

ABSTRACT
The stimulation of the immune system using oncolytic adenoviruses (OAds) has attracted significant interest and several studies suggested that OAds immunogenicity might be important for their efficacy. Therefore, we developed a versatile and rapid system to adsorb tumor-specific major histocompatibility complex class I (MHC-I) peptides onto the viral surface to drive the immune response toward the tumor epitopes. By studying the model epitope SIINFEKL, we demonstrated that the peptide-coated OAd (PeptiCRAd) retains its infectivity and the cross presentation of the modified-exogenous epitope on MHC-I is not hindered. We then showed that the SIINFEKL-targeting PeptiCRAd achieves a superior antitumor efficacy and increases the percentage of antitumor CD8+ T cells and mature epitope-specific dendritic cells in vivo. PeptiCRAds loaded with clinically relevant tumor epitopes derived from tyrosinase-related protein 2 (TRP-2) and human gp100 could reduce the growth of primary-treated tumors and secondary-untreated melanomas, promoting the expansion of antigen-specific T-cell populations. Finally, we tested PeptiCRAd in humanized mice bearing human melanomas. In this model, a PeptiCRAd targeting the human melanoma-associated antigen A1 (MAGE-A1) and expressing granulocyte and macrophage colony-stimulating factor (GM-CSF) was able to eradicate established tumors and increased the human MAGE-A1-specific CD8+ T cell population. Herein, we show that the immunogenicity of OAds plays a key role in their efficacy and it can be exploited to direct the immune response system toward exogenous tumor epitopes. This versatile and rapid system overcomes the immunodominance of the virus and elicits a tumor-specific immune response, making PeptiCRAd a promising approach for clinical testing.
KEYWORDS: Cancer vaccine, humanized mice, immunotherapy, melanoma, oncolytic adenovirus, oncolytic vaccine, tumor epitopes
Introduction
OAds selectively kill tumor cells 1,2 countering tumor growth and favoring the spreading of damage-associated molecular patterns (DAMPs) that can, under certain circumstances, lead to the activation of surrounding antigen-presenting cells (APCs) 3,4 and some degrees of expansion of tumor-specific T-lymphocytes.5 However, the lack of sufficient number of tumor-specific T cells and immunosuppressive mechanisms limits the efficacy of OAds.6 More sophisticated oncolytic vectors encode for immune-modulating molecules such as IL-23,7 TNFa,8 CD40L,9,10 or GM-CSF.5,11,12 In addition, to further improve the specificity of the immune response, adenoviruses encoding for tumor antigens have been designed.13 The efficacy of these oncolytic agents, however, depends on their persistence into the patients and their transduction efficiency, which is a common limitation for all adenoviral vectors used in clinical trials due to pre-existing immunity 14 and the rapid production of neutralizing antibodies.15 In addition, the time required for the genetic manipulation of the different viruses encoding for different antigens and the consequent re-evaluation by the competent authorities (FDA and/or EMEA) makes this approach incompatible with next generation of personalized approaches that rely on the identification of patient-specific antigenic signatures to adapt immunotherapeutic protocols.
To this end, we specifically developed a novel oncolytic vaccine platform in which tumor peptides are not expressed by the virus and are not part of the viral proteins, in contrast to the vast majority of current approaches. The peptides are instead adsorbed onto the viral capsid, allowing for the efficient co-delivery of adjuvant (virus) and tumor-specific epitopes (Fig. 1). Thus, the oncolytic virus acts as an “active” carrier that is able to kill tumor cells and to boost the immunological response against the chosen antigen. This method does not involve chemical or genetic modification of the virus, significantly increasing the rapidity and the versatility of the preparation. As a direct consequence, this system addresses the need for tumor-specific and even personalized therapies that could account for the expression of different antigens in different patients or different antigens in different stages of the same tumor.
Figure 1.
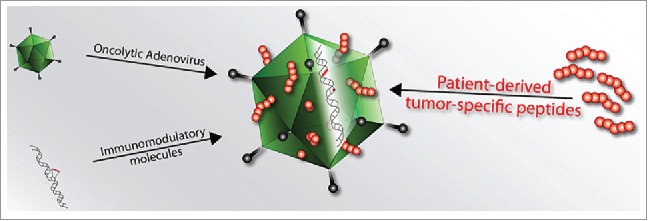
Schematic of PeptiCRAd (Peptide-coated Conditionally Replicating Adenovirus). PeptiCRAd is a novel cancer vaccine platform that exploits the natural immunogenicity of adenoviruses. OAds act as adjuvants for exogenous MHC-I tumor epitopes that are loaded onto the viral capsid by electrostatic interactions. These peptides could be known MHC-I epitopes or patient-derived tumor epitopes. Therefore, PeptiCRAd retains all the properties of a conventional oncolytic adenovirus (direct tumor-killing ability and possibility to express immune-stimulating molecules), however, it has a superior ability to stimulate a tumor-specific immune response.
In this study, we characterized the physical and biological properties of Peptide-coated Conditionally Replicating Adenoviruses (PeptiCRAds). We demonstrated that by absorbing tumor-specific MHC-I-restricted peptides onto the viral capsid, we can direct the immunity toward the tumor, leading to a significantly increased efficacy in different models of murine melanoma and human melanoma in humanized mice. PeptiCRAd represents a novel oncolytic vaccine platform that is able to fully exploit the immunogenicity of OAds and that can be rapidly adapted to different antigens and tumors without any genetic modification.
Results
The negative charge of the adenovirus capsid can be used to complex positively charged immunogenic peptides, forming PeptiCRAd
Adenovirus capsids is negatively charged 16 bearing mostly acidic/negative regions in the hexon protein (red and pale red regions in Fig. S1), thus we hypothesized that MHC-I-restricted peptides, modified to become positively charged, would bind to the capsid of the virus via electrostatic interactions. We tested our hypothesis by using the MHC-I epitope SIINFEKL derived from chicken ovalbumin (OVA).17 The addition of a poly-lysine (polyK) chain to the aminoacidic sequence increases the net charge of the peptide from 0 to +6 mV at neutral pH (Fig. 2A). Next, we coated APTES silica SiO2 sensor with OAds (Fig. S2A) and we injected increasing concentrations of SIINFEKL or polyK-SIINFEKL into the surface plasmon resonance (SPR) system (Fig. 2B). No virus-peptide interaction was observed with the unmodified neutral SIINFEKL (Fig. 2B, dashed line), whereas a concentration-dependent interaction was observed with the modified positive polyK-SIINFEKL (Fig. 2B, solid line). In these experimental settings, we observed a plateau when using solutions of peptide with a concentration above 7 uM. We found that the binding model for these electrostatic interactions is a complex one, since high goodness of fit (R2=0,997) is observed only when applying a co-operativity model to the data (Fig. S2B). In addition, according to this model, we were able to estimate a binding constant of 2.88 × 10−6 M and a Hill coefficient of +2.68 indicating a positive co-operativity.
Figure 2.

