

ABSTRACT
We conducted a phase II clinical trial of anti-CTLA-4 antibody (ipilimumab) and granulocyte-macrophage colony-stimulating factor (GM-CSF) in 22 patients with metastatic melanoma and determined clinical outcomes and immunologic responses. The treatment consisted of a 3-mo induction with ipilimumab at 10 mg/kg administered every 3 weeks for four doses in combination with GM-CSF at 125 µg/m2 for 14 d beginning on the day of the ipilimumab infusion and then GM-CSF for 3 mo on the same schedule without ipilimumab. This was followed by maintenance therapy with the combination every 3 mo for up to 2 y or until disease progression or unacceptable toxicity. Blood samples for determination of immune subsets were obtained before treatment, at week 3 (end of cycle 1) and at week 6 (end of cycle 2). Blood samples were also obtained from seven subjects who were cancer-free. The immune response disease control (irDC) rate at 24 weeks was 41% and the overall response rate (ORR) was 32%. The median progression free-survival (PFS) was 3.5 mo and the median overall survival (OS) was 21.1 mo. 41% of the patients experienced Grade 3 to 4 adverse events. We conclude that this combination is safe and the results suggest the combination may be more effective than ipilimumab monotherapy. Further, the results suggest that lower levels of CD4+ effector T cells but higher levels of CD8+ T cells expressing PD-1 at pre-treatment could be a potential biomarker for disease control in patients who receive immunotherapy with ipilimumab and GM-CSF. Further trials of this combination are warranted.
KEYWORDS: CD4+ effector T cells, CD8+ T cells, clinical trial, CTLA-4, GM-CSF, immunotherapy, ipilimumab, metastatic melanoma, PD-1
Abbreviations
- ACTH
adrenocorticotropic hormone
- AJCC
American Joint Committee on Cancer
- CBC
complete blood count
- CD
cluster of differentiation
- CI
confidence interval
- CNS
central nervous system
- CT
computed tomography
- CTCAE
Common Terminology Criteria for Adverse Events
- CTLA-4
cytotoxic T lymphocyte antigen-4
- ECOG
Eastern Cooperative Oncology Group
- FACS
fluorescence-activated cell sorting
- FOXP3
foxhead box P3
- GM-CSF
granulocyte macrophage colony-stimulating factor
- ir
immune-related
- irAE
immune-related adverse events
- irCR
Immune-related complete response
- irDC
immune-related disease control
- irPD
immune-related progressive disease
- irPR
immune-related partial response
- LDH
lactate dehydrogenase
- mCRPC
mestastatic castration resistant prostate cancer
- NCI
National Cancer Institute
- NIH
National Institute of Health
- ORR
overall response rate
- OS
overall survival
- PBMC
peripheral blood mononuclear cells
- PD-1
programmed death receptor-1
- PFS
progression free survival
- RECIST
Response Evaluation Criteria in Solid Tumors
- RR
response rate
- Teff
effector T cells
- Tregs
regulatory T cells
- WHO
World Health Organization
Introduction
Ipilimumab (Yervoy), an anti-CTLA-4 monoclonal antibody, was the first agent reported, in a randomized trial, to improve survival in patients with metastatic melanoma.1 and was approved by the FDA in 2011 for the treatment of unresectable or metastatic melanoma.2 Since its approval, it has been studied in combination therapy regimens in an effort to build on these promising results and to gain information about the immunologic mechanisms and improve clinical outcomes.
GM-CSF (sargramostim, Leukine) is a hematopoietic growth factor that triggers proliferation and differentiation of hematopoietic progenitor cells, mainly neutrophils, monocytes/macrophages and myeloid-derived dendritic cells, and is approved by the FDA for this purpose.3 It also has been shown to have an ability to activate macrophages which distinguish and destroy tumor cells from normal cells,4 be able to stimulate peripheral blood monocytes in vitro to become cytotoxic for human melanoma cells,5,6 produce monocyte activation and tumoricidal activity with in vivo administration,7,8 and to stimulate the production of an angiogenesis inhibitor by macrophages.9 GM-CSF also acts as a primary mediator of proliferation, maturation, and migration of dendritic cells,10-12 which are antigen-presenting cells involved in primary and secondary T-cell immune responses, particularly with tumors. In previous studies of adjuvant therapy of melanoma, GM-CSF was reported to prolong OS in patients with Stage III and IV disease as compared with matched historical controls,13 in patients with Stage IIIC melanoma in a single center study using contemporaneous but not randomized controls,14 and an exploratory analysis showed a trend toward improved OS melanoma in a prospective randomized trial with controls treated with placebo.15 GM-CSF has also shown encouraging results in combination with ipilimumab for patients with hormone refractory prostate cancer.16
We proposed that the combination of GM-CSF and ipilimumab, which enhance immune responses via different mechanisms might provide added clinical benefit as compared to monotherapy with either alone without added toxicity since these agents have different safety profiles. Further, we explored the immunologic responses in patients receiving the combination. During our trial, a study similar to ours was published showing an OS benefit and decreased toxicity in patients treated with the combination of GM-CSF and ipilimumab versus ipilimumab alone.17
Results
Patient characteristics
Twenty-two patients were enrolled in the study (Table 1). The median age among study patients was 65 (range 41–85 years). The majority of patients (68%) had M1c disease, including 10 patients (45.5%) with liver involvement and 4 patients (18%) with CNS involvement. Fifteen patients (68%) had elevated lactic dehydrogenase (LDH). The Eastern Cooperative Oncology Group (ECOG) performance status was 0 in 20 patients (91%) and 1 in 2 patients (9%). Thirteen patients (59%) had received prior therapy.
Table 1.
