

ABSTRACT
Programmed cell death (PD)-1/PD-1 ligand-1 (PD-L1)-targeted therapy has emerged as a promising therapeutic strategy for lung cancer. However, whether EML4-ALK regulates PD-L1 expression in lung cancer remains unknown. A total of 532 pulmonary adenocarcinomas (pADCs), including 58 ALK-translocated tumors, were immunohistochemically evaluated for PD-L1 and PD-1. H23 (EGFRWild-typeEML4-ALK−PD-L1Low) and H2228 (EGFRWild-typeEML4-ALK+PD-L1High) cells were transfected with EML4-ALK or ALK short interfering RNAs and used to investigate the alterations in PD-L1 expression. PD-L1 expression was detected in 81% of ALK-translocated pADCs; this value was significantly higher than those of pADCs with EGFR mutation, KRAS mutation or lacking ALK, EGFR or KRAS mutation (p <0.005 for all). Moreover, ALK-translocated pADC with PD-L1 expression showed significantly higher numbers of tumor-infiltrating PD-1+ cells. ALK knockdown or inhibition (crizotinib treatment) in H2228 cells downregulated PD-L1 expression. Transfection of H23 cells with EML4-ALK enhanced PD-L1 expression, which was compromised by crizotinib treatment. This ALK-dependent upregulation of PD-L1 expression was mediated by STAT3 and hypoxia-inducible factor (HIF)-1α under normoxia and hypoxia. Furthermore, EML4-ALK enhanced HIF-1α expression through increasing transcription and decreasing ubiquitination of HIF-1α. In ALK-translocated pADC tissues, significant positive correlations between PD-L1 and nuclear HIF-1α (p < 0.05) or pSTAT3 expression levels (p<0.005) were observed. Among patients with ALK-translocated pADC, strong PD-L1 expression was significantly associated with shorter progression-free (p = 0.001) and overall survival (p = 0.002) after crizotinib treatment. Collectively, our findings demonstrate that ALK-derived pADCs increase PD-L1 expression via HIF-1α and/or STAT3, thus providing a rationale for PD-1/PD-L1 pathway-targeted therapy in ALK-translocated lung cancer.
KEYWORDS: Adenocarcinoma, anaplastic lymphoma kinase, cancer immunotherapy, hypoxia-inducible factor-1, immune checkpoint, programmed cell death-1, programmed cell death-ligand 1
Abbreviations
- ALCL
anaplastic large cell lymphoma
- ALK
anaplastic lymphoma kinase
- ChIP
chromatin immunoprecipitation
- EGFR
epidermal growth factor receptor
- EML4
echinoderm microtubule-associated protein-like 4
- FFPE
formalin-fixed paraffin-embedded
- HIF-1α
hypoxia-inducible factor-1α
- IHC
immunohistochemistry
- mAb
monoclonal antibody
- NSCLC
non-small cell lung cancer
- OS
overall survival
- pADCs
pulmonary adenocarcinomas
- PD-1
programmed cell death-1
- PD-L1
programmed cell death-1 ligand-1
- PFS
progression-free survival
- qPCR
quantitative PCR
- qRT-PCR
quantitative reverse transcription PCR
- sc
scramble
- TILs
tumor-infiltrating lymphocytes
- TKI
tyrosine kinase inhibitor
- WT
wild-type
Introduction
Anaplastic lymphoma kinase (ALK) translocation has been observed in various cancers, including lymphoma, carcinoma and sarcoma.1 In carcinomas, ALK translocation occurs mostly with echinoderm microtubule-associated protein-like 4 (EML4), which plays an important oncogenic role in pADC in vitro and in vivo.2 EML4-ALK translocation is observed approximately in 5% of pADCs,3 and patients with ALK translocation tend to show an unfavorable prognosis.4 However, an ALK tyrosine kinase inhibitor (TKI) was shown to prolong the survival of pADC patients with ALK translocation,5,6 demonstrating superior to standard cytotoxic chemotherapy in patients with advanced ALK-translocated lung cancer.6,7 Nevertheless, most patients with ALK-translocated pADC eventually develop resistance to ALK TKI,8 indicating that the development of an alternative therapeutic reagent is urgent for the treatment of patients with ALK TKI resistance. To this end, a comprehensive understanding of the mechanisms underlying acquisition of TKI resistance in patients with ALK-translocated pADC is needed.
The programmed cell death-1 (PD-1)/PD-1 ligand (PD-L) pathway is an important immune checkpoint that contributes to the maintenance of self-tolerance and control of excessive immune responses.9 Recently, it has been shown that cancer cells also utilize the PD-1/PD-L pathway to evade host immune surveillance.9 PD-1 is expressed on immune cells, particularly in activated T cells, while PD-L1 and PD-L2 are expressed on antigen-presenting cells and non-immune cells in peripheral tissues.10 Engagement of PD-1 by PD-Ls suppresses T-cell proliferation and function by inducing T-cell apoptosis, anergy, exhaustion, and the production of immune-suppressive cytokines.11 However, interactions between PD-1 and PD-Ls are reversible, and blockade of PD-1/PD-L interactions restores effector T-cell functions and antitumor immune responses in vivo.12 Therefore, these findings suggest that blockade of the PD-1/PD-L1 pathway might affect immune-mediated tumor surveillance in vivo, enhancing tumor cell death. Consistently, antibodies inhibiting PD-1 or PD-L1 have yielded therapeutic benefits with an objective response and disease control in patients with non-small cell lung cancer (NSCLC) in early-phase clinical trials.13,14 Based on these findings, immunotherapy targeting the PD-1/PD-L1 pathway has emerged as a highly promising therapeutic strategy for NSCLC.