Physical characterization of the interaction between the modified MHC-I epitope SIINFEKL and OAd. (A) The net charge of SIINFEKL (dashed gray line, circles) or polyK-SIINFEKL (black line, triangles) is shown as a function of pH. (B) SPR was used to study the interaction between Ad5D24 oncolytic virus and increasing concentrations (0.15, 0.3, 0.6, 1.2, 2.4, 7.2, and 21.6 µM) of either SIINFEKL (dashed line) or polyK-SIINFEKL (solid line). (C) zeta potential (dashed gray line, left axis) and hydrodynamic diameter (solid black line, right axis) of virus-peptide complexes. Time-dependent study of complex´s zeta potential (D) and hydrodynamic diameter (E) after incubation at room temperature. Representative results from two different experiments are shown. The data are plotted as the mean ± SD (n = 3).
Next, we studied how the amount of peptide in the coating reaction could affect the OAds-peptide complexes (Fig. 2C). The lowest OAds: peptide ratio (1:5) was able to increase the charge of the viral particles from −29.7 ± 0.5 to +6.3 ± 0.06 mV, although under these conditions, heavy aggregation was observed, as indicated by an increase in the size of the complexes (800 ± 13.5 nm). Above 1:5, the net charge reached a plateau-like kinetic as we measured zeta potentials of +17.5 ± 0.2, +18.4 ± 0.1 and +18 ± 0.8 mV for the 1:50, 1:100 and 1:500 ratios, respectively. However, only at a ratio of 1:500 the hydrodynamic diameter of the complex decreased (reaching ˜120 nm), which represents the normal diameter of adenoviral particles. The complex presented good stability and no significant decrease of zeta potential occurred 30 and 45 min after incubation in the same conditions (Fig. 2D) compared to 15 min incubation. In addition, we report no aggregation at these time points (Fig. 2E) but only an increase in the hydrodynamic diameter which can be caused by the increased presence of water molecules on the particle. To prove that interaction is not restricted to polyK-SIINFEKL peptide, we complexed the adenovirus with another modified peptide, the polyK-MAGE A1 epitope. We observed an increase of the zeta potential (Fig. S2C) compared to the naked adenovirus and no aggregation (Fig. S2D) of the particles.
Modified MHC-I epitopes adsorbed onto peptiCRAd are efficiently cross presented
Next, we investigated whether the presence and the position of the polyK chain could affect the efficiency of cross presentation of the epitope on MHC-I. We pulsed ex vivo-cultured spleenocytes (from C57BL/6 mice) with two different lysine-extended versions: polyK-SIINFEKL (N-terminus extended) and SIINFEKL-polyK (C-terminus extended). As a negative control, we included extended SIINFEKL containing an amino caproic (AHX) residue, which is a well-known analog of lysine that can inhibit the proteolytic activity of the proteasome. We then assessed the cross presentation of the mature form of the epitope (SIINFEKL) on MHC-I by flow cytometry.18
The 94.5% of the spleenocytes pulsed with the N-terminus-extended peptide cross-presented SIINFEKL. In contrast, when the spleenocytes were pulsed with the C-terminus-extended SIINFEKL-polyK, the stained population decreased to 27.1% (Fig. 3A). Based on these findings, we chose the N-terminus-extended version (polyK-SIINFEKL) for further studies.
Figure 3.
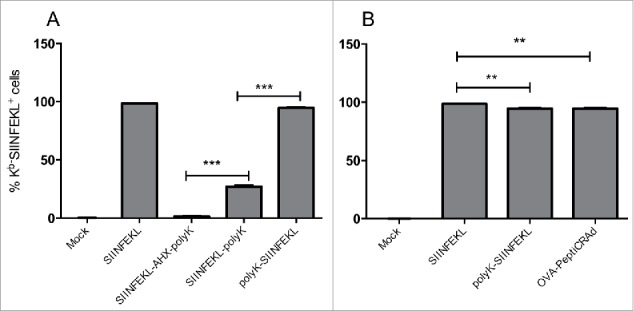
Cross-presentation of modified SIINFEKL analogs on MHC-I adsorbed or not adsorbed onto the viral capsid. (A) C57BL/6 fresh spleenocytes were incubated with SIINFEKL, the amino caproic acid-containing SIINFEKL-AHX-polyK, the C-terminus-extended SIINFEKL-polyK or the N-terminus-extended polyK-SIINFEKL. Cross presentation was determined with APC anti-H-2Kb bound to SIINFEKL or isotype control antibodies. (B) Similar to (A), spleenocytes were infected with of OVA-PeptiCRAd, or incubated with peptides SIINFEKL or polyK-SIINFEKL. The data are shown as the mean ± SD (n=2). Significance was assessed using the unpaired student´s t-test; ** p < 0.01, *** p < 0.001.
Next, we investigated if the adsorption of the modified SIINFEKL onto the viral capsid could affect its cross presentation. As in the previous experiment, we incubated mouse spleenocytes with the polyK-SIINFEKL or with OVA-PeptiCRAd (i.e. OAd coated with polyK-SIINFEKL). We found that all the conditions allowed for efficient MHC-I-restricted presentation of the SIINFEKL peptide (Fig. 3B).
PeptiCRAd shows increased infectivity compared with unmodified viruses
We investigated whether coating the viruses with modified peptides would affect their biological properties. We chose to study a human colorectal adenocarcinoma cell line (CACO-2) expressing low levels of coxsackie and adenovirus receptor (CAR) a human melanoma cell line expressing intermediate levels of CAR (A2058) and another human melanoma cell line expressing high levels of CAR (SK-MEL-2). PeptiCRAd showed unaltered oncolytic activity compared to naked Ad5D24 virus (Fig. 4A) in all cell lines; in addition we observed no toxic effect upon cell viability due to the modified polyK-SIINFEKL peptide.
Figure 4.
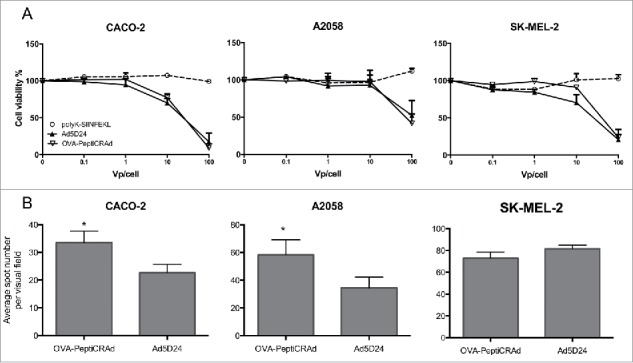
PeptiCRAd retains intact oncolytic activity and displays increased infectivity in cell lines with low CAR expression. (A) cell viability assay in different cell lines. The data are shown as the mean ± SD (n = 3). (B) Infectivity assay by ICC. Cells have been infected with 10 vp/cell of either naked OAd or OVA-PeptiCRAd. The average number of spots per visual field is presented (5 non-overlapping visual fields have been acquired and used for the generation of the means). Representative data from two independent experiments are shown as the mean ± SD (n = 2). Significance was assessed using the unpaired t-test with Welch's correction; * p < 0.05.
Next, we evaluated the infectivity of PeptiCRAd by immunocytochemistry (ICC; Fig. 4B). Whereas, we did not observe any significant difference in SK-MEL-2 cell line, when testing in vitro models with intermediate (A2058) and low (CACO-2) levels of CAR, PeptiCRAd showed a significant increase (p < 0.05) in infectivity compared with the naked adenovirus.