Baseline demographics and specific characteristics (n = 22).
| Characteristic |
n (%) |
|---|---|
| Sex | |
| Male | 12 (54.5%) |
| Female | 10 (45.5%) |
| LDH | |
| Normal | 7 (32%) |
| Elevated | 15 (68%) |
| ECOG performance status | |
| 0 | 20 (91%) |
| 1 | 2 (9%) |
| Stage | |
| IIIC, Unresectable | 1 (4.5%) |
| IV M1a | 1 (4.5%) |
| IV M1b | 5 (23%) |
| IV M1c | 15 (68%) |
| Sites of metastasis | |
| Lymph nodes | 14 (64%) |
| Lung | 14 (64%) |
| Liver | 10 (45.5) |
| Bone | 7 (32%) |
| Subcutaneous tissue | 6 (27%) |
| CNS | 4 (18%) |
| Skin | 3 (14%) |
| Adrenal | 3 (14%) |
| Intestine | 1 (4.5%) |
| Spleen | 1 (4.5%) |
| Retroperitoneum | 1 (4.5%) |
| Prior systemic therapy | |
| No | 15 (68%) |
| Yes | 7 (32%) |
| Prior therapy | |
| No | 9 (41%) |
| Yes | 13 (59%) |
Clinical outcomes
At 24 weeks, 7 of the 22 patients had a partial response (irPR), 2 patients had stable disease (irSD), and 13 patients had progressive disease (irPD) with 8 of the 13 having died by 24 weeks. Thus, the disease control rate (DCR: irCR, irPR, and irSD) at 24 weeks was 41% (primary endpoint). The objective response rate (ORR: irCR and irPR) was 32%. The range of time to objective response was 10.6 weeks to 24 weeks with median time of 12.3 weeks. One patient who had a partial response at 24 weeks developed a complete response (irCR) 1 y after start of treatment.
The median PFS was 3.5 mo (Fig. 2A) and the median OS was 21.1 mo (Fig. 2B). The median OS for those that developed irPD was 5.16 mo, while the median OS for those who experienced disease control has not been reached (Fig. 2C). As of the last follow-up date for this analysis, 10 patients out of 22 were still alive with a minimum follow-up of 7.0 mo for living patients.
Figure 2.

Kaplan–Meier plots of clinical outcomes (n = 22). (A) PFS. (B) OS as of analysis on the censor date. Dotted lines below and above the survival curve (solid line) show lower and upper 95% confidence intervals (CI) respectively. Vertical tick marks indicate OS of patients who were still alive as of the censor date. (C) OS in patients with irDC (n = 9, gray line) compared to OS in patients with irPD (n = 13, black line).
Objective responses were observed in patients receiving the study treatments and these were durable (Fig. 3A). Two patients demonstrated complete response of the index lesions from baseline scan but one of the two continued to have remaining non-index metastatic lesions on imaging and was therefore considered having irPR instead of irCR. Tumor regression is illustrated in a pre-treatment image of liver metastases in one patient and a follow-up image of the same area approximately 12 mo later showing resolution of the liver metastases.(Fig. 3B)
Figure 3.
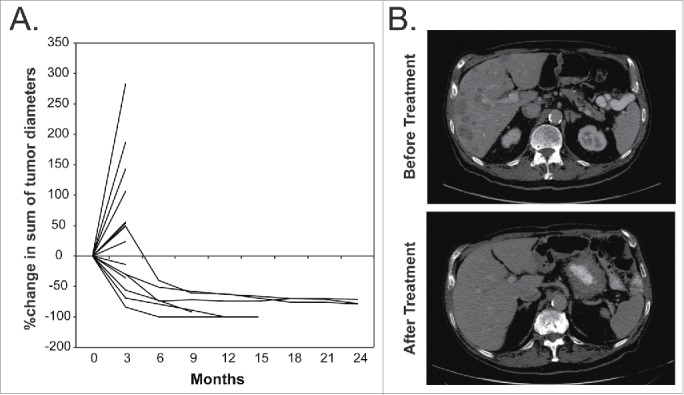
Illustration of Clinical Outcome. (A) The percentage change in the sum of the index tumor diameters for all patients that remained in the study long enough to have follow-up imaging. (B) A pre-treatment image of liver metastases for one patient and a follow-up image of the same area approximately 12 mo later showing resolution of the liver metastases. The remaining hypodense lesion is thought to represent a hepatic cyst.
Adverse events (AE)
The most frequent treatment-related adverse events were injection site reactions due to administration of GM-CSF (Table 2). Other common adverse events were those known to be associated with ipilimumab, including rash, pruritus, fatigue, diarrhea, colitis, anorexia and autoimmune phenomena including increased alanine aminotransferase (ALT), decreased adrenocorticotropic hormone (ACTH), retinitis, arthralgias, and hyperthyroidism. Most of the treatment-related adverse events were Grade 1–2 and were easily controlled; however, 41% of the patients experienced Grade 3–4 adverse events. There were no treatment-related deaths.
Table 2.