Although the predictive biomarkers for PD-1/PD-L1-targeted immunotherapy remain unclear, PD-L1 expression in tumor cells and immune cells was reported to be associated with therapeutic efficacy in patients with NSCLC.15,16 PD-L1 expression can be endogenously induced by activated oncogenic signaling pathways in tumor cells and/or exogenously (adaptively) by cytokines (e.g., interferon [IFN]γ) secreted from reactive immune cells.11 It was demonstrated that mutant EGFR in bronchial epithelial cells induced PD-L1 expression, and oncogenic EGFR signaling activated the PD-1/PD-L1 pathway in NSCLC in vivo.17 In human NSCLC tissues, the expression pattern of PD-L1 has been reported to vary according to genetic alterations and oncoprotein expression status or the amount of tumor-infiltrating immune cells.18-21 However, the association between the PD-l/PD-L1 pathway and ALK translocation in pADC remains unclear. In contrast to lung cancer, NPM-ALK fusion in anaplastic large cell lymphoma (ALCL) was demonstrated to induce PD-L1 expression.22 Moreover, ALK-positive lymphomas are immunogenic tumors that elicit immune responses against the ALK oncoantigen, which is aberrantly expressed by gene translocation in tumor cells.23-25 Based on these findings, we hypothesized that ALK translocation might be involved in the regulation of the PD-1/PD-L1 pathway in pADCs. Therefore, we performed this study with the following aims: (1) to characterize the expression status of PD-Ll and PD-1 in pADC patients with ALK translocation, (2) to elucidate the mechanism by which ALK regulates PD-L1 expression in pADC, and (3) to evaluate the association between PD-L1 expression and the clinical outcomes of patients with pADC after ALK inhibitor treatment.
Results
PD-L1 and PD-1 are frequently expressed in patients with ALK-translocated pADC
Our previous study demonstrated PD-L1 expression to be relatively frequent in ALK-translocated pADCs among 497 resected cases, but with marginal statistical significance (manuscript in press). To clarify this issue further, we collected an additional 35 cases of resected pADC harboring ALK translocation, thereby establishing a cohort of 532 cases of pADC, which included 46.5% (230/494) with EGFR mutation, 11.5% (26/226) with KRAS mutation, and 10.9% (58/532) with ALK translocation. The expression patterns of PD-L1 according to the molecular genetic status are summarized in Table S2. As shown in Fig. 1A–B, the percentages of PD-L1 expression were significantly higher in pADCs with ALK translocation (n = 58) than in those with EGFR mutation (n = 228), KRAS mutation (n = 25), or no genetic alteration of ALK, EGFR or KRAS (triple negative status, n = 60) (p < 0.005 for all). Of note, the proportion of strong (score 3) PD-L1 expression in the ALK-translocated group was 25.9%, which was higher than that (9.3%) observed in the others (p = 0.001) (Fig. 1B). Moreover, ALK-translocated pADC with PD-L1 expression exhibited a greater number of PD-1+ tumor-infiltrating lymphocytes (TILs) and a high ratio of PD-1+/CD8+ TILs (p = 0.025 and p < 0.005, respectively) (Fig. 1C and Table S3). These findings suggest that PD-1/PD-L1-dependent immune regulation might occur in the microenvironment of ALK-translocated pADC.
Figure 1.
