

ABSTRACT
Circulating tumor cells (CTCs) are cancer cells that are released from a tumor into the bloodstream. The presence of CTCs in peripheral blood has been associated with metastasis formation in patients with breast cancer. Therefore, the molecular characterization of CTCs may improve diagnostics and support treatment decisions.
We performed gene expression profiling to evaluate the enriched CTCs and peripheral blood mononuclear cells (PBMCs) of breast cancer patients using an expression panel of 55 breast cancer-associated genes. The study revealed several significantly differentially expressed genes in the CTC-positive samples, including a few that were exclusively expressed in these cells. However, the expression of these genes was barely detectable in the PBMC samples. Some genes were differentially expressed in PBMCs, and the expression of these genes was correlated with tumor grade and the formation of metastasis.
In this study, we have shown that the enriched CTCs of breast cancer patients overexpress genes involved in proteolytic degradation of the extracellular matrix (ECM) as well as genes that play important roles in the epithelial-mesenchymal transition (EMT) process that may occur in these cells.
KEYWORDS: Breast cancer, circulating tumor cells; gene expression profiling; peripheral blood mononuclear cells; prognostic
Abbreviations
- ADAM17
ADAM metallopeptidase domain 17
- CD24
CD24 molecule
- CD44
CD44 molecule
- cDNA
cDNA
- CTCs
circulating tumor cells
- ECM
extracellular matrix
- EMT
epithelial-mesenchymal transition
- EpCAM
epithelial cell adhesion molecule
- FDA
US Food and Drug Administration
- FOXO3
forkhead box O3
- HDAC2
histone deacetylase 2
- HER2
erb-b2 receptor tyrosine kinase 2
- H2AFZ
H2A histone family, member Z
- IGFR1
Insulin-like growth factor 1 receptor
- KIT
v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog
- KRAS
Kirsten rat sarcoma viral oncogene homolog
- MRP
ATP-binding cassette, sub-family C (CFTR/MRP)
- mTOR
mechanistic target of rapamycin
- MUC1
mucin1
- Myc
v-myc myelocytomatosis viral oncogene homolog
- OS
overall survival
- PARP
poly (ADP-ribose) polymerase 1
- PBMCs
peripheral blood mononuclear cells
- PCR
polymerase chain reaction
- PFS
progression-free survival
- PI3K
phosphatidylinositol 3′-kinase
- PI3KCA
phosphoinositide-3-kinase, catalytic, α polypeptide
- PTEN
phosphatase and tensin homolog
- PTPRC
protein tyrosine phosphatase receptor type C
- SATB1
SATB homeobox 1
- TGFβ
transforming growth factor β
- TP53
tumor protein p53
- UPA
urokinase plasminogen activator
- VEGFA
vascular endothelial growth factor A
- VEGFR1
Fms-related tyrosine kinase 1.
Introduction
Despite advances in the diagnosis and treatment of breast cancer, it remains one of the most common malignant diseases. In 2012, approximately 14.1 million new cancer cases and 8.2 million cancer-related deaths occurred, compared with 12.7 million and 7.6 million, respectively, in 2008 (International Agency for Research on Cancer). Metastasis is the leading cause of death in patients diagnosed with cancer.1 Cancer metastasis occurs when tumor cells dissociate from a primary tumor and migrate to distant organs through the peripheral bloodstream or lymphatic drainage system. The initiation of metastasis is a complex process that requires changes in cell phenotypes. Initially, primary tumor cells lose the capacity for cell-cell adhesion. Numerous proteins are known to be involved in the process of ECM degradation, such as proteolytic enzymes and matrix metalloproteases.2 The ECM provides mechanical support and transmits signals for cell survival. Once these signals cease after detachment, normal cells rapidly undergo a special form of apoptosis termed anoikis.3 In contrast, the biology of tumor cells is fundamentally different. Tumor cells have developed several mechanisms to avoid anoikis, primarily including the inhibition of the p53 apoptotic pathway.4,5 During the cell detachment, tumor cells lose epithelial properties and gain the mesenchymal properties. This process is described as the EMT6 and is under the strict control of multiple regulatory pathways, including those involving the TGFβ, NOTCH, and WNT proteins.7 Commonly used EMT molecular markers also include increased production of N-cadherin and vimentin.7 In 2011, Wicha and Hayes published a study showing that a specific subpopulation of CTCs may also undergo EMT.8
Metastasis formation in patients with breast cancer is related to the presence of CTCs in peripheral blood.9 CTCs are very rare cancer cells that are released from a primary tumor into the bloodstream. These cells are present at a ratio of 1 CTC to 106–107 peripheral blood cells.10 Their presence in peripheral blood is associated with significantly shorter progression-free survival (PFS) and overall survival (OS) in both early-stage and metastatic breast cancer patients.11 There are several methods used for the detection of CTCs; however, these assays must be highly sensitive and specific due to the scarcity of these cells. Current techniques mainly rely on an enrichment step that increases assay sensitivity. Novel PCR-based methods include an assay developed by AdnaGen AG (Langenhagen, Germany) that combines the immune-magnetic separation of EpCAM- and MUC1-positive blood cells with multiplex PCR to detect tumor-specific transcripts (HER2, MUC1, and EpCAM). AdnaGen's BreastCancerTest has high sensitivity (the detection limit is 2 CTCs in 5 mL of blood) and high specificity (> 90%). Data from several studies comparing the AdnaGen BreastCancerTest with the only FDA approved test available on the market, the CellSearch™ system (Veridex, USA), have indicated that both methods are at least equivalent in terms of the detection of CTCs in metastatic breast cancer patients.12
Most of the prognostic models are based on analyses of primary tumors and cancer cell lines; however, metastasis, and not the primary tumor, is the main determinant of the clinical outcome of cancer patients. It is already well known that primary tumors and CTCs do not always have the same genetic and molecular profiles.13-15 Therefore, the molecular characterization of CTCs should provide valuable insights into the behaviors of these cells in terms of their impacts on the formation of metastasis and subsequent resistance to chemotherapy.
The aim of this study was to compare the gene expression profiles of CTCs and PBMCs in breast cancer patients as well as healthy controls. Our analyses revealed genes that are exclusively expressed in CTCs; however, the expression of these genes was below the limit of detection, and they could not be detected in the background of PBMC gene expression. Interestingly, analyses of the genes expressed in PBMCs revealed genes that were correlated with tumor grade and metastasis formation.
Results
CTC enrichment and gene expression profiling
To characterize key genes associated with the presence of CTCs, we performed gene expression profiling of CTCs enriched from the blood of breast cancer patients and compared the results with those of CTC-negative samples prepared from healthy donors. In the breast cancer patient group (n=112), 3 out of 35 stage III patients (8.6%), and 1 out of 24 stage II patients (2.4%) were CTC-positive, which is in accordance with previous findings.16 Four months after the first CTC enrichment, we collected a second blood sample and performed CTC enrichment for the previous CTC-positive patients. Only two out of the four previously positive patients remained positive for the presence of CTCs. We performed further analysis using only those patients whose first and second blood samples were both CTC-positive. The primary tumors of the selected CTC-positive patients were negative for progesterone and HER2 receptors.
Although the EpCAM-based enrichment step eliminates a large number of leukocytes, we consistently observed contaminating leukocytes isolated together with CTCs.17 To ensure adequate control of the CTC-positive samples, 5 mL of blood from healthy female donors (n=4) was processed using an AdnaTest system.