Treatment-related adverse events.
| Adverse event | All grades | Grades 1–2 | Grades 3–4 |
|---|---|---|---|
| All | 122 | 99 | 23 |
| General and administration site | 39 | 35 | 4 |
| Injection site reaction | 15 | 13 | 2 |
| Fatigue | 12 | 11 | 1 |
| Fever | 6 | 6 | 0 |
| Infusion reaction | 3 | 2 | 1 |
| Skin and subcutaneous | 27 | 24 | 3 |
| Rash | 14 | 11 | 3 |
| Pruritus | 9 | 9 | 0 |
| Urticaria | 2 | 2 | 0 |
| Gastrointestinal | 20 | 12 | 8 |
| Diarrhea | 8 | 5 | 3 |
| Nausea | 6 | 6 | 0 |
| Colitis | 4 | 0 | 4 |
| Colon perforation | 1 | 0 | 1 |
| Metabolic | 11 | 10 | 1 |
| Anorexia | 8 | 7 | 1 |
| Investigational | 8 | 7 | 1 |
| Increased ALT | 3 | 3 | 0 |
| Weight loss | 2 | 2 | 0 |
| Decreased ACTH | 1 | 0 | 1 |
| Nervous system | 4 | 4 | 0 |
| Headache | 2 | 2 | 0 |
| Eye disorders | 4 | 0 | 4 |
| Retinitis | 1 | 0 | 1 |
| Optic nerve swelling | 1 | 0 | 1 |
| Blurred vision | 1 | 0 | 1 |
| Eye pain | 1 | 0 | 1 |
| Cardiac | 3 | 2 | 1 |
| Atrial fibrillation | 1 | 0 | 1 |
| Pericarditis | 1 | 1 | 0 |
| Musculoskeletal | 3 | 3 | 0 |
| Arthralgias | 2 | 2 | 0 |
| Endocrine | 2 | 1 | 1 |
| Hyperthyroidism | 2 | 1 | 1 |
The first two patients enrolled experienced infusion reactions during the second (but not the first) infusion of ipilimumab. These consisted of flushing, shortness of breath, and hypertension. They were treated by interrupting the infusion, administering diphenhydramine, and resuming the infusion at a slower rate. Thereafter, the protocol was amended so that patients were given premedication with diphenhydramine and famotidine prior to ipilimumab administration and the GM-CSF was given after the infusion, rather than before.
Study treatment was discontinued in five patients due to treatment-related adverse events: colitis (3), rash (1), and retinitis with optic nerve swelling (1). One of these patients completed 3 mo of therapy but then developed treatment-related colitis and colon perforation which was treated with a temporary colostomy. This patient had a durable CR, which is ongoing. Another of these patients, after the 9 mo of treatment, developed right eye pain and blurred vision due to retinitis and optic nerve swelling. The patient's vision diminished to 20/200 in the right eye which improved to 20/60 after treatment with topical steroid eye drops. Other notable adverse events included a single episode each of atrial fibrillation (Grade 3) and pericarditis (Grade 2).
Comparison of characteristics of study patients with response to treatment
Characteristics of study patients were compared between patients who experienced irDC (n = 9) with those in patients with irPD (n = 13). Our analyses showed that age, sex, LDH levels, stage, ECOG performance status, and prior therapy did not relate with response to treatment (Fig. S1). Moreover, the occurrence of immune-related adverse events designated either as less than Grade 3 or equal to or greater than Grade 3 did not relate with response. Of note, although 5 out of 13 patients with irPD received subsequent therapy after leaving the study, only 1 of the 9 patients who experienced irDC received subsequent therapy.
Treatment-induced immunological effects
One patient received only one cycle of treatment and had only a pretreatment blood sample available and was therefore excluded from these paired comparisons (n = 21 for these analyses). The absolute lymphocyte counts were significantly higher at week 3 and week 6 compared to week 0 (Fig. 4A). Among the total lymphocytes, the percentages of regulatory T cells (Tregs), CD4+ effector T (Teff) cells expressing PD-1, and CD8+ T cells expressing PD-1 were significantly higher at week 3 and week 6 compared to week 0 (Fig. 4D, 4H–I). The percentages of CD4+ Teff cells were only significantly higher at week 3 but not at week 6 compared to week 0 (Fig. 4B). The percentages of CD8+ T cells among the total lymphocytes were not significantly different from pre-treatment levels after either cycle of treatment (Fig. 4C). The ratios of both CD4+ Teff cells and CD8+ T cells to Tregs were significantly lower at both week 3 and week 6 compared to week 0.(Fig. 4E–F)
Figure 4.

Treatment-induced immunological time course for week 0, week 3 and week 6. (A) Absolute lymphocyte counts; (B) Percentage of CD4+ Teff cells of total lymphocytes; (C) Percentage of CD8+ T cells of total lymphocytes; (D) Percentage of Tregsof total lymphocytes; (E) Ratio of CD4+ Teff cells to Tregs; (F) Ratio of CD8+ T cells to Tregs; (G) Percentage change of Tregsfrom week 0; (H) Percentage of CD4+ Teff cells that expressed PD-1; (I) Percentage of CD8+ T cells that expressed PD-1. Connected dots show time course of the same patient. By Bonferroni correction, the statistical significance is declared if p value is < 0.025.
Comparison of immune subsets with patients' response to treatment
The results of the immune subset analyses were evaluated for relation with the patients' response to treatment (irDC, n = 9; irPD, n = 12). For most of these subsets, neither the baseline result nor the results at week 3 or week 6 during treatment related with the patients' response (Fig. 5, Table S1). A lower level of the percentage of CD4+ Teff cells expressing PD-1 at baseline and at week 6 related significantly with clinical benefit (irDC).(Fig. 5H)
Figure 5.

Comparisons of immune subsets between irDC and irPD. Scatter plots of the following immune subsets of patients with irDC versus patients with irPD at week 0, week 3 and week 6: (A) Absolute lymphocyte counts; (B) Percentage of CD4+ Teff cells of total lymphocytes; (C) Percentage of CD8+ T cells of total lymphocytes; (D) Percentage of Tregsof total lymphocytes; (E) Ratio of CD4+ Teff cells to Tregs; (F) Ratio of CD8+ T cells to Tregs; (G) Percentage change of Tregsfrom week 0; (H) Percentage of CD4+ Teff cells that expressed PD-1; (I) Percentage of CD8+ T cells that expressed PD-1. Error bars show standard deviations.