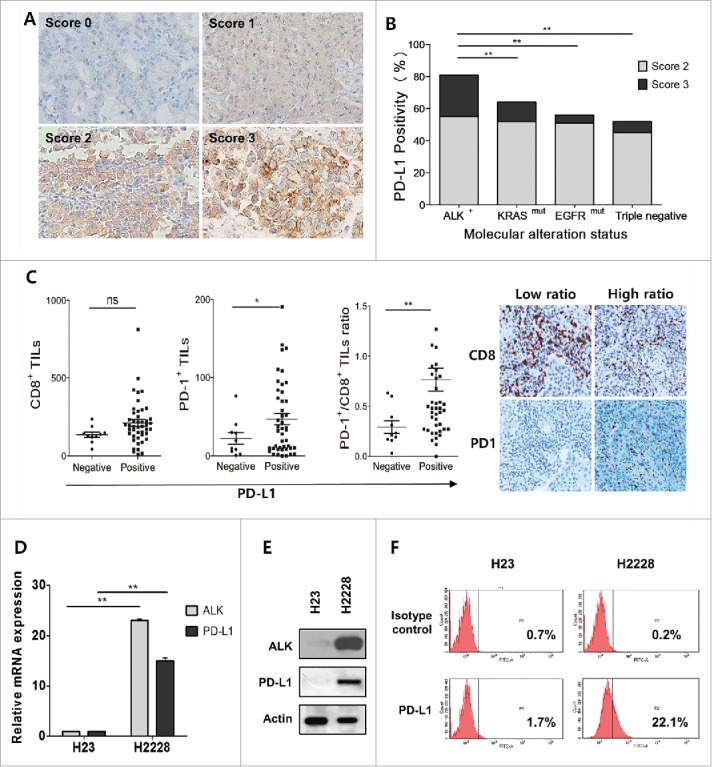
PD-L1 is frequently expressed in pulmonary adenocarcinomas (pADCs) harboring ALK translocation. (A) Representative immunohistochemical images for PD-L1 expression in pADCs with ALK translocation; none (score 0), weak (score 1), moderate (score 2) or strong (score 3) expression in tumor cells. (B) The percentages of PD-L1 expression were calculated among patients with ALK-translocated (n = 59), KRAS-mutated (n = 25), EGFR-mutated (n = 228) and triple negative (ALK, KRAS and EGFR all wild-type; n = 60) pADC. (C) The numbers of PD-1+ and CD8+ tumor-infiltrating lymphocytes (TILs) and PD-1+/CD8+ TIL ratios were estimated according to PD-L1 expression in patients (n = 58) with ALK-translocated pADC. The representative IHC images for PD-1 and CD8+ from cases showing a low or high ratio of PD-1+/CD8+ TIL are shown in the right panel. (D) The levels of mRNA expression of PD-L1 and ALK were evaluated in H23 (EGFRWild-typeEML4-ALK−) and H2228 (EML4-ALK+) cells using qRT-PCR. The bar graphs show the relative expression levels [2(-ddCt)] of ALK or PD-L1 in pADC cell lines compared with those in H23 cells as the reference. The values presented are the mean values ± standard deviation (SD) of at least three independent experiments. (E and F) The expression levels of ALK and PD-L1 were evaluated in H23 and H2228 cells by Western blotting, and the surface expression of PD-L1 was evaluated by flow cytometry. Statistically significant differences are indicated by * and **, which signify p < 0.05 and p < 0.005, respectively, as determined using the χ2 test (B) or Student's t-test (C, D). (ns: not significant).
EML4-ALK upregulates PD-L1 expression in pADC cell lines
Based on these observations, we hypothesized that ALK translocation might be involved in the regulation of PD-L1 expression in pADC. To address this, we selected H23 cells (EGFRWT/ALKWT/PD-L1Low) and H2228 cells (EML4-ALK+/PD-L1High) (Fig. 1D–F) by screening several pADC cell lines with variable genetic alterations (data not shown) and used them throughout the study. Upon transfection with three different ALK siRNAs or treatment with ALK inhibitor (crizotinib), total and cell surface expression levels of PD-L1 in H2228 were significantly decreased (Fig. 2A–D). Furthermore, transfection of H23 cells with the pcDNA-EML4-ALK variant 1 (EML4-ALK v1) or pcDNA-EML4-ALK variant 3 (EML4-ALK v3) construct significantly upregulated PD-L1 expression at the mRNA, total protein, and surface expression levels (Fig. 2E–G), which were attenuated by subsequent crizotinib treatment (Fig. 2H–I). IHC of tumor tissues from patients with ALK-translocated pADC revealed that tumors with PD-L1 expression tend to have higher phosphorylated ALK expression (Fig. S1). Taken together, these data indicate that EML4-ALK upregulates PD-L1 expression in pADC cell lines.
Figure 2.
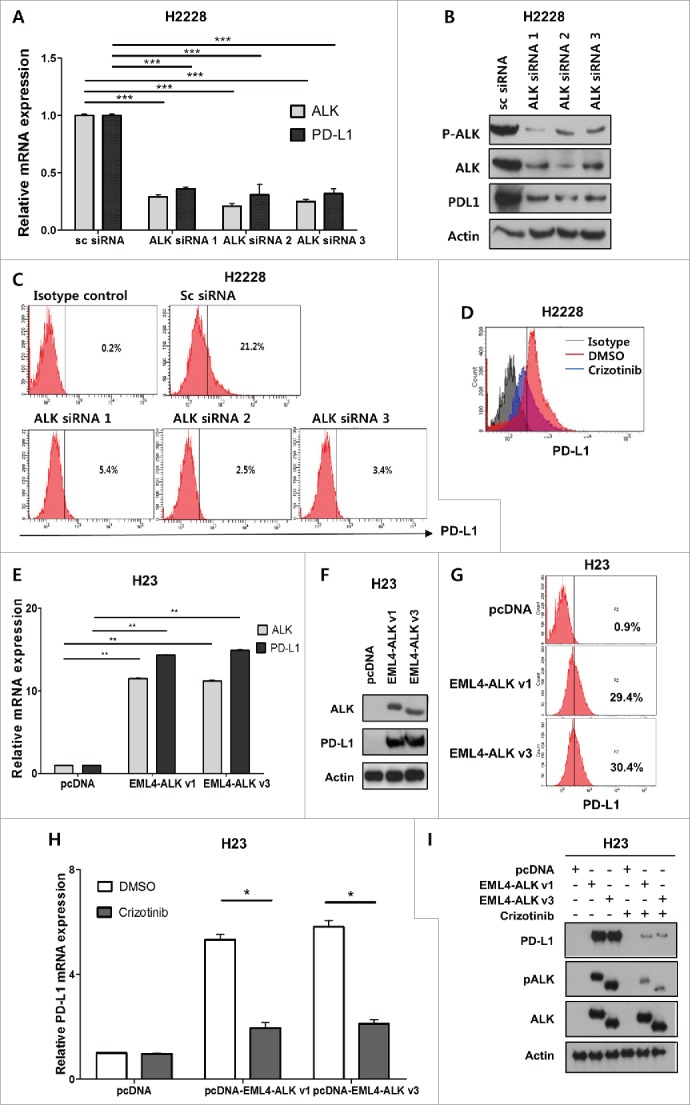