Characterization of the anti-tumor immunity and efficacy of PeptiCRAd in a murine model of melanoma
To thoroughly study the antitumor efficacy of PeptiCRAd, we first used a murine model of melanoma over-expressing chicken OVA (B16-OVA).17 A pilot experiment was performed using an OAd bearing the D24 deletion in E1A (Ad5D24) 2 coated with the modified poly-K-SIINFEKL. We observed a significantly reduced tumor growth in mice treated with PeptiCRAd (Fig. S3). Therefore, we investigated further this model by using a CpG-rich OAd (Ad5D24-CpG) 19 to further boost immunity (Fig. 5) through Toll-like receptor 9 activation. The study groups included mice treated with OVA-PeptiCRAd, with non-complexed Ad5D24-CpG and SIINFEKL (Ad5D24-CpG+SIINFEKL), with naked Ad5D24-CpG, with SIINFEKL peptide alone or with saline solution (mock).
Figure 5.
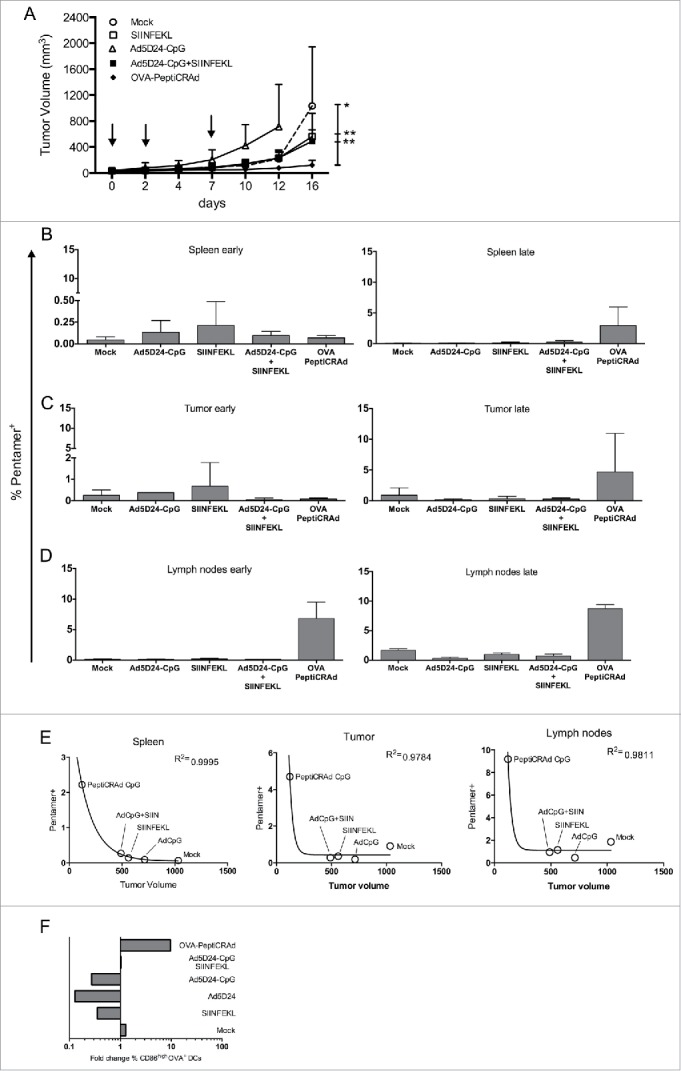
Antitumor efficacy of PeptiCRAd and immunological analysis of antigen-specific CD8+ T cells and DCs. C57BL/6 mice (n=8–9) received 3×105 B16-OVA cells in both flanks. Treatment was initiated nine days later and included saline solution (mock), peptide alone (SIINFEKL), virus alone (Ad5D24-CpG), a mixture of virus and SIINFEKL peptide (Ad5D24-CpG+SIINFEKL), and Ad5D24-polyK-SIINFEKL complex (OVA-PeptiCRAd). Mice were treated three times (on days 0, 2, and 7; black arrows). At day 7, before the third injection, mice from each group were sacrificed for early immunological analysis (n = 2–3). The late immunological analysis was performed on samples collected at the end of the experiment (tumors and spleens n = 3–4; lymph nodes n = 2–3). (A) Average tumor volume is represented excluding mice sacrificed at day 7. The percentage of SIINFEKL-Pentamer+ cells among CD19−CD8+ T-cells is reported for spleens (B), tumors (C) and draining lymph nodes (D) at early (left panels) and late (right panels) time points. Samples from Ad5D24-CpG group were collected at day 12. Data are presented as the mean ± SD. (E) The average tumor size at the end of the experiment was plotted against the average percentage of SIINFEKL-Pentamer+ CD8+ T cells at late time point. A correlation analysis was performed using a non-linear exponential model and the R square value is reported for each set of data. (E) Dendritic cells (CD19−CD3−CD11c+) showing a mature profile (CD86high) and cross-presenting SIINFEKL on their H2-Kb was determined in the spleens 7 and 16 d after the start of the treatment. The fold change between the two time points is presented. Statistical analysis was done using unpaired Mann–Whitney test; *p < 0.05, **p < 0.01.
Intratumoral injections of PeptiCRAd significantly reduced the tumor's growth compared with treatment with saline buffer, SIINFEKL peptide or the mixture of OAd and SIINFEKL. At the end of the experiment, the average volume of the tumors in the OVA-PeptiCRAd-treated mice was significantly lower than in all other groups (Fig. 5A).
Next, we studied the immunological background, hypothesizing that the increased antitumor efficacy could be explained by a more efficient CD8+ T cell response. To this end, we analyzed spleens (Fig. 5B), tumors (Fig. 5C) and draining lymph nodes (Fig. 5D) of mice 7 and 16 d after the start of the treatment (early and late time points respectively). At early time point, the CD8+ response against the SIINFEKL epitope was generally low in spleens (Fig. 5B, left) and tumors (Fig. 5C, left), with mice treated with SIINFEKL peptide alone showing an increased trend. Mice treated with OVA-PeptiCRAd did not show any increased SIINFEKL response in spleens and tumors while we detected a larger population of epitope-specific CD8+ T cells in the draining lymph nodes (Fig. 5D, left) compared to all other groups. At the end of the experiment (day 16), we observed that mice treated with OVA-PeptiCRAd showed an increased percentage of pentamer specific CD8+ T cells in spleens, tumors and lymph nodes (Figs. 5B, 5C and 5D on the right).
Next, we studied the correlation between the immunological response and the antitumor effect (Fig. 5E). We found that data would fit best to a non-linear model. According to the exponential model, a very good correlation between tumor volumes and CD8+-response was found in spleens (R2+0.9995); in tumors and lymph nodes the correlation was still high but slightly lower than in spleens. Interestingly, in the correlation analyses, the PeptiCRAd group consistently showed the smallest tumor volume and the greatest immunological response.
Finally, we evaluated the effect of PeptiCRAd vaccination on professional antigen presenting cells 7 and 16 d after the start of the treatment (early and late time points respectively). In particular, we were interested in the proportion of dendritic cells (DCs; CD19− CD3− CD11c+) showing a mature phenotype (CD86high) and presenting the SIINFEKL peptide on MHC-I. We hypothesized that these cells might be the ones responsible for direct CD8+ T cell activation through the cross-presentation mechanism. At the late time point, mice treated with PeptiCRAd showed a significantly higher percentage of mature SIINFEKL-presenting DCs (p < 0.05) than mice treated with the non-complexed Ad5D24-CpG+SIINFEKL (Fig. S4B). When both time points are considered, PeptiCRAd induced the biggest increase of CD86high OVA+ DCs, 9.67-fold change (Fig. 5F).