Cancer-free controls vs. study patients
Immune subset distributions of patients with irDC and irPD were compared with those of cancer-free controls. The percentages of CD4+ Teff cells, CD8+ T cells and Tregs of cancer-free controls were not significantly different from pre-treatment levels of patients with irDC or irPD (Fig. 6A–C, Table S2). However, patients with irPD had significantly higher pre-treatment percentages of CD4+ Teff cells expressing PD-1 compared to cancer-free controls, whereas the levels between patients with irDC and cancer-free controls did not differ (Fig. 6D). On the other hand, the pre-treatment percentages of CD8+ T cells expressing PD-1 were significantly higher in patients with irDC compared to cancer-free controls, whereas the levels between patients with irPD and cancer-free controls did not differ.(Fig. 6E)
Figure 6.

Comparisons of immune subsets between cancer-free controls and pre-treatment levels of patients with metastatic melanoma. Scatter plots of the following immune subsets for cancer-free controls, irDC and irPD: (A) Percentage of CD4+ Teff cells of lymphocytes; (B) Percentage of CD8+ T cells of lymphocytes; (C) Percentage of Tregs of lymphocytes; (D) Percentage of CD4+ Teff cells expressing surface PD-1; (E) Percentage of CD8+ T cells expressing surface PD-1. Error bars show standard deviations. Error bars show standard deviations. By Bonferroni correction, the statistical significance is declared if p value is < 0.017.
Discussion
The combination of ipilimumab and GM-CSF demonstrated clinical benefit in patients with metastatic melanoma despite the fact that the majority of the patients included in this study had characteristics indicating a poor prognosis and predicting a short duration of survival. These characteristics included Stage IV M1c disease.18,19 and multiple sites of metastases,20-22 including CNS and liver. In addition, 68% of the patients had elevated LDH levels, also associated with poor prognosis.23,24 Nonetheless, the results of this study suggest that this combination has significant activity in these patients. The DCR, ORR, and PFS in the study patients were similar to those reported previously for patients receiving second-line ipilimumab monotherapy.1 or first-line ipilimumab therapy with dacarbazine.25 However, the median OS in our study was 21.1 mo was more than double the 10.1 mo reported for second-line ipilimumab monotherapy.1 and almost double the 11.2 mo reported for first-line ipilimumab therapy with dacarbazine.25 Some patients enjoyed deep and durable clinical responses that were ongoing at last follow-up. It is unlikely that the survival advantage was due to follow-on therapies the patients were given, since only six patients received follow-on therapy of whom five had irPD.
During the period of this trial, a Phase II randomized trial was conducted in which patients with metastatic melanoma were randomized to receive ipilimumab plus GM-CSF or ipilimumab alone.17 The treatment regimen, reported by Hodi et. al. was similar to that used in the trial reported herein and the results were analogous. The median PFS reported herein was 3.5 mo and was 3.1 mo for treatment with the combination in the trial reported by Hodi et. al.; the median OS reported herein was 21.1 mo and was 17.5 mo with the combination in the trial reported by Hodi et. al.; the incidence of adverse events was 40.9% reported herein and was 44.9% reported for the combination in the trial reported by Hodi et. al. Follow-on therapies were not reported for patients participating in the trial reported by Hodi et. al., so the impact of these on survival is unknown. These promising results warrant follow-up in a larger trial. A randomized trial of nivolumab and ipilimumab with or without sargramostim in treating patients with unresectable metastatic melanoma is planned (NCT02339571).
There has been an intense search for patient characteristics clinical correlates, and biomarkers associated with clinical response to ipilimumab and other immunotherapies. Early studies suggested that the occurrence of irAEs was correlated with clinical response to ipilimumab therapy.26-28 There was no association between Grade 3 or 4 AEs in patients with irDC in our small study; however, the patient who had the most severe irAE (bowel perforation requiring temporary colostomy) was the only patient in the trial who had an irCR and remains in CR. It has been reported that the absolute lymphocyte counts after two ipilimumab treatments or early increase in lymphocyte counts correlate significantly with clinical benefit and OS in a clinical trial of ipilimumab treatment of melanoma.29,30 In our study, the lymphocyte counts did increase significantly from the baseline levels after 1 and 2 cycles of ipilimumab therapy, but there was no difference between patients with irDC or irPD. Further, it has been reported that an increase in circulating Tregs after treatment with two cycles (6 weeks) of ipilimumab monotherapy was associated with an improved PFS.31 Conversely, a decrease in Tregs at week 12 has also been observed to associate significantly with disease control rate at week 24 and survival.32 In our study, the percentages of Tregs among the total lymphocytes were significantly higher at week 3 and week 6 compared to week 0 but the distributions of the percentages of Tregs did not differ between patients with irDC and irPD.
Increase in the levels of immune subsets indicating activation of immune cells such as CD4+ Teff cells and Tregs have been similarly observed in patients with metastatic castration resistant prostate cancer (mCRPC) treated with ipilimumab and GM-CSF.33 However, neither baseline levels of immune subsets including CD4+ Teff cells, CD8+ T cells, Tregs, ratio of CD4+ T cells to Tregs, ratio of CD8+ T cells to Tregs, nor changes to the levels following therapy correlated with response to treatment (irDC or irPD) for patients with metastatic melanoma or with OS for mCRPC patients treated with the combination. Hodi et. al. reported a greater increase in CD4+ and CD8+ T cells that expressed ICOS in patients treated with ipilimumab and GM-CSF compared to the increase in patients treated with ipilimumab monotherapy,17 however, they did not report that this was related to clinical response.