Because of the very low amount of starting material, cDNA from the CTC-positive patients and controls was pre-amplified prior to qPCR analysis of 55 cancer-related genes (see the Methods section). The selection of genes was based on previous reports in the literature comprising breast cancer, stem cell, and EMT markers.18,19 A total of 27 genes were detected in the CTC-enriched samples (Table S1). We did not observe any significant differences in gene expression between the first and second blood samples collected from the CTC-positive patients (Fig. S1), and for further analysis, we included both data sets (both measurements). Principal component analysis of relative gene expression was performed to visualize the possible clustering of samples. The gene expression profile of the CTCs was so unique that principal component analysis clearly distinguished between the CTC-positive samples and CTC-negative controls (Fig. 1A). Hierarchical clustering and heat map imaging of the gene expression data of the 27 detected genes revealed high heterogeneity in the gene expression profiles of both the CTC-positive patients and healthy donors (Fig. 1B).
Figure 1.
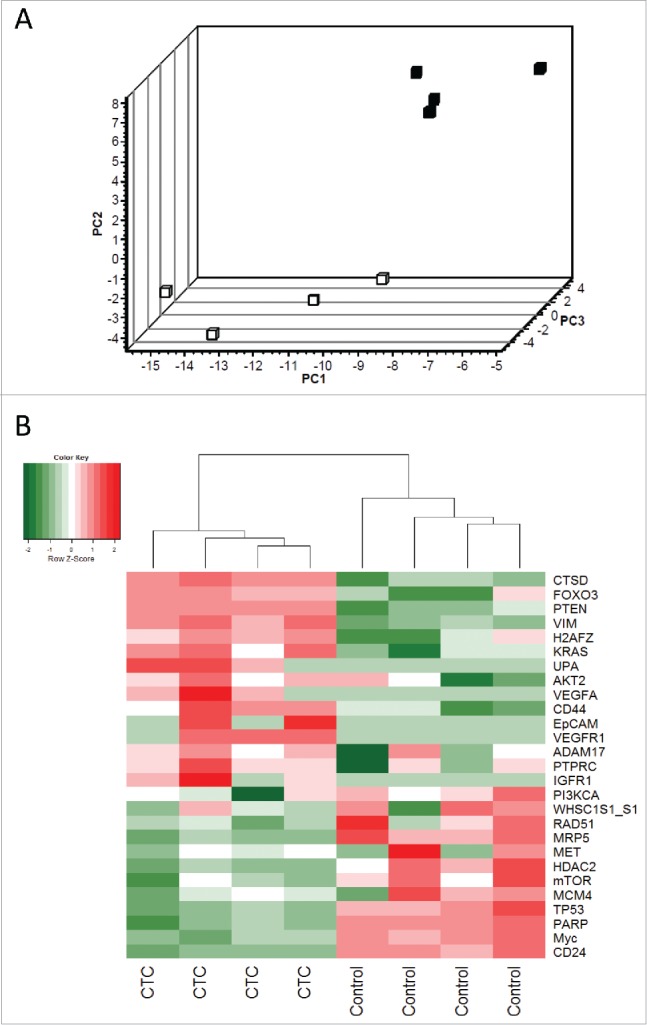
Principle component analysis and hierarchical clustering of relative gene expression in CTC-enriched samples. (A) Principal component analysis represents the differential gene expression pattern of CTC-enriched samples (black squares, n = 4) and healthy controls (white squares, n = 4). Axis: X = PC1: PCA Component 1 (71.98% variance); Y = PC2: PCA Component 2 (93.35% variance) Z = PC3: PCA Component 3 (96.94% variance). (B) Heat map depicting the expression levels of mRNAs in the CTC-enriched samples (CTC, n = 4) compared with their expression levels in healthy controls (n = 4). The red and green squares indicate high and low mRNA levels, respectively.
Gene expression profiling revealed 19 genes with significant differential expression between the CTC-positive group and CTC-negative (control) group. A total of seven genes were significantly upregulated in the CTC-enriched samples, and five (EpCAM, IGFR1, UPA, VEGFA, and VEGFR1) were expressed exclusively in CTCs (Fig. 2). Interestingly, gene expression profiling revealed a strong reduction in a gene cluster associated with tumor progression. These genes (CD24, MRP5, mTOR, Myc, and PARP) were significantly downregulated in the CTC-enriched samples compared with the healthy controls.
Figure 2.
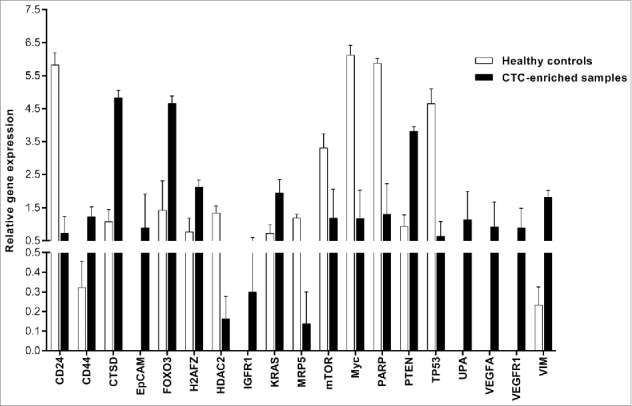
Gene expression profiling of selected genes in CTC-enriched samples. This figure shows the genes that were significantly differentially expressed. The PCR data were normalized to reference genes and expressed on a log2 scale. All values are presented as the mean ± SEM. White bars, healthy donors (n = 4); black bars, CTC-enriched samples (n = 4). The PCR data were analyzed by unpaired Student's t-test. Comparisons were considered significant at p ≤ 0.05.
Gene expression profiling of PBMCs
One of the main aims of this study was to determine the prognostic value of PBMCs from breast cancer patients. We hypothesized that a strong CTC gene signature could be detected in PBMCs. Thus, we performed gene expression profiling of 147 PBMC samples from breast cancer patients using the same panel of genes that was used for CTC profiling. Genes with > 50% missing data were excluded from the analysis. Out of the 55 genes included in the panel, a final group of 36 genes was further analyzed (Table S1). A total of 17 genes were significantly differentially expressed (p ≤ 0.05) between the groups (Fig. 3). We mainly focused on the genes identified by CTC profiling. We found that some of the genes exclusively expressed in CTCs had either no or very low expression in PBMCs. EpCAM and VEGFR1 were excluded from analysis due to a high percentage of missing data, and UPA exhibited a very low level of gene expression in PBMCs (Cq values > 34). The remainder of the upregulated genes (IGFR1 and VEGFA) in CTCs did not show the same regulatory pattern in PBMCs, exhibiting either no change or downregulation compared with the healthy controls. The genes that were downregulated in the CTC-enriched samples showed similar expression patterns in the PBMC samples. In accordance with the data obtained from CTC profiling, the expression levels of CD24, HDAC2, mTOR, Myc, PARP, and TP53 were significantly reduced in the patient group. Overall, the majority of genes detected in the PBMCs of breast cancer patients were downregulated or unchanged, except for MRP4, which was significantly overexpressed in the PBMCs of the breast cancer patients (Fig. 3).
Figure 3.