Multivalent PeptiCRAd shows enhanced antitumor activity toward distant, untreated melanomas
Next, we studied the efficacy of PeptiCRAd upon non-treated contralateral melanomas and whether targeting two tumor antigens (via multivalent PeptiCRAd) would increase the overall efficacy. Therefore, we chose two polyK-modified versions of the tumor-specific MHC-I-restricted epitopes SVYDFFVWL (TRP-2180-188; restricted to the murine MHC-I molecule H-2Kb) and KVPRNQDWL (human gp10025-33, or hgp100; restricted to the murine MHC-I molecule H-2Db 20), both expressed by B16-F10 cells.21
We first implanted 1×105 B16-F10 cells into the right flank of C57BL/6 mice (Fig. 6A). After 10 days, intratumoral treatments were initiated as follows: (i) saline solution (mock), (ii) naked oncolytic virus (Ad5D24-CpG), (iii) single-coated hgp100-PeptiCRAd, (iv) single-coated TRP-2-PeptiCRAd, and (v) double-coated TRP-2-hgp100-PeptiCRAd. Two days after the last treatment, we injected 3×105 B16-F10 cells into the left flank of the mice (Fig. 6A). The double-coated PeptiCRAd significantly reduced the growth of the primary tumors compared with the other control groups (Fig. 6B). When analyzing the size of the secondary-untreated tumors, we observed an increased efficacy of all three PeptiCRAds. The overall growth of the secondary tumors was significantly reduced by the double-coated PeptiCRAd (Fig. S5). In particular, the secondary tumors of mice treated with TRP-2-hgp100-PeptiCRAd were significantly smaller compared with those in the controls receiving saline solution (p < 0.01) or only Ad5D24-CpG (p < 0.05; Fig. 6C). Although not statistically significant, the improved antitumor efficacy on the secondary melanomas was noticed also when comparing the double-targeted PeptiCRAd to both the single-targeted ones.
Figure 6.
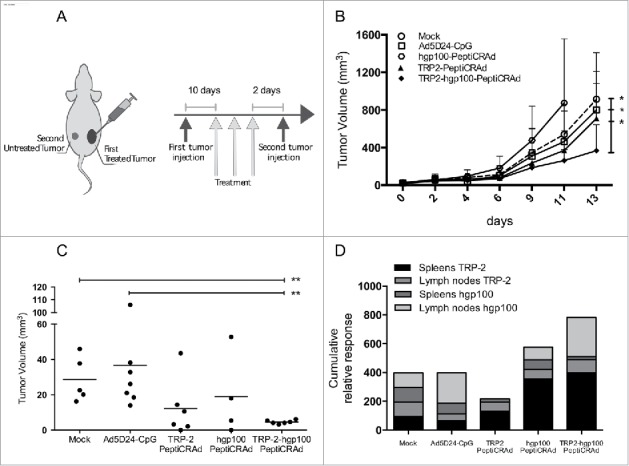
Targeting two tumor antigens with PeptiCRAd reduces the growth of both treated and untreated tumors. (A) C57BL/6 mice received 1×105 B16-F10 melanoma cells on the right flank and 3×105 B16-F10 cells on their left flank two days after the last treatment. Only the right tumor was treated. (B) The growth of the primary (right) tumor is presented as the mean ± SD (n = 6–7). (C) The size of the secondary (left) tumors at the end of the experiment is reported for each individual mouse. Tumor size oh hgp100-PeptiCRAd group is referred to day 11 before euthanasia of the group of mice. (D) The percentages of TRP-2- and hgp100-specific CD8+ T cells in spleens and inguinal lymph nodes form each mouse were normalized against mock, stacked into single columns and presented as the cumulative relative response for each experimental group. Samples from hgp100-PeptiCRAd group were collected at day 11. Significance was assessed by using the unpaired Mann–Whitney test; *p < 0.05; **p < 0.01.
To better clarify the mechanisms underpinning these results, we performed a flow cytometry analysis to study the specific CD8+ T cell populations. In mice treated with TRP-2-hgp100 PeptiCRAd, we observed the largest cumulative relative response of epitope-specific CD8+ T cells in mice treated with TRP-2-hgp100-PeptiCRAd (Fig. 6D).
Taken together, these results demonstrate that the PeptiCRAd approach is effective against a less immunogenic and more aggressive melanoma model. In addition, targeting multiple antigens might be important to increase the outcome of this therapeutic protocol.
PeptiCRAd displays enhanced efficacy and antitumor immunity in humanized mice bearing human tumors
Finally, we studied PeptiCRAd in a clinical-relevant model. To this end, we first engrafted triple-knockout mice (NOD.Cg-Prkdcscid-IL2rgtm1Wjl/SzJ, or NSG) with the human melanoma cell line SK-MEL-2 and then HLA-A3 matched human peripheral blood mononuclear cells (PBMCs) from a healthy donor were injected into the same mice intravenously. One day after, mice were treated with PeptiCRAd, uncoated virus or saline solution. We chose an epitope derived from MAGE-A196-104 (SLFRAVITK) and modified it to allow for interaction with the viral capsid (polyK-SLFRAVITK). To maximize the stimulation of the engrafted human immune system, we selected an OAd expressing human GM-CSF.5
As expected, both viruses performed better than the control mock, however, we found the MAGE-A1 PeptiCRAd to be significantly more effective when compared to the uncoated virus (Fig. 7A). In fact, we observed a rapid rejection of the tumors in mice treated with PeptiCRAd as shown by the area under the curve for each group of mice (Fig. 7B). Interestingly, when the same experiment was repeated in mice without a human immune system, no clear difference was observed between PeptiCRAd and the uncoated virus (Fig. 7C) suggesting the important role of the immune system in the efficacy of our cancer vaccine platform. To explore this hypothesis, we studied the presence of human MAGE-A196-104-specific CD8+ T cells by pentamer staining (Fig. 7D). We found the largest population in the spleens of humanized mice treated with MAGE-A1 PeptiCRAd.
Figure 7.
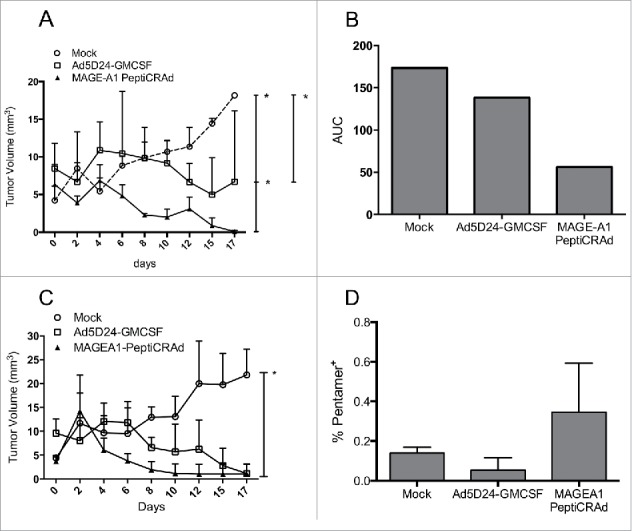
Efficacy of PeptiCRAd in humanized mice bearing human melanomas. Humanized and non-humanized mice bearing SK-MEL-2 human melanomas were treated with one of the following: (i) saline solution (mock), (ii) Ad5D24-GM-CSF, and (iii) MAGE-A1 PeptiCRAd. The tumor volume of the humanized mice (A) is presented as the mean ± SD (n = 3). (B) The area under the curve (AUC) relative to the size of the tumors of humanized-mice is presented. (C) The tumor volume of non-humanized mice (n = 2) is reported as the mean ± SD. (D) Percentage of MAGE-A1-specific CD8+ T cell population is presented as the mean ± SD (n = 2). Mann-Whitney test; *p < 0.05.