EML4-ALK upregulates PD-L1 expression in pADC cell lines. (A, B and C) H2228 (EML4-ALK+/PD-L1High) cells were transfected with ALK siRNA 1, 2, 3 or scramble (sc) siRNA (100 nM), and then harvested 48 h after transfection. The expression levels of ALK and PD-L1 in cells transfected with ALK siRNAs were compared with those transfected with the sc siRNA using qRT-PCR, Western blotting, and flow cytometry. (D) H2228 cells were treated with crizotinib (100 nM); 24 h later, the surface expression of PD-L1 was assessed on cells treated with dimethyl sulfoxide (DMSO) and crizotinib using flow cytometry. (E, F and G) H23 (EML4-ALK−/PD-L1Low) cells were transfected with pcDNA, pcDNA-EML4-ALK v1 or pcDNA-EML4-ALK v3 (1 μg), and harvested 24 h after transfection. The expression of ALK and PD-L1 was then estimated using qRT-PCR, Western blotting, and flow cytometry. (H and I) H23 cells were transfected with pcDNA, pcDNA-EML4-ALK v1, or pcDNA-EML4-ALK v3 and then incubated with the crizotinib (2 μM) for 24 h, followed by qRT-PCR for PD-L1 and Western blotting for PD-L1, pALK, and ALK. The values presented in the histogram are the mean values ± SD of at least three independent experiments. Statistically significant differences are indicated by *, ** and ***, which signify p < 0.05, p < 0.005 and p < 0.0005, respectively, as determined by Student's t-test.
EML4-ALK enhances PD-L1 expression in pADCs via STAT3
It has been reported that ALK exerts biological activities on cancer cells via various signaling pathways, including STAT3, AKT, and ERK pathways,1 and that the NPM-ALK fusion protein upregulates PD-L1 via STAT3 signaling in ALCL.22 Thus, we explored whether the STAT3 signaling pathway is involved in the EML4-ALK-mediated upregulation of PD-L1 in pADC cell lines. Suppression of EML4-ALK in H2228 cells by ALK siRNA 1 transfection attenuated pSTAT3 expression relative to STAT3 levels (Fig. 3A), suggesting that EML4-ALK might enhance PD-L1 expression in pADC cell lines by regulating STAT3 activity. Consistent with this suggestion, the treatment of H2228 cells with S3I-201 (a STAT3 inhibitor) downregulated PD-L1 expression (Fig. 3B–C). Furthermore, transfection of H23 cells with EML4-ALK v1 or v3 markedly upregulated pSTAT3 levels relative to STAT3 expression (Fig. 3D). In addition, EML4-ALK-mediated upregulation of PD-L1 in H23 cells was compromised by S3I-201 treatment (Fig. 3E). These findings suggest that activation of the STAT3 pathway might regulate EML4-ALK-mediated upregulation of PD-L1 in pADC cell lines. The protein-DNA binding assay revealed that pSTAT3 was bound to the PD-L1 promoter region in H23 cells transfected with EML4-ALK (Fig. 3F). Furthermore, PD-L1 expression showed a significantly positive correlation with the nuclear pSTAT3 levels by IHC of tumor tissues from patients with ALK-translocated pADC (p < 0.005) (Fig. 3G). Combined, these data suggest that EML4-ALK increases PD-L1 expression in pADCs via STAT3 by directly binding to the PD-L1 promoter.
Figure 3.