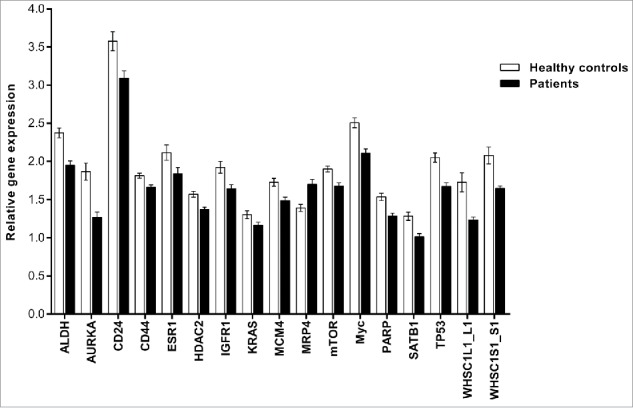
Gene expression profiling of significantly differentially expressed genes in PBMCs. This figure shows genes that were significantly differentially expressed in breast cancer patients only. The PCR data were normalized to the selected reference genes and expressed on a log2 scale. All values are presented as the mean ± SEM. White bars, control group (n = 43); black bars, breast cancer patients (n = 147). The PCR data were analyzed by unpaired Student's t-test. Comparisons were considered significant at p ≤ 0.05.
Next, we investigated the relationships between gene expression in PBMCs and clinicopathological parameters, including metastasis formation, tumor grade, and receptor status of the primary tumors (Fig. 4). ADAM17, FOXO3, HER2, KIT, KRAS, MRP1, MRP2, MRP4, PI3KCA, PTPRC, and VEGFA were significantly downregulated in the group of patients with metastasis (Fig. 4A). The reduced gene expression of CD44, HER2, and SATB1 in PBMCs was significantly associated with higher-grade primary tumors (Fig. 4B). Moreover, a negative HER2 receptor status was associated with higher expression of KRAS, MRP2, MRP4, PI3KCA, SATB1, and TP53 (Fig. 4C).
Figure 4.
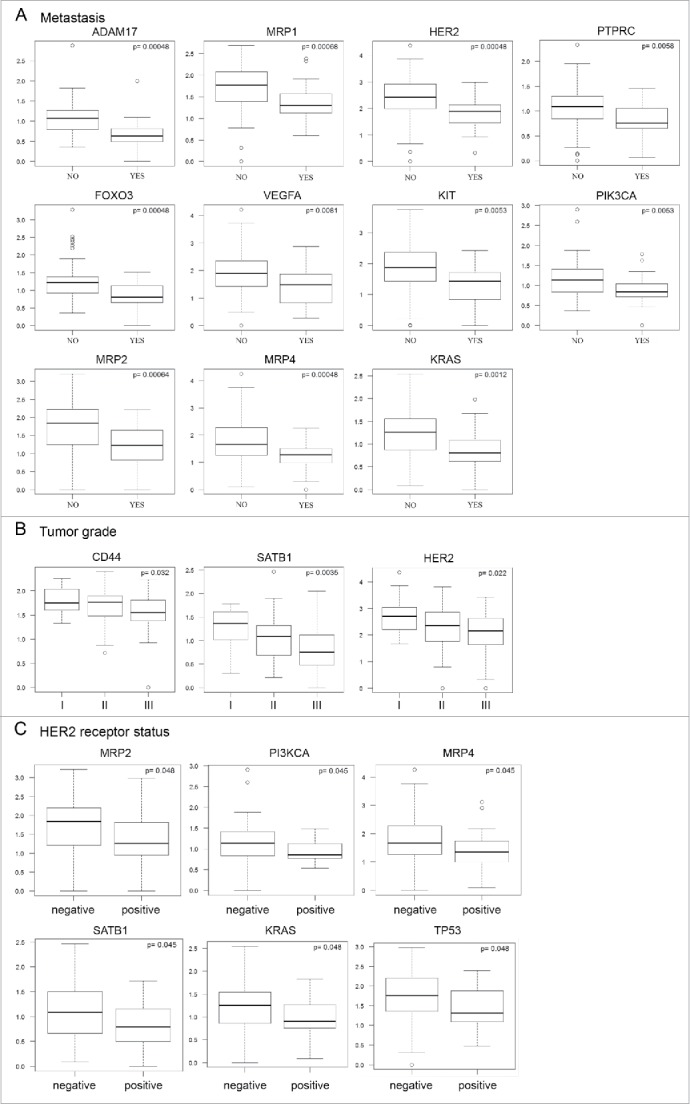
Correlation analyses of gene expression profiles and clinical parameters of patients. Each plot represents correlation of particular gene expression with formation of metastasis (A), tumor grade (B), and HER2/neu receptor status (C). We observed that downregulated expression of selected genes in the PBMCs of breast cancer patients (n = 147) is significantly associated (p ≤ 0.05, p-values are controlled for false positives) with formation of metastasis (A), tumor grade (B), and HER2/neu receptor status (C).
Discussion
Detection of CTCs has been correlated with decreased PFS and OS in patients with primary and metastatic breast cancer.11 In addition to the prognostic impact of the CTC count in breast cancer, the molecular characterization of these cells offers new perspectives that can enhance our understanding of their biology and may improve the prediction of metastasis formation, enabling better treatment decisions. Despite recent advances in isolation techniques, CTC detection in peripheral blood remains challenging. In contrast, the isolation of PBMCs is routinely performed. Therefore, the ability to predict tumor behavior and patient outcome based on the gene expression profiling of PBMCs would be a significant advance.
We screened a total of 112 blood specimens for the presence of CTCs in breast cancer patients at different stages of the disease. A total of 43 blood specimens from healthy donors were used as controls. In this study, we did not detect any CTCs in the peripheral blood of patients in the early stage of the disease. These cells were detected in the peripheral blood of approximately 2% of stage II and 9% of stage III patients. We then performed gene expression profiling of CTCs from breast cancer patients. Based on a comparison with the healthy control group, we identified a set of genes that were overexpressed in CTCs.
Interestingly, most of the identified genes play important roles in cell motility, the EMT process and metastasis formation. Because the ECM is a major physical barrier, cancer cells must increase the production of proteolytic enzymes to penetrate surrounding tissue and establish a new metastatic site. Many studies have shown that the urokinase plasminogen system has a key role in the proteolytic degradation of the ECM.20 Activation of the urokinase plasminogen system has been observed in many cancer types and has been associated with poor prognosis in breast cancer patients.21 Furthermore, this system plays a key role in cell survival. The data presented by Alfano et al. clearly demonstrate the role of urokinase signaling in the protection of tumor cells against the special type of programmed cell death termed anoikis.4 Similar results have been found in invasive MDA-MB-231 breast cancer cells by Ma et al.22.
In this study, we found that the gene expression of urokinase plasminogen activator (UPA) was overexpressed in the CTC-enriched samples. Importantly, this gene was one of the five genes that were exclusively expressed in these samples. These results confirm previous findings that the urokinase plasminogen system is activated in CTC-enriched samples. We found that VEGFA and IGFR1 expression was increased in these samples. Several groups have indicated that the urokinase plasminogen system may be activated by a variety of growth factors, including vascular endothelial growth factor (VEGF) and insulin-like growth factor.23,24 Histone deacetylation is also involved in the regulation of UPA gene transcription. Indeed, it has been reported that histone deacetylase (HDAC) inhibitors induce UPA expression and cancer cell invasion.25 In our study, HDAC2 was significantly downregulated in the CTC-enriched samples. IGFR1 has an additional role in tumor cell survival. The activation of IGFR1 signaling partially contributes to breast cancer recurrence by improving cell survival via the suppression of anoikis.5 UPA was not the only ECM-degrading protease detected in CTC-enriched samples. Overexpression of CTSD, which encodes the protease cathepsin D, was also detected. This protein contributes to the poor prognosis of breast cancer patients26 and is involved in tumor invasion and the formation of metastasis.27
VEGFR1 was also found to be expressed in the CTCs. This gene encodes a protein that functions as one of the three tyrosine kinase receptors for VEGF. VEGFR1 has a well-described role in tumor progression as a positive regulator of angiogenesis.28 Interestingly, the activation of VEGFR1 may also lead to the EMT process,29 which is mainly associated with the overexpression of vimentin.7 Our findings revealed the significant upregulation of vimentin gene expression in the CTC-enriched samples. H2A histone family member Z (H2AFZ) was another gene that was deregulated in these samples. The product of this gene plays a major role in critical biological processes, such as chromosome segregation, cell cycle progression, and maintenance of the heterochromatin/euchromatin status.30 Its expression is significantly associated with the invasion of tumor cells into lymph nodes, the formation of metastasis and decreased patient survival.31
Another important process in the formation of metastasis is the adhesion of CTCs to endothelial cells within a vessel. E-selectin is a cell adhesion molecule expressed in endothelial cells. Interestingly, it is expressed in human bone marrow microvascular endothelium,32 which is one of the most frequent sites of cancer metastasis.33 CD44 is a ligand for E-selectin,34 and its expression has been detected in numerous breast cancer cell lines and primary tumors.35 We found that its expression was significantly increased in the CTC-enriched samples, which is in accordance with the findings of Theodorpoulos et al.36.