These data confirm our previous findings: PeptiCRAd platform is more effective than uncoated viruses as the oncolytic activity cooperates synergistically with immune system activation, hence improving the efficacy of oncolytic vaccine in a clinically relevant model.
Discussion
Personalized approaches are necessary to increase the success rate and, most importantly, to avoid the side effects associated with powerful 22-24 but unspecific immunotherapies.25 OAds are an attractive platform thanks to their natural immunogenicity. However, the time required for the genetic and structural manipulation of these viruses represent a major limitation for their use in personalized approaches. Therefore, we focused on the development of a new and versatile cancer vaccine platform based on OAds coated with MHC-I epitopes.
Coating the viral capsid with different polymers and materials is a widely used approach to increase the infectivity of adenoviruses 26 or to reduce the interaction with neutralizing antibodies. The rationale behind our system is different since we aim at improving conventional OAds by using them as tumor killing adjuvants for the peptides used to coat their surface. We first demonstrated that positive tumor epitopes bind the negative viral capsid electrostatically and that the modified MHC-I epitope (i.e. polyK-SIINFEKL) can be cross presented. Interestingly, the position of the polyK chain in the peptide sequence has an important effect on the cross presentation. In fact, C-terminus-extended epitopes are cross presented with a significantly lower efficiency compared with their N-terminus-extended counterparts. These results can be explained by considering that classical and non-classical cross-presentation mechanisms cooperate inside APCs. In fact, while the C-terminus-extended epitope need to undergo the classical proteasome-dependent cross-presentation pathway,27 the N-terminus extended versions can take advantage of a faster alternative pathway thanks to the presence of amino peptidases into the phagosomes from the endoplasmic reticulum.28 Hence, our results are consistent with other studies showing that the sequences flanking a mature epitope affect its cross presentation.29
We found that the infectivity of PeptiCRAd is enhanced in cell lines with low CAR expression. Previous studies demonstrated that the surface modification could affect the transduction efficacy of adenoviral vectors.16,30,31 In the case of PeptiCRAd, the positive peptides bring multiple residues of lysine that can neutralize the acidic/negative zones of the viral capsid. Hence, with less interference, the remaining positive areas could now favor the interaction of the capsid with the negative cell membrane and this phenomenon becomes important when the expression of CAR is limited.
To study the antitumor efficacy of the immunogenicity of OAds dissociated from the oncolysis, we chose a murine model of melanoma in which the human adenovirus serotype 5 is unable to replicate.8 Mice treated with OVA-PeptiCRAd showed a significantly decreased tumor growth and a larger antigen-specific immune response compared to control. Similar results have been reported for naked armed oncolytic viruses.8 However, in all our experiments PeptiCRAd was more effective than naked OAds. Our data suggest that the increased efficacy of PeptiCRAd could be due to mediated by a modulation of APCs that could uptake the virus-epitope complex. This would eventually cause the maturation of these cells, mainly due to the adenoviral-danger signals, and the cross presentation of the epitopes that are bound to the viral surface. In fact, we showed in vivo that the proportion of DCs that were activated and cross-presenting SIINFEKL was significantly increased in the group of mice treated with PeptiCRAd. These results are consistent with current knowledge of other vaccine systems 32 and with a recent work showing that adjuvant-antigen complexes are more effective than the single components because of superior targeting of APCs and, in particular, DCs.33 These results clearly show that the immunogenicity of OAds provides antitumor efficacy in absence of oncolysis, which was thought to be the only mode of action of OAds.
Next, we studied a more relevant tumor model by using B16-F10 tumors and targeting real tumor antigens rather than an artificial one (i.e. chicken OVA). We found that targeting two tumor antigens results in a broader immunological response that could explain the increased efficacy of TRP-2-hgp100-PeptiCRAd in controlling the growth of distant untreated tumors. Targeting multiple tumor antigens is important to limit the escape of malignant cells from immune surveillance 34 and to manage the variability of tumor cells.35 In fact, clinical trials have shown that patients who responded to multiple tumor epitopes are significantly more likely to experience stable disease or a partial response.36 In addition, the multivalent approach is particularly useful if the tumor down regulates one antigen or one type of HLA during the therapy.37
Finally, we used a clinically relevant in vivo model: mice bearing human melanomas and engrafted with a human immune system. This was still not an optimal solution as the development of graft-versus-host responses and the decrease of functional engrafted immune cells restricted the experimental window. For this reason, we started the treatment even if tumors showed a smaller volume compared to other experiments. Nevertheless, humanized mice represent the only model that allows for the study of both immunological properties and oncolytic activity of human OAds. We observed that PeptiCRAd could completely cure melanomas of all humanized mice, whereas in non-humanized mice, PeptiCRAd lost its advantage over the normal OAd. These interesting results highlight once again the complex interaction between OAds and the immune system. We have to consider that the immunity attempts to clear the OAds from the host, playing in this sense against the oncolytic virus and in favor of the tumor. Therefore, in absence of the immune system, both viruses are effective, whereas in presence of the immune system PeptiCRAd has a significant advantage. In support of this, the analysis of the epitope-specific CD8+ T cell population revealed an increased anti-MAGE-A1 response in mice treated with MAGE-A1 PeptiCRAd compared to other control groups. In this sophisticated model, we could appreciate the synergistic relationship between oncolysis and the immunogenicity of oncolytic viruses. In 2006, it was shown that cells infected by OAds undergo a particular cell death program that is characterized by autophagy and spreading of immunogenic signals (ATP and HMGB1 release and calreticulin exposure).38 Along the same line of research, recent studies attempted to exploit the immunogenic cell death (ICD) promoted by OAds to increase the efficacy of cancer virotherapy.8,39 We conclude that PeptiCRAd system largely benefits from the oncolytic activity of OAds but does not relies on it completely.
In summary, tumor-specific MHC-I-restricted epitopes can be complexed to an OAd, and these particles can act as a novel oncolytic vaccine platform. The possibility to target multiple antigenic entities simultaneously, improves upon the lack of specificity of most current immunotherapies. The clear advantage of this approach is its versatility, as no genetic and structural modifications of OAds are needed foster a powerful tumor-specific response and eventually re-direct it to other antigens. This gives the PeptiCRAd platform a clear advantage upon the classic antigen-expressing oncolytic vectors when considering highly personalized cancer vaccine immunotherapies.
Materials and methods
Cell lines, reagents, and human samples
The human lung carcinoma cell line A549, the human CACO-2, the human malignant melanoma cell line SK-MEL-2, the human melanoma cell line HS294T and the mouse melanoma cell line B16-F10 were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). The cell line B16-OVA,17 a mouse melanoma cell line expressing chicken OVA, was kindly provided by Prof. Richard Vile (Mayo Clinic, Rochester, MN, USA). All cell lines were cultured under appropriate conditions.