In this study, we observed that levels of CD4+ Teff cells expressing PD-1 were significantly higher at baseline in patients who subsequently had irPD on treatment than were the baseline levels in patients who had irDC on treatment or levels in cancer-free controls. Pre-treatment levels of CD8+ T cells expressing PD-1 were significantly higher than those in cancer-free controls in patients who experienced irDC on treatment. The levels of CD4+ Teff and CD8+ Tcells expressing PD-1 increased significantly at week 3 and week 6 of therapy with ipilimumab and GM-CSF. At week 6, the level of CD4+ Teff cells expressing PD-1 remained significantly higher in patients with irPD than in patients with irDC. In summary, lower pre-treatment levels of CD4+ Teff and higher pre-treatment levels of CD8+ T cells expressing PD-1 related significantly with irDC. This relationship for CD4+ Teff expressing PD-1 at baseline but not at week 6 was similarly observed for mCRPC patients with longer OS.33 Although the number of patients in this cohort is still small, this result suggests PD-1 expression in CD4+ Teff cells and in CD8+ T cells as potential pre-treatment biomarkers for clinical outcome of immunotherapy with ipilimumab and GM-CSF. We suggest further evaluation of levels of CD4+ Teff and CD8+ T cells expressing PD-1 as potential biomarkers of response in a larger cohort of patients undergoing treatment with GM-CSF and ipilimumab and comparison of these levels in PBMC from cancer-free controls.
Methods
Patients
Eligible patients were adults with histologically confirmed unresectable metastatic melanoma. Additional eligibility criteria included least one measurable lesion according to Immune-Related Response Criteria (irRC),34 ECOG performance status of 0–1; LDH ≤ 4× the upper limit of normal; adequate hematologic, kidney and liver function, and no history of autoimmune disease. Patients with brain metastases were eligible if they had been controlled for at least one month. Patients were allowed to have prior systemic therapy with other agents for metastatic disease but could not have concomitant therapy while on the study.
The protocol was approved by the Institutional Review Board of each participating institution and was conducted in accordance with the ethical principles of the Declaration of Helsinki and within the Good Clinical Practice guidelines as defined by the International Conference on Harmonization. All patients gave written informed consent for participation in the study. The trial was registered on ClinicalTrials.gov with Identifier NCT01363206.
Study design
The primary objective was to determine the irDC rate at 24 weeks. The irDC is defined as the sum of the immune response Complete Response (irCR) + immune response Partial Response (irPR) + immune response Stable Disease (irSD). Secondary objectives were: assessment of immunologic responses, overall survival OS, progression free survival (PFS), objective response rate (ORR: irCR and irPR), time to objective response (irCR and irPR), duration of objective response and safety of the combination as defined by the NCI Common Terminology Criteria for Adverse Events (CTCAE) criteria, version 4.0 (NIH, May 28, 2009). The planned accrual goal was 43 patients, but due to cessation of sponsor funding during the study, only 22 patients were accrued.
Treatment regimen
At the initiation of treatment (months 1–3), patients were treated with four cycles of GM-CSF and ipilimumab administered every 3 weeks (Fig. 1A). Ipilimumab was administered intravenously at a dose of 10 mg/kg on day 1 of each 21 d cycle. GM-CSF was administered subcutaneously daily for 14 d at a dose of 125 µg/m2 beginning on day 1 of each cycle. After the first four cycles of treatment, GM-CSF administration without ipilimumab continued for four more cycles on the same schedule and dose for the first 14 d of every 21 d cycle until month 6. Maintenance therapy began at month 6 and consisted of ipilimumab in the same dose (10 mg/kg) combined with 14 d of GM-CSF (Fig. 1B). This combination was administered every 3 mo thereafter for up to 2 y or until disease progression or unacceptable toxicity. At 12 weeks after treatment initiation, patients were kept on the study if there was progressive disease unless there was rapid clinical deterioration. Beginning at the assessment at 24 weeks, they were removed from the study if they had progressive disease on any follow-up imaging.
Figure 1.
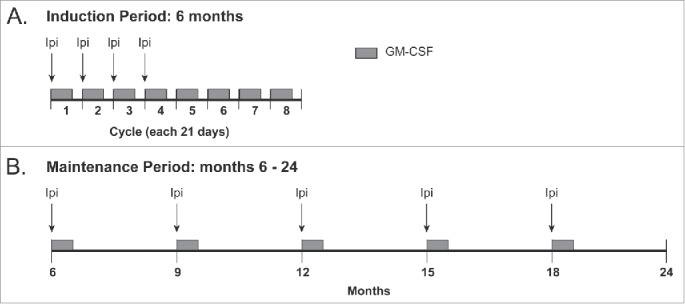
Treatment schema for induction period (6 mo) and maintenance period (months 6–24).
If one or more doses of ipilimumab were held because of side effects, then the corresponding doses of GM-CSF were also held to assure constant administration of GM-CSF relative to ipilimumab. If treatment with ipilimumab was held because of an adverse event, it was restarted at the next scheduled dosing when the adverse event resolved to ≤ Grade 1 severity. Infusion reactions were treated with antihistamines and/or famotidine at the discretion of the treating physician. Immune-related adverse events due to ipilimumab developing during the course of treatment were treated according to published standardized recommended algorithms. The study protocol was amended during the trial period to require pre-medication with famotidine and diphenhydramine prior to treatment to ipilimumab to decrease the incidence of infusion reactions which had been observed with the combination. If ipilimumab treatment was to be discontinued due to side effects, administration of GM-CSF was also discontinued. However, treatment with ipilimumab was allowed to continue per schedule if treatment with GM-CSF was discontinued due to side effects.