However, this study also raises several questions about the roles of the other genes that were overexpressed in the CTC-enriched samples, namely FOXO3, PTEN, and KRAS. FOXO3 and PTEN are known tumor suppressor genes that are involved in the PI3K-Akt signaling pathway. Two of the main roles of Akt are the promotion of cell proliferation and the inhibition of apoptosis. FOXO3 is a key downstream target of the PI3K-Akt signaling pathway and promotes tumor suppression, cell cycle arrest, the repair of damaged DNA, and apoptosis, and it also plays a pivotal role in promoting longevity.37 PTEN regulates the activation of PI3K by dephosphorylating phosphatidylinositol 3,4,4-triphosphate (PIP3) to form phosphatidylinositol 4,5-bisphosphate (PIP2). PIP3 is an important second messenger in tumorogenesis that activates Akt and other signaling molecules involved in a variety of cellular events, such as survival, proliferation, cell motility, and invasion.38
There are two possible explanations for the overexpression of FOXO3 and PTEN in CTC-enriched samples. First, the functions of both tumor suppressor genes predominantly depend on specific posttranslational modifications rather than on gene expression levels. Several other studies have reported that FOXO3 and PTEN activities can be controlled by posttranslational regulation mechanisms, including phosphorylation, acetylation, methylation, ubiquitin-mediated proteasomal degradation, and alteration of subcellular localization.39,40 Second, it has been demonstrated that PTEN and KRAS are frequently mutated and can either lose their function or gain oncogenic properties.41,42
Gene expression profiling of CTCs also revealed several surprising findings. Genes that are known to be expressed in tumors were downregulated in the CTC-enriched samples compared with the control group, including mTOR, Myc, and PARP, which are known for their roles in the regulation of DNA repair43,44 and tumor progression.45-47 These surprising results must be further investigated.
Due to the low number of CTC-positive patients in this study we performed an additional data analysis using the TCGA data package (The Cancer Genome Atlas, freely accessible database) to compare the gene signature of CTCs (our data) with the gene signature of metastasis and primary tumors (TCGA data). In total, we analyzed 1,090 primary tumors and 7 metastasis, all from breast cancer patients. The analysis revealed similar differential expression of HDAC2, TP53, and UPA (Fig. 5). HDAC2 and TP53 were downregulated in metastasis compared with primary tumors, which is consistent with our data obtained from CTC-positive patients. UPA was over-expressed in primary tumors compared to metastasis and also overexpressed in our CTC-positive patients.
Figure 5.
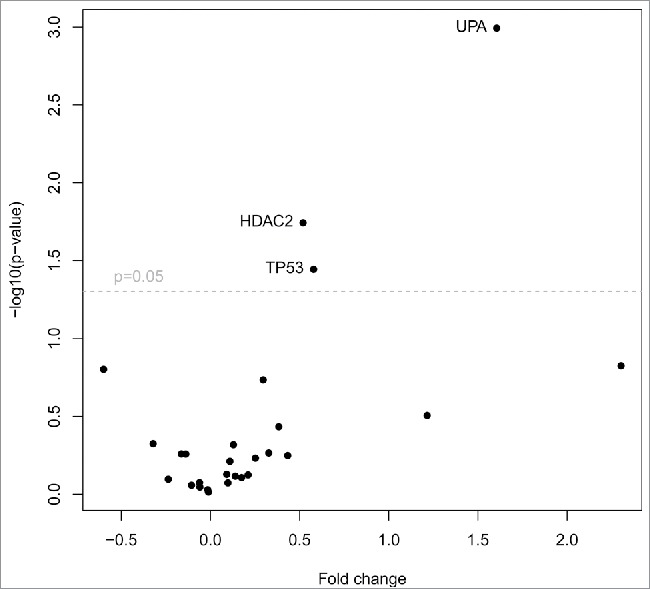
TCGA data analysis of primary tumors and metastasis. Volcano plot filtering shows differentially expressed genes between primary tumors (n = 1,090) and metastasis (n = 7). Three genes (HDAC2, TP53, UPA), that are highlighted above the horizontal line (statistical significance boundary of p < 0.05) were significantly upregulated in primary tumors.
In order to evaluate the prognostic relevance of the gene signature of CTCs we correlated high expression of these genes and patients survival in the independent cohorts of patients (TGCA data set), by performing Kaplan–Meier analyses using the third quartile of the gene's expression as a cut-off value. Among the 27 genes examined, high expression of 5 genes (CD24, HDAC2, KRAS, PIK3CA, RAD51), was associated with significantly shorter overall-survival (p < 0.05, log-rank test), and high expression of RAD51 was significantly associated with shorter relapse-free survival (Fig. 6). Strikingly, not a single gene examined was associated with a favorable outcome.
Figure 6.
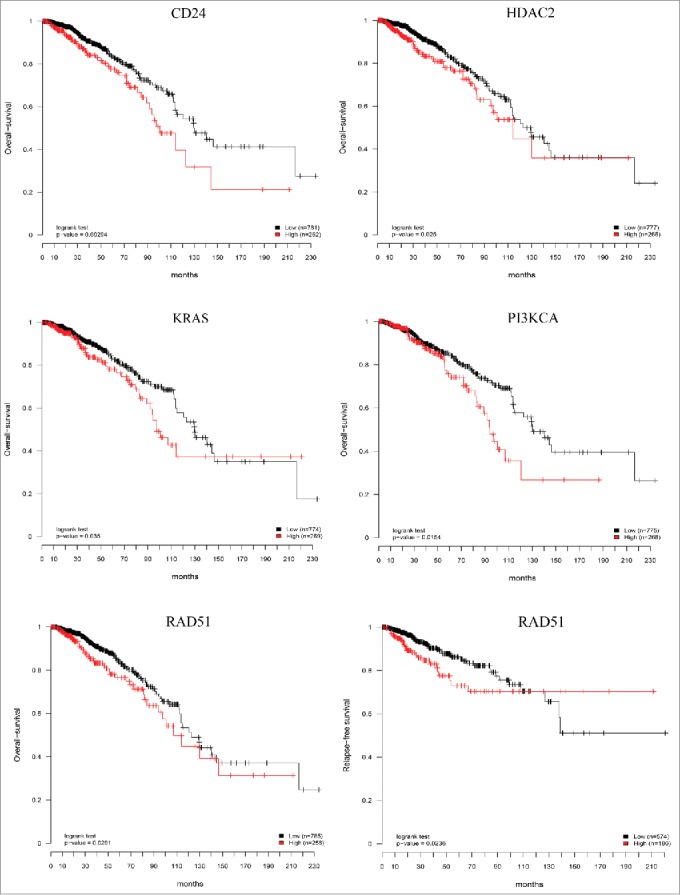
Prognostic relevance of the CTCs gene signature. Six independent Kaplan–Meier curves show the comparison of overall-survival and relapse-free survival in independent cohort of breast cancer patients (TCGA data set) and confirmed that high expression of the genes studied is associated with poor patient's outcome. Red curves: high gene expression; black curves: low gene expression; p values from log-rank tests comparing the two KM curves are shown at the bottom of each plot.