SIINFEKL (OVA257-264), polyK-SIINFEKL, SIINFEKL-polyK, polyK-AHX-SIINFEKL, polyK-SVYDFFVWL (TRP-2180-188), polyK-KVPRNQDWL (hgp10025-33), and polyK-SLFRAVITK (MAGE-A196-104) peptides were purchased from Zhejiang Ontores Biotechnologies Co. (Zhejiang, China).
The net charge of peptides was calculated by the Peptide Property Calculator Ver. 3.1 online tool.40
The HLA genotype of the SK-MEL-2 cell line was HLA-A*03 - *26; B*35 - *38; C*04 - *12. Buffy coat from a healthy donor was also obtained from the Finnish Red Cross service, and the genotype was determined as HLA-A*03 - *03; B*07 - *27; C*01 - *07.
OAd preparation
All OAds were generated, propagated, and characterized using standard protocols, as previously described.41
All viruses used in this study have been previously reported: Ad5D24 is an adenovirus that features a 24-base-pair deletion (D24) in the E1A gene,2 Ad5D24-CpG is an OAd bearing a CpG-enriched genome in the E3 gene,19 and Ad5D24-GM-CSF is an OAd expressing GM-CSF under the control of the viral E3 promoter.5
PeptiCRAd complex formation
All PeptiCRAd complexes described in this work were prepared by mixing oncolytic viruses and polyK-epitopes at a 1:500 ratio according to the following protocol: (i) for each microliter of viral preparation used, the corresponding number of micrograms of protein present was calculated; (ii) then, for each microgram of viral protein, 500 µg of peptide was added; (iii) after vortexing, the mixture was incubated at room temperature (RT) for 15 min; and (iv) the solution was vortexed and used for assays or animal injections. New PeptiCRAds were prepared before each experiment using fresh reagents. All dilutions of virus and peptides required before incubation were performed in sterile Milli-Q water adjusted to pH 7.4. The PeptiCRAds were then diluted with the buffer required by the assay.
Zeta potential and dynamic light scattering (DLS) analysis
Samples were prepared as described in the previous section. Each sample was then vortexed and diluted to a final volume of 700 µL with sterile Milli-Q water adjusted to pH 7.4, after which the sample was transferred to a polystyrene disposable cuvette to determine the size of the complexes. Afterward, the sample was recovered from the cuvette and transferred to a DTS1070 disposable capillary cell (Malvern, Worcestershire, UK) for zeta potential measurements. All measurements were performed at 25°C with a Zetasizer Nano ZS (Malvern).
Surface plasmon resonance
Measurements were performed using a multi-parametric SPR Navi™ 220A instrument (Bionavis Ltd, Tampere, Finland). Milli-Q water with its pH adjusted to 7.4 was used as a running buffer. A constant flow rate of 30 µL/min was used throughout the experiments, and temperature was set to +20°C. Laser light with a wavelength of 670 nm was used for surface plasmon excitation.
A sensor slide with a silicon dioxide surface was activated by 3 min of plasma treatment followed by coating with APTES ((3-aminopropyl)triethoxysilane) by incubating the sensor in 50 mM APTES in toluene solution for 1 h. The sensor was then placed into the SPR device, and the OAds were immobilized in situ on the sensor surface of the test channel by injecting 50 µg/mL OAds in Milli-Q water (pH 7.4) for approximately 12 min, followed by a 3 min wash with 20 mM CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate). The second flow channel was used as a reference and was injected with Milli-Q water (pH 7.4), followed by washing with CHAPS. The baseline was observed for at least 10 min before sample injections. PolyK-SIINFEKL or SIINFEKL was then injected into both flow channels of the flow cell in parallel, with increasing concentrations.
Viability assay
MTS assay was performed according to the manufacturer's protocol (CellTiter 96 AQueous One Solution Cell Proliferation Assay; Promega, Nacka, Sweden). Spectrophotometric data were acquired with Varioskan Flash Multimode Reader (Thermo Scientific, Carlsbad, CA, USA).
Infectivity assay by ICC
Tumor cells were seeded at 2.0×105 cells per well on 24-well plates in three or five replicates. The following day, the cells were infected with 10 vp/cell. The plates were then centrifuged for 90 min at 1,000 rcf at 37°C, followed by incubation for 48 h. Infectivity analysis was performed using the anti-hexon monoclonal antibody (Novus Biologicals, Littleton, CO, USA), diluted 1:2,000. For each well, five images of non-overlapping fields were acquired using an AMG EVOS XL microscope (AMG group, Life Technologies). The following formula was used to determine the infectious titer:
For the infectivity comparisons, the data are presented as the average number of spots in each field.
Cross-presentation experiment
2×106 spleenocytes in 800 µL of 10% RPMI-1640 culture media were incubated with 200 µL of SIINFEKL, polyK-SIINFEKL, SIINFEKL-polyK, or SIINFEKL-AHX-polyK peptide dilution (0.19 μg/μL). Alternatively, 7.9×109 vp mixed with 37.5 µg of polyK-SIINFEKL (OVA-PeptiCRAd) in 200 µL of 10% RPMI-1640 was applied. The PeptiCRAd complex was prepared as described previously. After 2 h of incubation cells were washed and stained with either APC anti-mouse H-2Kb bound to SIINFEKL or APC Mouse IgG1, κ Isotype Ctrl (BioLegend, San Diego, CA, USA), and the samples were analyzed by flow cytometry.
Animal experiments and ethical permits
All animal experiments were reviewed and approved by the Experimental Animal Committee of the University of Helsinki and the Provincial Government of Southern Finland. C57BL/6 mice were obtained from Scanbur (Karlslunde, Denmark), and immunodeficient triple-knockout NOD.Cg-Prkdcscid-IL2rgtm1Wjl/SzJ mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA).
For the efficacy experiments 3×105 B16-OVA or 1×105 B16-F10 or 2×106 SK-MEL-2 cells were injected subcutaneously on the flanks of mice. Three treatment injections were performed on established tumors.
Flow cytometry analysis
Flowcytometry analysis was performed using a BD LSR II (BD Biosciences) or a Gallios (Beckman Coulter) flow cytometer and FlowJo software (Tree Star, Ashland, OR, USA). Epitope-specific T cells were studied using MHC Class I Pentamers (ProImmune, Oxford, UK). Other antibodies used included the following: murine and human Fc block CD16/32 (BD PharMingen); FITC anti-mouse CD8+ and FITC anti-human CD8+ (ProImmune); PE/Cy7 anti-mouse CD3ε, PE/Cy7 anti-mouse CD19, FITC anti-mouse CD11c, PerCp/Cy5.5 anti-mouse CD86, APC anti-mouse H-2Kb bound to SIINFEKL and APC Mouse IgG1, κ Isotype Ctrl (BioLegend). All staining procedures were performed according to the manufacturer's recommendations.
Statistical analyses and correlation models
Statistical analysis was performed using GraphPad Prism 6 (GraphPad Software, Inc.., La Jolla, CA, USA). A detailed description of the statistical methods used to analyze the data from each experiment can be found in each figure caption. For correlation analysis presented in Fig. 5A, a non-linear regression was used: exponential one phase decay. The model is described by the equation
Y0 is the Y value when X (time) is zero. It is expressed in the same units as Y. Plateau is the Y value at infinite times, expressed in the same units as Y. K is the rate constant, expressed in reciprocal of the X axis time units. If X is in minutes, then K is expressed in inverse minutes.