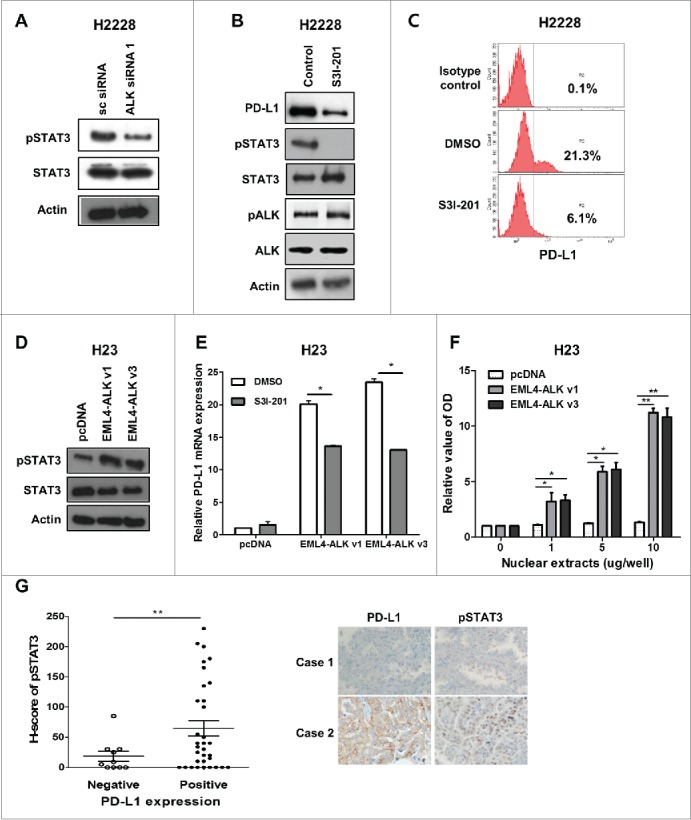
EML4-ALK-induced PD-L1 expression is dependent on STAT3 activity. (A) H2228 cells were transfected with ALK siRNA 1 or scramble siRNA, and the expression levels of pSTAT3 and STAT3 were evaluated by Western blotting. (B and C) H2228 cells were treated with S3I-201 (100 μM) for 24 h, and then the expression levels of PD-L1, pSTAT3, STAT3, pALK, and ALK were evaluated by Western blotting and flow cytometry. (D) The expression levels of pSTAT3 and STAT3 were evaluated in H23 cells transfected with pcDNA, pcDNA-EML4-ALK v1 or pcDNA-EML4-ALK v3 by Western blotting. (E) H23 cells were transfected with pcDNA, pcDNA-EML4-ALK v1 or pcDNA-EML4-ALK v3 and then were incubated with S3I-201 (100 μM). Twenty-four hours after incubation, qRT-PCR was performed to determine the levels of PD-L1 expression. (F) H23 cells were transfected with pcDNA, pcDNA-EML4-ALK v1 or pcDNA-EML4-ALK v3, and nuclear extracts from these cells were then submitted for protein-DNA binding assays to evaluate STAT3 binding to the PD-L1 promoter. The colorimetric activities were determined using a luminometer. The relative optical density values of cells transfected with pcDNA-EML4-ALK v1 or pcDNA-EML4-ALK v3 versus those of cells transfected with pcDNA according to the amount of nuclear extract are shown. (G) The dot plot represents nuclear pSTAT3 expression using the H score according to PD-L1 expression in tumor tissues from patients with ALK-translocated pADC. The representative IHC images are shown in the right panel. The values presented in the histogram are the mean values ± SD of at least three independent experiments. Statistically significant differences are indicated by * and **, which signify p < 0.05 and p < 0.005, respectively, as determined by Student's t-test.
EML4-ALK upregulates PD-L1 expression in pADC through HIF-1α in a hypoxic environment
Cancer cells are likely to exist under hypoxic conditions in vivo, suggesting that a hypoxic microenvironment might affect EML4-ALK-mediated enhancement of PD-L1 in pADCs. Consistently, a recent study reported that PD-L1 expression in cancer cells is induced by hypoxia, indicating that PD-L1 is a novel transcriptional target of HIF-1α.26,27 Furthermore, it was demonstrated in ALK-translocated lymphoma that hypoxia-related molecular pathways were activated by inducing HIFs.28 Thus, we investigated whether HIF-1α is involved in EML4-ALK-mediated enhancement of PD-L1 expression in pADC under hypoxia.
Upon treatment of H2228 cells with CoCl2, the expression levels of PD-L1, as well as that of HIF-1α, were increased at the mRNA and protein levels (Fig. 4A–C). Notably, CoCl2 treatment of H2228 cells also increased ALK phosphorylation (Fig. 4B). ChIP assay demonstrated increased binding of HIF-1α to HRE1 and 2 within the promoter region of PD-L1 in H2228 cells under hypoxia (Fig. 4D–E). Moreover, enhancement of PD-L1 expression in H2228 cells under hypoxic conditions was inhibited by PX-12 (a HIF-1α inhibitor) treatment (Fig. 4F). In contrast to H2228 cells, H23 cells exhibited upregulation of HIF-1α but minimal expression of PD-L1 under hypoxia (Fig. 5A–B). However, upon transfection of H23 cells with EML4-ALK, PD-L1 expression was enhanced at much higher mRNA and protein levels under hypoxia than normoxia (Fig. 5A–B). Notably, HIF-1α expression showed a much greater increase in EML4-ALK-transfected H23 cells under hypoxia, compared with pcDNA (mock vector)-transfected H23 cells, at both the transcriptional and protein levels (Fig. 5A–B). These findings suggest that EML4-ALK might increase the expression levels of PD-L1 under hypoxia via upregulation of HIF-1α in pADCs. Meanwhile, HIF-1α expression in cancer cells is regulated via transcription or post-translational modification such as ubiquitin-mediated degradation.29 To explore this, we performed an ubiquitination assay using H23 cells. As shown in Fig. 5C–D, polyubiquitination of HIF-1α and interaction of HIF-1α and VHL were decreased more in EML4-ALK-transfected H23 cells than in mock vector-transfected H23 cells under normoxic or hypoxic conditions. Consistently, the degradation of HIF-1α was delayed after treatment with cycloheximide in EML4-ALK-transfected H23 cells compared with mock vector-transfected H23 cells under hypoxic conditions (Fig. 5E). Moreover, IHC demonstrated a significant positive correlation between nuclear HIF-1α and PD-L1 expression in tumor tissues from patients with ALK-translocated pADCs (p < 0.05) (Fig. 5F). Taken together, these findings suggest that EML4-ALK might increase HIF-1α expression under hypoxia by enhancing transcription and inhibiting ubiquitination of HIF-1α, resulting in the stabilization of HIF-1α, consequently contributing to HIF-1α-mediated upregulation of PD-L1 under hypoxia.