Epithelial to mesenchymal transition is not the only prerequisite for a successful formation of metastasis. CTCs need to have mechanisms that allow them to survive both in the blood stream and at the site of implantation. Little is known about the immunomodulatory capacity of CTCs that would allow them to escape the attack of the immune response. We observed a significant downregulation of mTOR and Myc expression in the CTC-enriched samples. It has been published that the transcription factor Myc is essentially involved in T cell activation-induced cell growth and proliferation, while mTOR deficiency is associated with regulatory T cell differentiation.49 Besides that, the transcription factor FOXO3 regulates dendritic cells function. Increased expression of FOXO3 is associated with the induction of immune tolerance.50 Our CTC-enriched samples had FOXO3 significantly overexpressed. Similarly, expression of known immunosuppressive molecules such as VEGFA and its receptor VEGFR151 was exclusively detected in CTCs and not in PBMCs. Finally, PARP functions as a negative regulator of Foxp3+ regulatory T cells.52 PARP downregulation in CTC-enriched samples may thus also contribute to the immuno-suppressive effect of CTCs.
In the second part of the study, we examined whether the gene expression profile of CTCs could be detected in the PBMC fraction in breast cancer patients. Therefore, we performed gene expression profiling of the same panel of 55 cancer-related genes in PBMCs from 147 breast cancer patients at different stage of the disease. Unfortunately, most of the genes did not show the same regulatory patterns that were detected in the CTC-enriched samples and following interesting findings may have been caused by the excessive background gene expression in the haematopoietic cells. Some of the genes that were significantly differentially expressed in patient's PBMCs were also detected in CTCs (CD24, CD44, HDAC2, IGFR1, KRAS, mTOR, Myc, PARP, and TP53), however all of them were downregulated in patient's PBMCs including the CD44, HDAC2, and KRAS genes which were in contrast upregulated in CTCs. MRP4 was the only upregulated gene in patient's PBMCs, however this gene was not detectable in CTCs.
This study also showed that the specific gene expression profile of PBMCs in breast cancer patients may have a prognostic value, although its significance differs from that of the CTC profile. We identified genes associated with tumor grade and the formation of metastasis differentially expressed in PBMCs.
In summary, most prognostic models rely on information derived from primary tumors or cancer cell lines; however, the clinical outcome of cancer patients is largely determined by metastasis rather than primary tumors. CTCs play a key role in the metastatic spread of primary tumors; therefore, the molecular characterization of CTCs may improve the prediction of metastasis formation and lead to better treatment decisions. We showed that CTCs from breast cancer patients mainly overexpress genes involved in proteolytic degradation of the ECM as well as genes that play important roles in the EMT process in cancer cells. In addition, the urokinase plasminogen system was found to be preferentially activated in CTCs. Despite all of the challenges encountered, such as leukocyte contamination and a low frequency of CTCs, we were successfully able to perform expression profiling of cancer-related genes in CTC-enriched samples and in PBMCs of breast cancer patients. However, our findings should be interpreted with caution due to the small number of CTC-positive patients evaluated.
Patients and methods
Patient characteristics
The study has received ethical approval by the Multicentric Ethics Committee of the University Hospital Motol (approved 13.1.2010; ref. 4.1.1) and all patients signed a written informed consent form.
A total of 147 breast cancer patients were enrolled in this study between 2011 and 2014. The presence of a tumor was confirmed histologically. The patients were distributed into four groups according to tumor stage (stage I to IV), which was determined by a pathologist in compliance with common standards. The median patient age at the time of diagnosis was 47 y (ranging from 24 to 84 y). Patients in the advanced stage of the disease were treated with neoadjuvant chemotherapy (a combination of anthracycline/cyclophosphamide/taxanes) before surgery, and blood samples were collected. At the time of sample collection, 83% of the patients did not have any metastasis. A total of 105 patients had a positive estrogen receptor status, 92 patients were progesterone receptor-positive, and 32 had HER2/neu overexpression, as determined by pathology reports. Details of the patient clinicopathological parameters are presented in Table 1.
Table 1.
Clinicopathological parameters of all patients enrolled in the study. Patient group (n = 147); median age: 47 years; age range: 24–84 years.
| No. of patients | % | |
|---|---|---|
| Time between primary treatment and CTC sampling | ||
| ≤ 1 year | 70 | 62.5 |
| > 1 year | 42 | 37.5 |
| Grading (Bloom-Richardson) | ||
| I (well differentiated) | 22 | 15 |
| II (moderately differentiated) | 77 | 52.4 |
| III (poorly differentiated) | 48 | 32.6 |
| Cancer stage | ||
| I | 62 | 42.2 |
| II | 28 | 19 |
| III | 43 | 29.3 |
| IV | 14 | 9.5 |
| Lymph node involvement | ||
| Yes | 67 | 45.6 |
| No | 80 | 54.4 |
| Estrogen receptor status | ||
| Positive | 105 | 71.4 |
| Negative | 42 | 28.6 |
| Progesterone receptor status | ||
| Positive | 92 | 62.6 |
| Negative | 55 | 37.4 |
| HER2/neu status | ||
| Positive | 32 | 21.8 |
| Negative | 115 | 78.2 |
| Chemotherapy | ||
| Yes | 67 | 45.6 |
| No | 80 | 54.4 |
| Metastasis | ||
| Yes | 26 | 17.7 |
| No | 121 | 82.3 |
| Disease progression | ||
| Yes | 31 | 21.1 |
| No | 115 | 78.2 |
Blood collection
Peripheral blood samples for the enrichment of CTCs and isolation of PBMCs were collected at the Department of Obstetrics and Gynecology, 2nd Faculty of Medicine, Charles University in Prague, University Hospital Motol (112 blood samples in total) and at the Department of Oncology, 1st Faculty of Medicine, Charles University in Prague, General University Hospital (35 blood samples in total). For PBMC isolation, 8 mL of peripheral blood was drawn from each patient and healthy donor and collected into a K2EDTA BD Vacutainer (Becton Dickinson Diagnostics, USA). For CTC enumeration, 7 mL of peripheral blood was drawn into an AdnaCollect tube (AdnaGen, Germany) and processed within 4 h after collection, according to the manufacturer's instructions.
Isolation and detection of CTCs
CTC enrichment from peripheral blood samples was performed with AdnaTest BreastCancer Select and AdnaTest BreastCancer Detect kits (AdnaGen AG, Germany). The AdnaTest system enables the immunomagnetic enrichment of tumor cells using antibody-linked Dynabeads, which specifically bind to the tumor markers EpCAM (GA733–2) and MUC1. Multiplex PCR is then performed to identify tumor-specific transcripts (HER2, MUC1, and EpCAM). Briefly, peripheral blood collected in an AdnaCollect tube (AdnaGen AG, Germany) was preserved at 4–8°C, transported to the laboratory and processed for up to 4 h after collection. A 5-mL aliquot of collected blood was incubated with a pre-prepared mixture of antibodies against EpCAM and MUC1, followed by magnetic separation, lysis of the separated cell fraction, mRNA isolation and reverse transcription (Sensiscript® Reverse Transcriptase, Qiagen, Germany). cDNA was then used as a template for multiplex PCR, and the PCR products were analyzed with an Agilent 2100 Bioanalyzer (Agilent Technologies, USA). The AdnaTest was considered to be positive when a PCR fragment of at least one tumor-specific transcript was clearly detected and the densitometrically calculated PCR product concentration was > 0.15 ng/μL. β-actin was used as an internal PCR control, according to the manufacturer's instructions.