For models of virus-peptide interaction presented in Fig. S2 the following equations have been used:
Y = SPR response, Ymax = SPR response at saturation, [P]= concentration of poly K-SIINFEKL, K = 1/Kco = binding constant, α = Hill coefficient describing co-operativity of the interaction (α > 1, positive co-operativity; α < 1, negative co-operativity).
Disclosure of potential conflicts of interest
Vincenzo Cerullo, Cristian Capasso, and Mari Hirvinen are co-inventors in patent application based on the present work.
Acknowledgments
We thank Dr Petri Ihalainen (Åbo Akademi University) for assistance with atomic field microscopy and Victor Cervera Carrascon for his precious technical help.
Funding
This research was supported by the three-year research grant (University of Helsinki), K. Albin Johansson Foundation, Foundation for the Finnish Cancer Institute, CDR and Division of Pharmaceutical Biosciences (University of Helsinki), and a TEKES-TUTL grant (Team Finland Network). C.C. was supported by the Drug Research Doctoral Program, M.H. was supported by the Finnish Cultural Foundation, and A.V. was supported by a CREME grant.
References
- 1.Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, Ng L, Nye JA, Sampson-Johannes A, Fattaey A et al.. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996; 274:373-6; PMID:8832876; http://dx.doi.org/ 10.1126/science.274.5286.373 [DOI] [PubMed] [Google Scholar]
- 2.Heise C, Hermiston T, Johnson L, Brooks G, Sampson-Johannes A, Williams A, Hawkins L, Kirn D. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat Med 2000; 6:1134-9; PMID:11017145; http://dx.doi.org/ 10.1038/80474 [DOI] [PubMed] [Google Scholar]
- 3.Zhang SN, Choi IK, Huang JH, Yoo JY, Choi KJ, Yun CO. Optimizing DC vaccination by combination with oncolytic adenovirus coexpressing IL-12 and GM-CSF. Mol Ther 2011; 19:1558-68; PMID:21468000; http://dx.doi.org/ 10.1038/mt.2011.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seiler MP, Gottschalk S, Cerullo V, Ratnayake M, Mane VP, Clarke C, Palmer DJ, Ng P, Rooney CM, Lee B. Dendritic cell function after gene transfer with adenovirus-calcium phosphate co-precipitates. Mol Ther 2007; 15:386-92; PMID:17235318; http://dx.doi.org/ 10.1038/sj.mt.6300029 [DOI] [PubMed] [Google Scholar]
- 5.Cerullo V, Pesonen S, Diaconu I, Escutenaire S, Arstila PT, Ugolini M, Nokisalmi P, Raki M, Laasonen L, Sarkioja M et al.. Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients. Cancer Res 2010; 70:4297-309; PMID:20484030; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-3567 [DOI] [PubMed] [Google Scholar]
- 6.Cerullo V, Diaconu I, Kangasniemi L, Rajecki M, Escutenaire S, Koski A, Romano V, Rouvinen N, Tuuminen T, Laasonen L et al.. Immunological effects of low-dose cyclophosphamide in cancer patients treated with oncolytic adenovirus. Mol Ther 2011; 19:1737-46; PMID:21673660; http://dx.doi.org/ 10.1038/mt.2011.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi IK, Li Y, Oh E, Kim J, Yun CO. Oncolytic adenovirus expressing IL-23 and p35 elicits IFN-gamma- and TNF-alpha-co-producing T cell-mediated antitumor immunity. PLoS One 2013; 8:e67512; PMID:23844018; http://dx.doi.org/ 10.1371/journal.pone.0067512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirvinen M, Rajecki M, Kapanen M, Parviainen S, Rouvinen-Lagerstrom N, Diaconu I, Nokisalmi P, Tenhunen M, Hemminki A, Cerullo V. Immunological effects of a tumor necrosis factor alpha-armed oncolytic adenovirus. Hum Gene Ther 2015; 26(3):134-44; PMID:25557131; http://dx.doi.org/ 10.1089/hum.2014.069 [DOI] [PubMed] [Google Scholar]
- 9.Diaconu I, Cerullo V, Hirvinen ML, Escutenaire S, Ugolini M, Pesonen SK, Bramante S, Parviainen S, Kanerva A, Loskog AS et al.. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res 2012; 72:2327-38; PMID:22396493; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2975 [DOI] [PubMed] [Google Scholar]
- 10.Liljenfeldt L, Dieterich LC, Dimberg A, Mangsbo SM, Loskog AS. CD40L gene therapy tilts the myeloid cell profile and promotes infiltration of activated T lymphocytes. Cancer Gene Ther 2014; 21:95-102; PMID:24481488; http://dx.doi.org/ 10.1038/cgt.2014.2 [DOI] [PubMed] [Google Scholar]
- 11.Pesonen S, Diaconu I, Cerullo V, Escutenaire S, Raki M, Kangasniemi L, Nokisalmi P, Dotti G, Guse K, Laasonen L et al.. Integrin targeted oncolytic adenoviruses Ad5-D24-RGD and Ad5-RGD-D24-GMCSF for treatment of patients with advanced chemotherapy refractory solid tumors. Int J Cancer 2012; 130:1937-47; PMID:21630267; http://dx.doi.org/ 10.1002/ijc.26216 [DOI] [PubMed] [Google Scholar]
- 12.Koski A, Kangasniemi L, Escutenaire S, Pesonen S, Cerullo V, Diaconu I, Nokisalmi P, Raki M, Rajecki M, Guse K et al.. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther 2010; 18:1874-84; PMID:20664527; http://dx.doi.org/ 10.1038/mt.2010.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sorensen MR, Holst PJ, Pircher H, Christensen JP, Thomsen AR. Vaccination with an adenoviral vector encoding the tumor antigen directly linked to invariant chain induces potent CD4(+) T-cell-independent CD8(+) T-cell-mediated tumor control. Eur J Immunol 2009; 39:2725-36; PMID:19637230; http://dx.doi.org/ 10.1002/eji.200939543 [DOI] [PubMed] [Google Scholar]
- 14.Nwanegbo E, Vardas E, Gao W, Whittle H, Sun H, Rowe D, Robbins PD, Gambotto A. Prevalence of neutralizing antibodies to adenoviral serotypes 5 and 35 in the adult populations of The Gambia, South Africa, and the United States. Clin Diagn Lab Immunol 2004; 11:351-7; PMID:15013987; http://dx.doi.org/ 10.1128/CDLI.11.2.351-357.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaiss AK, Machado HB, Herschman HR. The influence of innate and pre-existing immunity on adenovirus therapy. J Cell Biochem 2009; 108:778-90; PMID:19711370; http://dx.doi.org/ 10.1002/jcb.22328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fasbender A, Zabner J, Chillon M, Moninger TO, Puga AP, Davidson BL, Welsh MJ. Complexes of adenovirus with polycationic polymers and cationic lipids increase the efficiency of gene transfer in vitro and in vivo. J Biol Chem 1997; 272:6479-89; PMID:9045673; http://dx.doi.org/ 10.1074/jbc.272.10.6479 [DOI] [PubMed] [Google Scholar]
- 17.Moore MW, Carbone FR, Bevan MJ. Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell 1988; 54:777-85; PMID:3261634; http://dx.doi.org/ 10.1016/S0092-8674(88)91043-4 [DOI] [PubMed] [Google Scholar]