Figure 4.
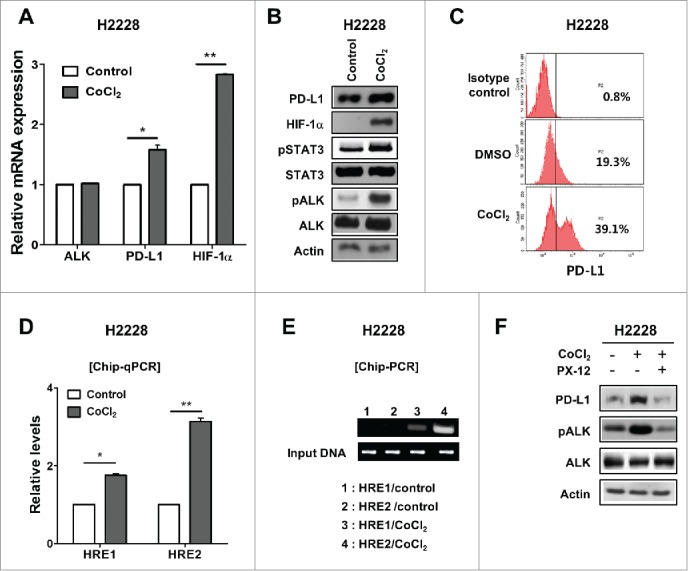
PD-L1 expression under hypoxia is upregulated via HIF-1α in EML4-ALK-translocated pADC cell lines. (A) Expression of HIF-1α was estimated in H2228 cells treated with CoCl2 (100 μM) for 3 h, while those of ALK and PD-L1 were measured in these cells 24 h later using qRT-PCR. (B and C) The expression of PD-L1, HIF-1α, pSTAT3, STAT3, pALK and ALK was evaluated in H2228 cells treated with CoCl2 by Western blotting, while the surface expression of PD-L1 was determined in these cells by flow cytometry. (D and E) H2228 cells were treated with CoCl2, and the cellular extracts were subjected to ChIP assay for hypoxia response element (HRE)1 and HRE2 within the PD-L1 promoter followed by real-time qPCR (D) and semi-quantitative PCR (E). (F) The levels of PD-L1, pALK and ALK expression were assessed in H2228 cells treated with CoCl2 and/or subsequent PX-12 (HIF-1α specific inhibitor) (100 nM, for 24 h) by Western blotting. The values presented in the histogram are the mean values ± SD of at least three independent experiments. Statistically significant differences are indicated by * and **, which signify p < 0.05 and p < 0.005, respectively, as determined by Student's t-test.
Figure 5.
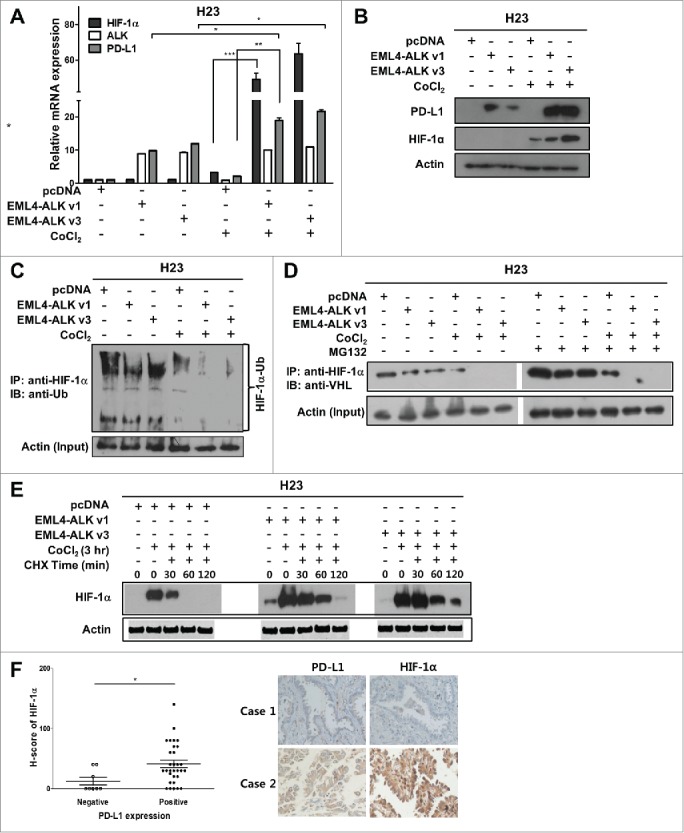
EML4-ALK upregulates PD-L1 expression under hypoxia by enhancing HIF-1α expression in pADC cells. (A and B) H23 cells were transfected with pcDNA, pcDNA-EML4-ALK v1, or pcDNA-EML4-ALK v3. After 24 h, cells were treated with CoCl2 (100 μM, 3 h) and subjected to qRT-PCR and Western blotting to determine the levels of HIF-1α, ALK and PD-L1. The relative values in the histogram are the mean values ± SD of at least three independent experiments. (C) H23 cells transfected with pcDNA, pcDNA-EML4-ALK v1, or pcDNA-EML4-ALK v3 were treated with CoCl2 or control. Total cell lysates of these cells were immunoprecipitated with anti-HIF-1α antibody and immunoblotted with anti-ubiquitin antibody. (D) H23 cells transfected with pcDNA, pcDNA-EML4-ALK v1, or pcDNA-EML4-ALK v3 were treated with the MG132 (10 μM for 3 h) and then CoCl2 or control. Total cell lysates of these cells were immunoprecipitated with anti-HIF-1α antibody and immunoblotted with anti-VHL antibody. (E) H23 cells transfected with pcDNA, pcDNA-EML4-ALK v1, or pcDNA-EML4-ALK v3 were treated with CoCl2 and subsequently incubated with cycloheximide (CHX, 100 μM) for the indicated time, and then the HIF-1α protein levels were monitored by Western blotting. (F) The dot plot describes nuclear HIF-1α expression using H scores according to PD-L1 expression in tumor tissues from patients with ALK-translocated pADC. The representative IHC images are shown in the right panel. Statistically significant differences are indicated by *, ** and ***, which signify p < 0.05, p < 0.005 and p < 0.0005, respectively, as determined by Student's t-test.