Response and toxicity assessments
The patients underwent staging studies with CT scans of the chest, abdomen and pelvis and MRI of the brain at screening (within 28 d of the first treatment cycle) and every 3 mo thereafter. Tumor assessments were made using the irRC guidelines.34 Laboratory analysis with a complete blood count (CBC), serum chemistry panel, LDH urinalysis, thyroid studies, pregnancy test, autoimmune panel, and ACTH level were performed at screening. CBC, serum chemistry, thyroid studies, and urinalysis were performed every three weeks during months 1 to 6 (induction phase) and then every 6 mo thereafter (maintenance phase). An autoimmune panel and ACTH level were performed every 6 mo during the study. Monitoring of the safety data was done by the investigator(s) every three weeks during the induction phase (months 1 to 6) and then every 6 mo during the maintenance phase (months 6 to 24).
Immunologic analysis
Blood samples to assess treatment effects on circulating immune cells were obtained before treatment, at week 3 (end of cycle 1) and at week 6 (end of cycle 2) and were cryopreserved for subsequent analysis by flow cytometry. In addition to study participants, PBMC were also obtained from seven cancer-free controls. Flow cytometry was carried out as previously described.33 Briefly, cell surface staining was performed in fluorescence-activated cell sorting (FACS) buffer for 30 min at 4°C. Intracellular foxhead box P3 (FoxP3) was performed using the FoxP3 fix/perm buffer set (Biolegend, Inc.) according to the manufacturer's protocol. CD8+ T cells were defined as CD4−CD3+; CD4+ Teff cells were defined as CD4+CD3+FoxP3−; and Tregs were defined as CD4+CD3+FoxP3+CD127lowCD25+. Gating strategies for CD4+ Teff cells, CD8+ T cells, Tregs and for PD-1+ CD4+ Teff cells and PD-1+ CD8+ T cells are shown in Fig. S2. The following anti-human antibodies were used: (Alexa Fluor 700)-CD3 (clone HIT3a), (Brilliant violet 570)-CD4 (clone RPA-T4), (Brilliant violet 650)-CD25 (clone BC96), (Alexa Fluor 647)-CD127 (clone A019D5), (Alexa Fluor 488)-FoxP3 (clone 206D), and (Brilliant violet 421)-PD-1 (clone EH12.2H7). All antibodies were purchased from Biolegend, Inc. Stained cells were fixed with Fluorofix buffer (Biolegend, Inc.) according to manufacturer's instructions and analyzed with an LSR II flow cytometer (BD Biosciences). Data analysis was performed with FlowJo software (FLowjo, LLC.). The percentage (%) of positive cells was gated based on appropriate isotype control.
Statistical analysis
All analyses were based on the intent-to-treat principle, that is, all study participants were included in the analyses, regardless of whether or not they were protocol violators or completed the study. Descriptive analyses were used for assessment of baseline patient demographics for safety parameters.
The primary study outcome measure was the rate of disease control 24 weeks after the start of protocol therapy. PFS was defined as the number of months from the start of protocol treatment to disease progression; OS was defined as the number of months from the start of treatment to death from any cause. PFS and OS analyses were carried out using the Kaplan–Meier method.35 The censored date for the last follow-up for these analyses was on February 25th 2015. OS of cancer patients who experienced irDC (n = 9) were compared with OS of patients with irPD (irPD) (n = 13) using the log-rank test.
Categorical patient's characteristics such as sex, stage of disease, ECOG performance status, prior therapy, prior systemic therapy, and LDH (where patients were categorized as within reference range or not) were compared between patients with irDC and patients with irPD using Fisher's exact test.
Continuous patient's characteristics such as age and LDH levels between patients with irDC and irPD were compared using Mann–Whitney U-test. One patient had a different reference range of LDH levels from the other patients and the values were thus not included in this analysis.
The distributions of the percentage of paired immune subsets at week 0 (pre-treatment) were compared with week 3 (end of cycle 1) or with week 6 (end of cycle 2) using Wilcoxon matched-pairs signed rank test. Only patients who had paired samples for at least two data points were included in this analysis (n = 21). The distributions of percentage of immune subsets between patients with irDC and irPD at week 0, week 3, or week 6 were compared using Mann–Whitney U-test. The distribution of immune subsets from cancer-free controls (n = 7) were similarly compared with those in patients with irDC or irPD only at the pre-treatment time point. Bonferroni corrections for multiple comparisons were carried out where applicable.
Disclosure of potential conflicts of interest
L.E.S. has received research funding from Bristol-Myers Squibb and Genzyme and has been a Consultant to Genzyme/Sanofi Aventis; R.W. is on the Speaker's Bureau for Bristol-Myers Squibb. The remaining authors declare no conflicts of interest.
Supplementary Material
Funding
This work was supported in part by funds from the Melanoma Research Institute, Joyce N. Furman Memorial Trust, and Bristol-Myers Squibb. Ipilimumab was provided by Bristol-Myers Squibb and GM-CSF was provided by Genzyme, a Sanofi Company. S.S.K. is funded by the Pelican cancer fellowship from the Peter Michael Foundation.