PBMC isolation
PBMCs were isolated from 8 mL of peripheral blood by Ficoll-Paque PLUS (GE Healthcare, Sweden) gradient centrifugation. Briefly, 4 mL of Ficoll was pipetted into 15-mL centrifuge tubes. Heparinized blood was diluted at a 1:1 ratio with phosphate-buffered saline containing EDTA (PBS-EDTA pH 7.5, Lonza, Belgium) and carefully layered over a Ficoll-Paque gradient (8 mL/tube). Samples were centrifuged at 600 × g for 30 min at 20°C. The PBMC layer was collected, and the cells were washed twice in PBS-EDTA (10 min/270 × g/4°C and 10 min/220 × g/4°C), followed by a final wash step in PBS without EDTA. The cells were then resuspended and counted using a Bürker chamber. Cell lysates were prepared from 2×106 PBMCs and 350 μL of RLT buffer with 1% β-mercaptoethanol (Qiagen, Germany). The samples were stored at −80°C.
Isolation of RNA from PBMCs and reverse transcription
Total RNA was isolated using an RNeasy Mini Kit (Qiagen, Germany). Each sample containing RLT buffer and cell lysate was quickly thawed and processed in accordance with the manufacturer's protocol; including a DNase I digestion step. The RNA concentration and purity were determined using a NanoDrop 2000c (Thermo Scientific, Germany), and the RNA integrity was assessed using an Agilent 2000 Bioanalyzer (Agilent, USA). Purified RNA samples were stored at −80°C until further use. cDNA was synthesized from 100 ng of total RNA using an iScript cDNA Synthesis Kit (BioRad, USA).
cDNA pre-amplification of CTC-enriched samples
The pre-amplification of CTC cDNA was performed with a T100 Thermal Cycler (Bio-Rad, USA). Each reaction (50 µL) contained primers (50 nM each; IDT, USA), RNase-free water, 5 µL of CTC cDNA (including the beads) and GrandMaster Preamp Mix (TATAA Biocenter, Sweden). All pre-amplification assays performed are listed in the Table S2. Pre-amplification was carried out under the following conditions: 1 min at 95°C followed by 18 cycles of pre-amplification (95°C for 5 s, 60°C for 120 s, and 72°C for 60 s). The pre-amplification product was placed on dry ice and diluted 8× with RNase-free water prior to use.
Gene expression profiling of CTCs and PBMCs
Quantitative real-time PCR (qPCR) was performed to assess the CTC samples with a LightCycler480 (Roche, Switzerland) and the PBMC samples with QuantStudio (Life Technologies, USA). In both cases, 384-well blocks and ZEN hydrolysis probe detection chemistry (IDT, USA) were used. Each reaction (10 µL) contained primers (400 nM each; IDT, USA), ZEN probe (200 nM; IDT, USA), RNase-free water, 2 µL of 8x-diluted pre-amplification product and GrandMaster Probe mix (TATAA Biocenter, Sweden). The beads were removed from the CTC samples. For PBMC profiling, 1 μL of 5x-diluted cDNA was used per qPCR. Diluted cDNA was mixed with GrandMaster Probe mix and dispensed in a 384-well plate, and the primers, probes and water were added. The temperature cycling protocol was as follows: 1 min at 95°C, followed by 45 cycles of amplification (95°C for 5 s and 60°C for 30 s). Liquid handling was carried out with an EpMotion 5070 (Eppendorf, Germany). qPCR was performed in duplicate. Cycle of quantification (Cq) values were obtained using the 2nd derivative maximum method with LightCycler and the global threshold method of QuantStudio. The specificities of all assays were previously validated by melting curve analysis, and the formation of PCR products of the expected lengths was confirmed by agarose gel electrophoresis. All assays were designed to span introns, and the BLAST algorithm was used to check for potential pseudogenes. During validation, all assays resulted in more than five cycles of difference between the normal cDNA sample and the RT negative control that contained only residual genomic DNA, the contribution of which was subtracted using ValidPrime.53 All qPCR assays were optimized so that primer-dimer signals did not appear within 45 cycles of amplification, and the PCR efficiencies for all assays were 90–100%. Standard curves were analyzed with GenEx (MultiD Analyses). An interplate calibrator (TATAA Biocenter, Sweden) was used to compensate for instrument variation between qPCR runs, and reactions were performed in quadruplicate. All experiments were conducted according to the Minimum Information for Publication of qPCR Experiments guidelines.54
Statistical analysis
PCR data were analyzed by unpaired Student's t-test using GenEx software (MultiD Analyses). Relative gene expression levels were calculated using the ΔΔCt method and were normalized to the expression levels of reference genes selected by Normfinder. All values are presented as the mean ± SEM. Differences were considered significant at p ≤ 0.05. The differential expression of genes according to clinical parameters was assessed by Student's t-test or ANOVA.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by IGA grant CR: NT 11404–5 and by GAUK grant 5734/2012.