STAT3 and HIF-1α cooperatively enhance PD-L1 expression in EML4-ALK- translocated pADC cells under hypoxia
Upregulation of PD-L1 in pADCs via both STAT3 and HIF-1α led to the hypothesis that these proteins might cooperatively contribute to increasing PD-L1 expression in EML4-ALK-translocated pADCs under hypoxia. Co-immunoprecipitation assay demonstrated that HIF-1α was bound to pSTAT3 in H2228 cells under hypoxia (Fig. 6A), suggesting that the interaction between the two proteins might be critical to enhance PD-L1 expression in pADCs under hypoxia. Moreover, the enhancement of PD-L1 expression in H2228 cells under hypoxia was suppressed by STAT3 inhibitor treatment (Fig. 6B). Collectively, these data suggest that STAT3 and HIF-1α might be cooperatively involved in PD-L1 upregulation in EML4-ALK-translocated lung cancer as schematically illustrated in Fig. 6C.
Figure 6.
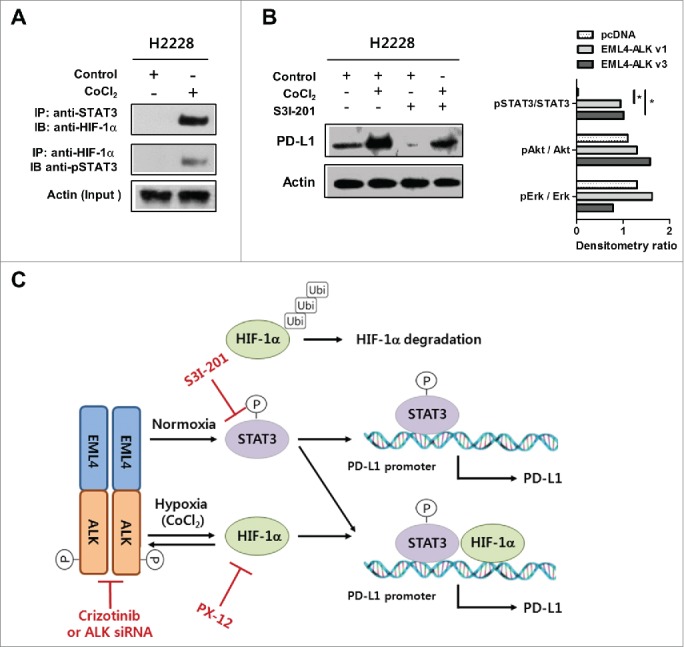
STAT3 and HIF-1α cooperatively enhance PD-L1 expression in EML4-ALK-translocated pADC cells under hypoxia. (A) Immunoprecipitation and immunoblotting were performed using H2228 cells treated with CoCl2 to estimate the interaction between HIF-1α and STAT3 as indicated. (B) The levels of PD-L1 expression were measured in H2228 cells treated with CoCl2 in the presence or absence of S3I-201 for 24 h by Western blotting. (C) Diagram of our current model for the EML4-ALK-mediated regulation of PD-L1 expression in pADC under normoxia or hypoxia.
Clinical correlation between PD-L1 expression in tumors and outcome of ALK inhibitor therapy in patients with ALK-translocated pADC
To investigate the clinical relevance of PD-L1 expression in ALK-translocated pADC, we analyzed the clinicopathological features and clinical outcomes of patients with ALK-translocated pADC according to the PD-L1 expression status. In patients with ALK-translocated pADC (n = 148) who underwent surgical resection and advanced/metastasis of tumors, those of older age (≥60 y) frequently showed PD-L1 expression in tumors. Otherwise, there was no statistically significant difference in gender, smoking status, or TNM stage between patients with and without PD-L1 expression in tumors (Table S4).