References
- 1.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al.. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711-23; PMID:20525992; http://dx.doi.org/ 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lipson EJ, Drake CG. Ipilimumab: an anti-CTLA-4 antibody for metastatic melanoma. Clin Cancer Res 2011; 17:6958-62; PMID:21900389; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nightingale SL. From the Food and Drug Administration. JAMA 1991; 265:2315; PMID:21900389; http://dx.doi.org/6379124 10.1001/jama.1991.03460180021009 [DOI] [PubMed] [Google Scholar]
- 4.Fidler IJ, Kleinerman ES. Lymphokine-activated human blood monocytes destroy tumor cells but not normal cells under cocultivation conditions. J Clin Oncol 1984; 2:937-43; PMID:6379124 [DOI] [PubMed] [Google Scholar]
- 5.Grabstein KH, Urdal DL, Tushinski RJ, Mochizuki DY, Price VL, Cantrell MA, Gillis S, Conlon PJ. Induction of macrophage tumoricidal activity by granulocyte-macrophage colony-stimulating factor. Science 1986; 232:506-8; PMID:3083507; http://dx.doi.org/ 10.1126/science.3083507 [DOI] [PubMed] [Google Scholar]
- 6.Thomassen MJ, Barna BP, Rankin D, Wiedemann HP, Ahmad M. Differential effect of recombinant granulocyte macrophage colony-stimulating factor on human monocytes and alveolar macrophages. Cancer Res 1989; 49:4086-9; PMID:2545332 [PubMed] [Google Scholar]
- 7.Chachoua A, Oratz R, Hoogmoed R, Caron D, Peace D, Liebes L, Blum RH, Vilcek J. Monocyte activation following systemic administration of granulocyte-macrophage colony-stimulating factor. J Immunother Emphasis Tumor Immunol 1994; 15:217-24; PMID:8032545; http://dx.doi.org/ 10.1097/00002371-199404000-00008 [DOI] [PubMed] [Google Scholar]
- 8.Demir G, Klein HO, Tuzuner N. Low dose daily rhGM-CSF application activates monocytes and dendritic cells in vivo. Leukemia Res 2003; 27:1105-8; PMID:12921948; http://dx.doi.org/ 10.1016/S0145-2126(03)00097-3 [DOI] [PubMed] [Google Scholar]
- 9.Dong Z, Kumar R, Yang X, Fidler IJ. Macrophage-derived metalloelastase is responsible for the generation of angiostatin in Lewis lung carcinoma. Cell 1997; 88:801-10; PMID:9118223; http://dx.doi.org/ 10.1016/S0092-8674(00)81926-1 [DOI] [PubMed] [Google Scholar]
- 10.Young JW, Szabolcs P, Moore MA. Identification of dendritic cell colony-forming units among normal human CD34+ bone marrow progenitors that are expanded by c-kit-ligand and yield pure dendritic cell colonies in the presence of granulocyte/macrophage colony-stimulating factor and tumor necrosis factor α. J Exp Med 1995; 182:1111-9; PMID:7561684; http://dx.doi.org/ 10.1084/jem.182.4.1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szabolcs P, Moore MA, Young JW. Expansion of immunostimulatory dendritic cells among the myeloid progeny of human CD34+ bone marrow precursors cultured with c-kit ligand, granulocyte-macrophage colony-stimulating factor, and TNF-α. J Immunol 1995; 154:5851-61; PMID:75385348639819 [PubMed] [Google Scholar]
- 12.Szabolcs P, Avigan D, Gezelter S, Ciocon DH, Moore MA, Steinman RM, Young JW. Dendritic cells and macrophages can mature independently from a human bone marrow-derived, post-colony-forming unit intermediate. Blood 1996; 87:4520-30; PMID:8639819; http://dx.doi.org.ucsf.idm.oclc.org/ [PubMed] [Google Scholar]
- 13.Spitler LE, Grossbard ML, Ernstoff MS, Silver G, Jacobs M, Hayes FA, Soong SJ. Adjuvant therapy of stage III and IV malignant melanoma using granulocyte-macrophage colony-stimulating factor. J Clin Oncol 2000; 18:1614-21; PMID:10764421 [DOI] [PubMed] [Google Scholar]
- 14.Grotz TE, Kottschade L, Pavey ES, Markovic SN, Jakub JW. Adjuvant GM-CSF improves survival in high-risk stage iiic melanoma: a single-center Study. Am J Clin Oncol 2014; 37:467-72; PMID:23428946; http://dx.doi.org/ 10.1097/COC.0b013e31827def82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawson DH, Lee S, Zhao F, Tarhini AA, Margolin KA, Ernstoff MS, Atkins MB, Cohen GI, Whiteside TL, Butterfield LH et al.. Randomized, Placebo-Controlled, Phase III Trial of Yeast-Derived Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) Versus Peptide Vaccination Versus GM-CSF Plus Peptide Vaccination Versus Placebo in Patients With No Evidence of Disease After Complete Surgical Resection of Locally Advanced and/or Stage IV Melanoma: A Trial of the Eastern Cooperative Oncology Group-American College of Radiology Imaging Network Cancer Research Group (E4697). J Clin Oncol 2015; 33(34):4066-76; PMID:26351350; http://dx.doi.org/ 10.1200/JCO.2015.62.0500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fong L, Kwek SS, O'Brien S, Kavanagh B, McNeel DG, Weinberg V, Lin AM, Rosenberg J, Ryan CJ, Rini BI et al.. Potentiating endogenous antitumor immunity to prostate cancer through combination immunotherapy with CTLA4 blockade and GM-CSF. Cancer Res 2009; 69:609-15; PMID:19147575; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-3529 [DOI] [PubMed] [Google Scholar]
- 17.Hodi FS, Lee S, McDermott DF, Rao UN, Butterfield LH, Tarhini AA, Leming P, Puzanov I, Shin D, Kirkwood JM. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: a randomized clinical trial. JAMA 2014; 312:1744-53; PMID:25369488; http://dx.doi.org/ 10.1001/jama.2014.13943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.AJCC Cancer Staging Manual. New York: Springer, 2010 [Google Scholar]