- 18.Deng Y, Gibbs J, Bacik I, Porgador A, Copeman J, Lehner P, Ortmann B, Cresswell P, Bennink JR, Yewdell JW. Assembly of MHC class I molecules with biosynthesized endoplasmic reticulum-targeted peptides is inefficient in insect cells and can be enhanced by protease inhibitors. J Immunol 1998; 161:1677-85; PMID:9712031 [PubMed] [Google Scholar]
- 19.Cerullo V, Diaconu I, Romano V, Hirvinen M, Ugolini M, Escutenaire S, Holm SL, Kipar A, Kanerva A, Hemminki A. An oncolytic adenovirus enhanced for toll-like receptor 9 stimulation increases antitumor immune responses and tumor clearance. Mol Ther 2012; 20:2076-86; PMID:22828500; http://dx.doi.org/ 10.1038/mt.2012.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, Carroll MW, Liu C, Moss B, Rosenberg SA et al.. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med 1998; 188:277-86; PMID:9670040; http://dx.doi.org/ 10.1084/jem.188.2.277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Overwijk WW, Restifo NP. B16 as a mouse model for human melanoma. Curr Protoc Immunol 2001; Chapter 20:Unit 20 1; PMID:18432774; http://dx.doi.org/ 10.1002/0471142735.im2001s39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vacchelli E, Eggermont A, Fridman WH, Galon J, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: Immunostimulatory cytokines. Oncoimmunology 2013; 2:e24850; PMID:24073369; http://dx.doi.org/ 10.4161/onci.24850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aranda F, Vacchelli E, Eggermont A, Galon J, Sautes-Fridman C, Tartour E, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: Peptide vaccines in cancer therapy. Oncoimmunology 2013; 2:e26621; PMID:24498550; http://dx.doi.org/ 10.4161/onci.26621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aranda F, Vacchelli E, Obrist F, Eggermont A, Galon J, Herve Fridman W, Cremer I, Tartour E, Zitvogel L, Kroemer G et al.. Trial Watch: Adoptive cell transfer for anticancer immunotherapy. Oncoimmunology 2014; 3:e28344; PMID:25050207; http://dx.doi.org/ 10.4161/onci.28344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al.. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443-54; PMID:22658127; http://dx.doi.org/ 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nigatu AS, Vupputuri S, Flynn N, Neely BJ, Ramsey JD. Evaluation of cell-penetrating peptide/adenovirus particles for transduction of CAR-negative cells. J Pharm Sci 2013; 102:1981-93; PMID:23592439; http://dx.doi.org/ 10.1002/jps.23556 [DOI] [PubMed] [Google Scholar]
- 27.Heath WR, Carbone FR. Cross-presentation in viral immunity and self-tolerance. Nat Rev Immunol 2001; 1:126-34; PMID:11905820; http://dx.doi.org/ 10.1038/35100512 [DOI] [PubMed] [Google Scholar]
- 28.Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol 2012; 12:557-69; PMID:22790179; http://dx.doi.org/ 10.1038/nri3254 [DOI] [PubMed] [Google Scholar]
- 29.Ma X, Serna A, Xu RH, Sigal LJ. The amino acid sequences flanking an antigenic determinant can strongly affect MHC class I cross-presentation without altering direct presentation. J Immunol 2009; 182:4601-7; PMID:19342634; http://dx.doi.org/ 10.4049/jimmunol.0803806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Capasso C, Garofalo M, Hirvinen M, Cerullo V. The evolution of adenoviral vectors through genetic and chemical surface modifications. Viruses 2014; 6:832-55; PMID:24549268; http://dx.doi.org/ 10.3390/v6020832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wonganan P, Croyle MA. PEGylated Adenoviruses: From Mice to Monkeys. Viruses 2010; 2:468-502; PMID:21994645; http://dx.doi.org/ 10.3390/v2020468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clapp T, Siebert P, Chen D, Jones Braun L. Vaccines with aluminum-containing adjuvants: optimizing vaccine efficacy and thermal stability. J Pharm Sci 2011; 100:388-401; PMID:20740674; http://dx.doi.org/ 10.1002/jps.22284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li KY, Peers-Adams A, Win SJ, Scullion S, Wilson M, Young VL, Jennings P, Ward VK, Baird MA, Young SL. Antigen Incorporated In Virus-like Particles Is Delivered to Specific Dendritic Cell Subsets That Induce An Effective Antitumor Immune Response In Vivo. J Immunother 2013; 36:11-9; PMID:23211625; http://dx.doi.org/ 10.1097/CJI.0b013e3182787f5e [DOI] [PubMed] [Google Scholar]
- 34.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK et al.. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012; 482:400-4; PMID:22318521; http://dx.doi.org/ 10.1038/nature10755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andersen BM, Ohlfest JR. Increasing the efficacy of tumor cell vaccines by enhancing cross priming. Cancer Lett 2012; 325:155-64; PMID:22809568; http://dx.doi.org/ 10.1016/j.canlet.2012.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich PY, Mendrzyk R et al.. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med 2012; 18(8):1254-61; PMID:22842478; http://dx.doi.org/ 10.1038/nm.2883 [DOI] [PubMed] [Google Scholar]
- 37.Carretero R, Romero JM, Ruiz-Cabello F, Maleno I, Rodriguez F, Camacho FM, Real LM, Garrido F, Cabrera T. Analysis of HLA class I expression in progressing and regressing metastatic melanoma lesions after immunotherapy. Immunogenetics 2008; 60:439-47; PMID:18545995; http://dx.doi.org/ 10.1007/s00251-008-0303-5 [DOI] [PubMed] [Google Scholar]
- 38.Ito H, Aoki H, Kuhnel F, Kondo Y, Kubicka S, Wirth T, Iwado E, Iwamaru A, Fujiwara K, Hess KR et al.. Autophagic cell death of malignant glioma cells induced by a conditionally replicating adenovirus. J Natl Cancer Inst 2006; 98:625-36; PMID:16670388; http://dx.doi.org/ 10.1093/jnci/djj161 [DOI] [PubMed] [Google Scholar]
- 39.Liikanen I, Ahtiainen L, Hirvinen ML, Bramante S, Cerullo V, Nokisalmi P, Hemminki O, Diaconu I, Pesonen S, Koski A et al.. Oncolytic adenovirus with temozolomide induces autophagy and antitumor immune responses in cancer patients. Mol Ther 2013; 21:1212-23; PMID:23546299; http://dx.doi.org/ 10.1038/mt.2013.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biosynthesis Peptide Property Calculator Version 3.1. 612 East Main Street Lewisville, TX 75057 Bio-Synthesis Inc. [Google Scholar]
- 41.Kanerva A, Zinn KR, Chaudhuri TR, Lam JT, Suzuki K, Uil TG, Hakkarainen T, Bauerschmitz GJ, Wang M, Liu B et al.. Enhanced therapeutic efficacy for ovarian cancer with a serotype 3 receptor-targeted oncolytic adenovirus. Mol Ther 2003; 8:449-58; PMID:12946318; http://dx.doi.org/ 10.1016/S1525-0016(03)00200-4 [DOI] [PubMed] [Google Scholar]