References
- 1.Weigelt B, Peterse JL, van 't Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer 2005; 5:591-602; PMID:16056258; http://dx.doi.org/ 10.1038/nrc1670 [DOI] [PubMed] [Google Scholar]
- 2.Bernstein LR, Liotta LA. Molecular mediators of interactions with extracellular matrix components in metastasis and angiogenesis. Curr Opin Oncol 1994; 6:106-13; PMID:7515692; http://dx.doi.org/ 10.1097/00001622-199401000-00015 [DOI] [PubMed] [Google Scholar]
- 3.Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol 2001; 13:555-62; PMID:11544023; http://dx.doi.org/ 10.1016/S0955-0674(00)00251-9 [DOI] [PubMed] [Google Scholar]
- 4.Alfano D, Iaccarino I, Stoppelli MP. Urokinase signaling through its receptor protects against anoikis by increasing BCL-xL expression levels. J Biol Chem 2006; 281:17758-67; PMID:16632475; http://dx.doi.org/ 10.1074/jbc.M601812200 [DOI] [PubMed] [Google Scholar]
- 5.Ravid D, Maor S, Werner H, Liscovitch M. Caveolin-1 inhibits cell detachment-induced p53 activation and anoikis by upregulation of insulin-like growth factor-I receptors and signaling. Oncogene 2005; 24:1338-47; PMID:15592498; http://dx.doi.org/ 10.1038/sj.onc.1208337 [DOI] [PubMed] [Google Scholar]
- 6.Mego M, Mani SA, Cristofanilli M. Molecular mechanisms of metastasis in breast cancer–clinical applications. Nat Rev Clin Oncol 2010; 7:693-701; PMID:20956980; http://dx.doi.org/ 10.1038/nrclinonc.2010.171 [DOI] [PubMed] [Google Scholar]
- 7.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014; 15:178-96; PMID:24556840; http://dx.doi.org/ 10.1038/nrm3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wicha MS, Hayes DF. Circulating tumor cells: not all detected cells are bad and not all bad cells are detected. J Clin Oncol 2011; 29:1508-11; PMID:21422428; http://dx.doi.org/ 10.1200/JCO.2010.34.0026 [DOI] [PubMed] [Google Scholar]
- 9.Braun S, Naume B. Circulating and disseminated tumor cells. J Clin Oncol 2005; 23:1623-6; PMID:15755968; http://dx.doi.org/ 10.1200/JCO.2005.10.073 [DOI] [PubMed] [Google Scholar]
- 10.Ross AA, Cooper BW, Lazarus HM, Mackay W, Moss TJ, Ciobanu N, Tallman MS, Kennedy MJ, Davidson NE, Sweet D et al.. Detection and viability of tumor cells in peripheral blood stem cell collections from breast cancer patients using immunocytochemical and clonogenic assay techniques. Blood 1993; 82:2605-10; PMID:8219214. [PubMed] [Google Scholar]
- 11.Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW et al.. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med 2004; 351:781-91; PMID:15317891; http://dx.doi.org/ 10.1056/NEJMoa040766 [DOI] [PubMed] [Google Scholar]
- 12.Andreopoulou E, Yang LY, Rangel KM, Reuben JM, Hsu L, Krishnamurthy S, Valero V, Fritsche HA, Cristofanilli M. Comparison of assay methods for detection of circulating tumor cells in metastatic breast cancer: AdnaGen AdnaTest BreastCancer Select/Detect versus Veridex CellSearch system. Int J Cancer 2012; 130:1590-7; PMID:21469140; http://dx.doi.org/ 10.1002/ijc.26111 [DOI] [PubMed] [Google Scholar]
- 13.Pestrin M, Bessi S, Galardi F, Truglia M, Biggeri A, Biagioni C, Cappadona S, Biganzoli L, Giannini A, Di Leo A. Correlation of HER2 status between primary tumors and corresponding circulating tumor cells in advanced breast cancer patients. Breast Cancer Res Treat 2009; 118:523-30; PMID:19597704; http://dx.doi.org/ 10.1007/s10549-009-0461-7 [DOI] [PubMed] [Google Scholar]
- 14.Aktas B, Muller V, Tewes M, Zeitz J, Kasimir-Bauer S, Loehberg CR, Rack B, Schneeweiss A, Fehm T. Comparison of estrogen and progesterone receptor status of circulating tumor cells and the primary tumor in metastatic breast cancer patients. Gynecol Oncol 2011; 122:356-60; PMID:21605893; http://dx.doi.org/ 10.1016/j.ygyno.2011.04.039 [DOI] [PubMed] [Google Scholar]
- 15.Banys M, Krawczyk N, Becker S, Jakubowska J, Staebler A, Wallwiener D, Fehm T, Rothmund R. The influence of removal of primary tumor on incidence and phenotype of circulating tumor cells in primary breast cancer. Breast Cancer Res Treat 2012; 132:121-9; PMID:21562707; http://dx.doi.org/ 10.1007/s10549-011-1569-0 [DOI] [PubMed] [Google Scholar]
- 16.Fehm T, Hoffmann O, Aktas B, Becker S, Solomayer EF, Wallwiener D, Kimmig R, Kasimir-Bauer S. Detection and characterization of circulating tumor cells in blood of primary breast cancer patients by RT-PCR and comparison to status of bone marrow disseminated cells. Breast Cancer Res 2009; 11:R59; PMID:19664291; http://dx.doi.org/ 10.1186/bcr2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sieuwerts AM, Kraan J, Bolt-de Vries J, van der Spoel P, Mostert B, Martens JW, Gratama JW, Sleijfer S, Foekens JA. Molecular characterization of circulating tumor cells in large quantities of contaminating leukocytes by a multiplex real-time PCR. Breast Cancer Res Treat 2009; 118:455-68; PMID:19115104; http://dx.doi.org/ 10.1007/s10549-008-0290-0 [DOI] [PubMed] [Google Scholar]
- 18.Obermayr E, Sanchez-Cabo F, Tea MK, Singer CF, Krainer M, Fischer MB, Sehouli J, Reinthaller A, Horvat R, Heinze G et al.. Assessment of a six gene panel for the molecular detection of circulating tumor cells in the blood of female cancer patients. BMC Cancer 2010; 10:666; PMID:21129172; http://dx.doi.org/ 10.1186/1471-2407-10-666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lauss M, Kriegner A, Vierlinger K, Visne I, Yildiz A, Dilaveroglu E, Noehammer C. Consensus genes of the literature to predict breast cancer recurrence. Breast Cancer Res Treat 2008; 110:235-44; PMID:17899371; http://dx.doi.org/ 10.1007/s10549-007-9716-3 [DOI] [PubMed] [Google Scholar]
- 20.Andreasen PA, Kjoller L, Christensen L, Duffy MJ. The urokinase-type plasminogen activator system in cancer metastasis: a review. Int J Cancer 1997; 72:1-22; PMID:9212216; http://dx.doi.org/ 10.1002/(SICI)1097-0215(19970703)72:1%3c1::AID-IJC1%3e3.0.CO;2-Z [DOI] [PubMed] [Google Scholar]
- 21.de Witte JH, Foekens JA, Brunner N, Heuvel JJ, van Tienoven T, Look MP, Klijn JG, Geurts-Moespot A, Grebenchtchikov N, Benraad T et al.. Prognostic impact of urokinase-type plasminogen activator receptor (uPAR) in cytosols and pellet extracts derived from primary breast tumours. Br J Cancer 2001; 85:85-92; PMID:11437407; http://dx.doi.org/ 10.1054/bjoc.2001.1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma Z, Webb DJ, Jo M, Gonias SL. Endogenously produced urokinase-type plasminogen activator is a major determinant of the basal level of activated ERK/MAP kinase and prevents apoptosis in MDA-MB-231 breast cancer cells. J Cell Sci 2001; 114:3387-96; PMID:11591826 [DOI] [PubMed] [Google Scholar]
- 23.Alexander RA, Prager GW, Mihaly-Bison J, Uhrin P, Sunzenauer S, Binder BR, Schutz GJ, Freissmuth M, Breuss JM. VEGF-induced endothelial cell migration requires urokinase receptor (uPAR)-dependent integrin redistribution. Cardiovasc Res 2012; 94:125-35; PMID:22287577; http://dx.doi.org/ 10.1093/cvr/cvs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xue A, Scarlett CJ, Jackson CJ, Allen BJ, Smith RC. Prognostic significance of growth factors and the urokinase-type plasminogen activator system in pancreatic ductal adenocarcinoma. Pancreas 2008; 36:160-7; PMID:18376307; http://dx.doi.org/ 10.1097/MPA.0b013e31815750f0 [DOI] [PubMed] [Google Scholar]