- 19.Balch CM, Soong SJ, Gershenwald JE, Thompson JF, Reintgen DS, Cascinelli N, Urist M, McMasters KM, Ross MI, Kirkwood JM et al.. Prognostic factors analysis of 17,600 melanoma patients: validation of the American Joint Committee on Cancer melanoma staging system. J Clin Oncol 2001; 19:3622-34; PMID:11504744 [DOI] [PubMed] [Google Scholar]
- 20.Balch CM, Soong SJ, Murad TM, Smith JW, Maddox WA, Durant JR. A multifactorial analysis of melanoma. IV. Prognostic factors in 200 melanoma patients with distant metastases (stage III). J Clin Oncol 1983; 1:126-34; PMID:6668496 [DOI] [PubMed] [Google Scholar]
- 21.Brand CU, Ellwanger U, Stroebel W, Meier F, Schlagenhauff B, Rassner G, Garbe C. Prolonged survival of 2 years or longer for patients with disseminated melanoma. An analysis of related prognostic factors. Cancer 1997; 79:2345-53; PMID:9191522; http://dx.doi.org/ [DOI] [PubMed] [Google Scholar]
- 22.Manola J, Atkins M, Ibrahim J, Kirkwood J. Prognostic factors in metastatic melanoma: a pooled analysis of Eastern Cooperative Oncology Group trials. J Clin Oncol 2000; 18:3782-93; PMID:11078491 [DOI] [PubMed] [Google Scholar]
- 23.Bedikian AY, Millward M, Pehamberger H, Conry R, Gore M, Trefzer U, Pavlick AC, DeConti R, Hersh EM, Hersey P et al.. Bcl−2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: the Oblimersen Melanoma Study Group. J Clin Oncol 2006; 24:4738-45; PMID:16966688; http://dx.doi.org/ 10.1200/JCO.2006.06.0483 [DOI] [PubMed] [Google Scholar]
- 24.Hersh EM, O'Day SJ, Ribas A, Samlowski WE, Gordon MS, Shechter DE, Clawson AA, Gonzalez R. A phase 2 clinical trial of nab-paclitaxel in previously treated and chemotherapy-naive patients with metastatic melanoma. Cancer 2010; 116:155-63; PMID:19877111; http://dx.doi.org/ 10.1002/cncr.24720 [DOI] [PubMed] [Google Scholar]
- 25.Robert C, Thomas L, Bondarenko I, O'Day S, M DJ, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ et al.. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011; 364:2517-26; PMID:21639810; http://dx.doi.org/ 10.1056/NEJMoa1104621 [DOI] [PubMed] [Google Scholar]
- 26.Bhatia S, Huber BR, Upton MP, Thompson JA. Inflammatory enteric neuropathy with severe constipation after ipilimumab treatment for melanoma: a case report. J Immunother 2009; 32:203-5; PMID:19238020; http://dx.doi.org/ 10.1097/CJI.0b013e318193a206 [DOI] [PubMed] [Google Scholar]
- 27.Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, Restifo NP, Haworth LR, Seipp CA, Freezer LJ et al.. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A 2003; 100:8372-7; PMID:12826605; http://dx.doi.org/ 10.1073/pnas.1533209100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol 2012; 30:2691-7; PMID:22614989; http://dx.doi.org/ 10.1200/JCO.2012.41.6750 [DOI] [PubMed] [Google Scholar]
- 29.Delyon J, Mateus C, Lefeuvre D, Lanoy E, Zitvogel L, Chaput N, Roy S, Eggermont AM, Routier E, Robert C. Experience in daily practice with ipilimumab for the treatment of patients with metastatic melanoma: an early increase in lymphocyte and eosinophil counts is associated with improved survival. Ann Oncol 2013; 24:1697-703; PMID:23439861; http://dx.doi.org/ 10.1093/annonc/mdt027 [DOI] [PubMed] [Google Scholar]
- 30.Ku GY, Yuan J, Page DB, Schroeder SE, Panageas KS, Carvajal RD, Chapman PB, Schwartz GK, Allison JP, Wolchok JD. Single-institution experience with ipilimumab in advanced melanoma patients in the compassionate use setting: lymphocyte count after 2 doses correlates with survival. Cancer 2010; 116:1767-75; PMID:20143434; http://dx.doi.org/ 10.1002/cncr.24951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tarhini AA, Edington H, Butterfield LH, Lin Y, Shuai Y, Tawbi H, Sander C, Yin Y, Holtzman M, Johnson J et al.. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PloS one 2014; 9:e87705; PMID:24498358; http://dx.doi.org/ 10.1371/journal.pone.0087705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simeone E, Gentilcore G, Giannarelli D, Grimaldi AM, Caraco C, Curvietto M, Esposito A, Paone M, Palla M, Cavalcanti E et al.. Immunological and biological changes during ipilimumab treatment and their potential correlation with clinical response and survival in patients with advanced melanoma. Cancer Immunol Immunother 2014; 63:675-83; PMID:24695951; http://dx.doi.org/ 10.1007/s00262-014-1545-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwek SS, Lewis J, Zhang L, Weinberg V, Greaney S, Harzstark A, Lin A, Ryan C, Small EJ, Fong L. Pre-existing levels of CD4 T cells expressing PD-1 are related to overall survival in prostate cancer patients treated with ipilimumab. Cancer Immunol Res 2015; 3(9):1008-16; PMID:25968455; http://dx.doi.org/ 10.1158/2326-6066.CIR-14-0227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolchok JD, Hoos A, O'Day S, Weber JS, Hamid O, Lebbé C, Maio M, Binder M, Bohnsack O, Nichol G. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 2009:15: 1742; PMID:19934295; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-1624 [DOI] [PubMed] [Google Scholar]
- 35.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Statistical Assoc 1958; 53:457-81; http://dx.doi.org/ 10.1080/01621459.1958.10501452 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.