Invest 2000; 105:1045-7; PMID:10772648; http://dx.doi.org/ 10.1172/JCI9872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Browder T, Butterfield CE, Kräling BM, Shi B, Marshall B, O'Reilly MS, Folkman J. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res 2000; 60:1878-86; PMID:10766175 [PubMed] [Google Scholar]
- 65.Tongu M, Harashima N, Monma H, Inao T, Yamada T, Kawauchi H, Harada M. Metronomic chemotherapy with low-dose cyclophosphamide plus gemcitabine can induce anti-tumor T cell immunity in vivo. Cancer Immunol Immun 2012; 62:383-91; http://dx.doi.org/ 10.1007/s00262-012-1343-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Berd D, Mastrangelo MJ. Effect of low dose cyclophosphamide on the immune system of cancer patients: depletion of CD4+, 2H4+ suppressor-inducer T-cells. Cancer Res 1988; 48:1671-75; PMID:2830969 [PubMed] [Google Scholar]
- 67.Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, Chauffert B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol, Immunother: CII 2007; 56:641-8; PMID:16960692; http://dx.doi.org/ 10.1007/s00262-006-0225-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ghiringhelli F, Apetoh L, Housseau F, Kroemer G, Zitvogel L. Links between innate and cognate tumor immunity. Curr Opin Immunol 2007; 19:224-31; PMID:17303400; http://dx.doi.org/ 10.1016/j.coi.2007.02.003 [DOI] [PubMed] [Google Scholar]
- 69.Rutella S, Danese S, Leone G. Tolerogenic dendritic cells: cytokine modulation comes of age. Blood 2006; 108:1435-40; PMID:16684955; http://dx.doi.org/ 10.1182/blood-2006-03-006403 [DOI] [PubMed] [Google Scholar]
- 70.Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res 2003; 63:4490-6; PMID:12907622 [PubMed] [Google Scholar]
- 71.Khong A, Nelson DJ, Nowak AK, Lake RA, Robinson BWS. The use of agonistic anti-CD40 therapy in treatments for cancer. Int Rev Immunol 2012; 31:246-66; PMID:22804570; http://dx.doi.org/ 10.3109/08830185.2012.698338 [DOI] [PubMed] [Google Scholar]
- 72.Matyszak MK, Citterio S, Rescigno M, Ricciardi-Castagnoli P. Differential effects of corticosteroids during different stages of dendritic cell maturation. Eur J Immunol 2000; 30:1233-42; PMID:10760813; http://dx.doi.org/ 10.1002/(SICI)1521-4141(200004)30:4%3c1233::AID-IMMU1233%3e3.0.CO;2-F [DOI] [PubMed] [Google Scholar]
- 73.Löwenberg M, Verhaar AP, Bilderbeek J, van Marle J, Buttgereit F, Peppelenbosch MP, van Deventer SJ, Hommes DW. Glucocorticoids cause rapid dissociation of a T-cell-receptor-associated protein complex containing LCK and FYN. EMBO Rep 2006; 7:1023-9; PMID:16888650; http://dx.doi.org/ 10.1038/sj.embor.7400775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harr MW, Rong Y, Bootman MD, Roderick HL, Distelhorst CW. Glucocorticoid-mediated inhibition of Lck modulates the pattern of T cell receptor-induced calcium signals by down-regulating inositol 1,4,5-trisphosphate receptors. J Biol Chem 2009; 284:31860-71; PMID:19776014; http://dx.doi.org/ 10.1074/jbc.M109.005579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Byrne MJ, Nowak AK. Modified RECIST criteria for assessment of response in malignant pleural mesothelioma. Ann Oncol 2004; 15:257-60; PMID:14760119; http://dx.doi.org/ 10.1093/annonc/mdh059 [DOI] [PubMed] [Google Scholar]