- 25.Pulukuri SM, Gorantla B, Rao JS. Inhibition of histone deacetylase activity promotes invasion of human cancer cells through activation of urokinase plasminogen activator. J Biol Chem 2007; 282:35594-603; PMID:17923479; http://dx.doi.org/ 10.1074/jbc.M705867200 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Thorpe SM, Rochefort H, Garcia M, Freiss G, Christensen IJ, Khalaf S, Paolucci F, Pau B, Rasmussen BB, Rose C. Association between high concentrations of Mr 52,000 cathepsin D and poor prognosis in primary human breast cancer. Cancer Res 1989; 49:6008-14; PMID:2790815 [PubMed] [Google Scholar]
- 27.Glondu M, Liaudet-Coopman E, Derocq D, Platet N, Rochefort H, Garcia M. Downregulation of cathepsin-D expression by antisense gene transfer inhibits tumor growth and experimental lung metastasis of human breast cancer cells. Oncogene 2002; 21:5127-34; PMID:12140763; http://dx.doi.org/ 10.1038/sj.onc.1205657 [DOI] [PubMed] [Google Scholar]
- 28.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol 2005; 23:1011-27; PMID:15585754; http://dx.doi.org/ 10.1200/JCO.2005.06.081 [DOI] [PubMed] [Google Scholar]
- 29.Yang AD, Camp ER, Fan F, Shen L, Gray MJ, Liu W, Somcio R, Bauer TW, Wu Y, Hicklin DJ et al.. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res 2006; 66:46-51; PMID:16397214; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3086 [DOI] [PubMed] [Google Scholar]
- 30.Dhillon N, Oki M, Szyjka SJ, Aparicio OM, Kamakaka RT. H2A.Z functions to regulate progression through the cell cycle. Mol Cell Biol 2006; 26:489-501; PMID:16382141; http://dx.doi.org/ 10.1128/MCB.26.2.489-501.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hua S, Kallen CB, Dhar R, Baquero MT, Mason CE, Russell BA, Shah PK, Liu J, Khramtsov A, Tretiakova MS et al.. Genomic analysis of estrogen cascade reveals histone variant H2A.Z associated with breast cancer progression. Mol Syst Biol 2008; 4:188; PMID:18414489; http://dx.doi.org/ 10.1038/msb.2008.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schweitzer KM, Drager AM, van der Valk P, Thijsen SF, Zevenbergen A, Theijsmeijer AP, van der Schoot CE, Langenhuijsen MM. Constitutive expression of E-selectin and vascular cell adhesion molecule-1 on endothelial cells of hematopoietic tissues. Am J Pathol 1996; 148:165-75; PMID:8546203 [PMC free article] [PubMed] [Google Scholar]
- 33.Wagers AJ, Lowe JB, Kansas GS. An important role for the α 1,3 fucosyltransferase, FucT-VII, in leukocyte adhesion to E-selectin. Blood 1996; 88:2125-32; PMID:8822932 [PubMed] [Google Scholar]
- 34.Draffin JE, McFarlane S, Hill A, Johnston PG, Waugh DJ. CD44 potentiates the adherence of metastatic prostate and breast cancer cells to bone marrow endothelial cells. Cancer Res 2004; 64:5702-11; PMID:15313910; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-0389 [DOI] [PubMed] [Google Scholar]
- 35.Olsson E, Honeth G, Bendahl PO, Saal LH, Gruvberger-Saal S, Ringner M, Vallon-Christersson J, Jonsson G, Holm K, Lovgren K et al.. CD44 isoforms are heterogeneously expressed in breast cancer and correlate with tumor subtypes and cancer stem cell markers. BMC Cancer 2011; 11:418; PMID:21957977; http://dx.doi.org/ 10.1186/1471-2407-11-418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Theodoropoulos PA, Polioudaki H, Agelaki S, Kallergi G, Saridaki Z, Mavroudis D, Georgoulias V. Circulating tumor cells with a putative stem cell phenotype in peripheral blood of patients with breast cancer. Cancer Lett 2010; 288:99-106; PMID:19619935; http://dx.doi.org/ 10.1016/j.canlet.2009.06.027 [DOI] [PubMed] [Google Scholar]
- 37.Calnan DR, Brunet A. The FoxO code. Oncogene 2008; 27:2276-88; PMID:18391970; http://dx.doi.org/ 10.1038/onc.2008.21 [DOI] [PubMed] [Google Scholar]
- 38.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006; 441:424-30; PMID:16724053; http://dx.doi.org/ 10.1038/nature04869 [DOI] [PubMed] [Google Scholar]
- 39.Singh G, Chan AM. Post-translational modifications of PTEN and their potential therapeutic implications. Curr Cancer Drug Targets 2011; 11:536-47; PMID:21486223; http://dx.doi.org/ 10.2174/156800911795655930 [DOI] [PubMed] [Google Scholar]
- 40.Zhao Y, Wang Y, Zhu WG. Applications of post-translational modifications of FoxO family proteins in biological functions. J Mol Cell Biol 2011; 3:276-82; PMID:21669942; http://dx.doi.org/ 10.1093/jmcb/mjr013 [DOI] [PubMed] [Google Scholar]
- 41.Califano R, Landi L, Cappuzzo F. Prognostic and predictive value of K-RAS mutations in non-small cell lung cancer. Drugs 2012; 72 Suppl 1:28-36; PMID:22712795; http://dx.doi.org/ 10.2165/1163012-S0-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 42.Bonneau D, Longy M. Mutations of the human PTEN gene. Hum Mutat 2000; 16:109-22; PMID:10923032; http://dx.doi.org/ 10.1002/1098-1004(200008)16:2%3c109::AID-HUMU3%3e3.0.CO;2-0 [DOI] [PubMed] [Google Scholar]
- 43.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C et al.. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434:917-21; PMID:15829967; http://dx.doi.org/ 10.1038/nature03445 [DOI] [PubMed] [Google Scholar]
- 44.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005; 434:913-7; PMID:15829966; http://dx.doi.org/ 10.1038/nature03443 [DOI] [PubMed] [Google Scholar]
- 45.Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol 2001; 17:615-75; PMID:11687500; http://dx.doi.org/ 10.1146/annurev.cellbio.17.1.615 [DOI] [PubMed] [Google Scholar]
- 46.Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis 2004; 9:667-76; PMID:15505410; http://dx.doi.org/ 10.1023/B:APPT.0000045801.15585.dd [DOI] [PubMed] [Google Scholar]
- 47.Deming SL, Nass SJ, Dickson RB, Trock BJ. C-myc amplification in breast cancer: a meta-analysis of its occurrence and prognostic relevance. Br J Cancer 2000; 83:1688-95; PMID:11104567; http://dx.doi.org/ 10.1054/bjoc.2000.1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J et al.. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011; 35:871-82; PMID:22195744; http://dx.doi.org/ 10.1016/j.immuni.2011.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 2009; 30:832-44; PMID:19538929; http://dx.doi.org/ 10.1016/j.immuni.2009.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watkins SK, Hurwitz AA. FOXO3: A master switch for regulating tolerance and immunity in dendritic cells. Oncoimmunology 2012; 1:252-4; PMID:22720261; http://dx.doi.org/ 10.4161/onci.1.2.18241 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51.Johnson BF, Clay TM, Hobeika AC, Lyerly HK, Morse MA. Vascular endothelial growth factor and immunosuppression in cancer: current knowledge and potential for new therapy. Expert Opin Biol Ther 2007; 7:449-60; PMID:17373897; http://dx.doi.org/ 10.1517/14712598.7.4.449 [DOI] [PubMed] [Google Scholar]
- 52.Zhang P, Maruyama T, Konkel JE, Abbatiello B, Zamarron B, Wang ZQ, Chen W. PARP-1 controls immunosuppressive function of regulatory T cells by destabilizing Foxp3. PloS One 2013; 8:e71590; PMID:23977081; http://dx.doi.org/ 10.1371/journal.pone.0071590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laurell H, Iacovoni JS, Abot A, Svec D, Maoret JJ, Arnal JF, Kubista M. Correction of RT-qPCR data for genomic DNA-derived signals with ValidPrime. Nucleic Acids Res 2012; 40:e51; PMID:22228834; http://dx.doi.org/ 10.1093/nar/gkr1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL et al.. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 2009; 55:611-22; PMID:19246619; http://dx.doi.org/ 10.1373/clinchem.2008.112797 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.