In patients who underwent surgical resection for primary lung tumor, PD-L1 expression had no influence on disease-free survival (data not shown). By contrast, among patients (n = 90) who received crizotinib, those with PD-L1 expression (score 2, 3) in tumor cells before crizotinib treatment tended to exhibit a shorter progression-free survival (PFS) and overall survival (OS) than those without PD-L1 expression (score 0, 1) (median PFS and OS: 20.7 and 52.9 mo, respectively, in the PD-L1-positive group vs. 28.1 and 96.0 mo, respectively in the PD-L1-negative group). Moreover, patients with strong (score 3) PD-L1 expression showed significantly shorter PFS and OS than those of the others (p = 0.001 and 0.002, respectively) (Fig. 7). Paired tumor tissues from patients before crizotinib treatment and after acquiring resistance to crizotinib were available in seven patients. In six patients, IHC revealed that PD-L1 expression was sustained or increased in pADC after the acquisition of crizotinib resistance compared with before crizotinib treatment (Fig. S2).
Figure 7.
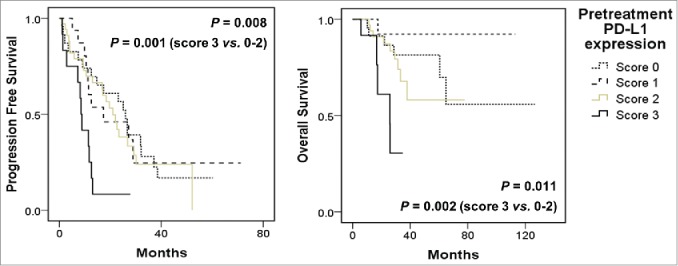
PD-L1 expression is associated with poor clinical outcome in patients with ALK-translocated pADC after ALK TKI therapy. Progression-free survival (left) and overall survival (right) after crizotinib treatment were plotted according to the PD-L1 IHC scores (score 0 [n = 25], 1 [n = 17], 2 [n = 36] or 3 [n = 12]) of tumor tissues from patients with ALK-translocated and advanced pADC before crizotinib treatment. Comparison of the survival times of all patients or those with PD-L1 IHC score 3 vs. others was performed using the Kaplan–Meier analysis method with the log-rank test.
Discussion
NSCLC is one of the leading cancers implicated in molecular and immune checkpoint-targeted therapy, thereby achieving significant improvements in the clinical outcome of patients.13,14,30 Among the immune checkpoints, the PD-1/PD-L1 pathway has emerged as a strong immune target for NSCLC. Thus, understanding the mechanism by which cancer cells regulate PD-L1 expression is invaluable for developing therapeutic strategies and predicting the clinical outcome of patients with cancer immunotherapy.15,16 Recently, several studies have demonstrated that oncogenic signals derived from mutation or loss of tumor suppressor genes upregulate the expression of immune checkpoint molecules in cancer cells during immune escape.11,17,22,31,32 In lung cancer, mutated EGFR has been reported to enhance PD-L1 expression, resulting in the suppression of T-cell function via activation of the PD-1/PD-L1 pathway.17 However, it is unclear whether other oncogenic drivers are involved in the regulation of PD-L1 expression in lung cancer cells. Our study demonstrated that ALK-driven pADCs in patients are characterized by high expression levels of PD-L1. Furthermore, EML4-ALK, an oncogenic driver, upregulated PD-L1 expression via HIF-1α and STAT3 in pADC cell lines. To the best of our knowledge, this study provides the first demonstration that EML4-ALK increases the levels of PD-L1 expression through the regulation of transcription factors in pADCs, although another type of ALK fusion protein, NPM-ALK, was demonstrated to induce PD-L1 expression in ALCL cells.22
Our experiments demonstrated that HIF-1α transcriptionally upregulated PD-L1 by binding to HREs within the PD-L1 promoter region in ALK-translocated lung cancer, similar to murine tumor cells, myeloid-derived suppressor cells, and human breast and prostate cancer cells.26,27 Moreover, it was demonstrated that EML4-ALK increased PD-L1 expression in pADC cell lines by regulating HIF-1α and STAT3 activities under hypoxia, but STAT3 alone under normoxia, suggesting that EML4-ALK might differentially regulate PD-L1 expression in pADCs, depending on the tumor microenvironment in terms of normoxia versus hypoxia. Given that solid tumors, including lung cancer, are more prone to exist under a hypoxic microenvironment, it is more likely that EML4-ALK-mediated enhancement of PD-L1 expression might occur under hypoxia rather than under normoxia in vivo. Consistently, it was reported that ALK activity upregulated HIF-1α and HIF-2α in ALK-translocated ALCL and NSCLC in vitro and in vivo, thereby mediating tumor growth and metastasis.28 Therefore, it is conceivable that HIF-1α might play a more critical role in the regulation of PD-L1 expression in pADCs under hypoxia.
Meanwhile, nuclear HIF-1α and pSTAT3 expression was significantly correlated with PD-L1 expression in tumor tissues from patients with ALK-translocated pADCs. Moreover, a significant positive correlation was also observed between nuclear HIF-1α and pSTAT3 (linear correlation coefficient = 0.412; p = 0.036) (data not shown). These results suggest that ALK-mediated STAT3 and HIF-1α pathways regulate the expression of PD-L1 cooperatively. Consistent with this suggestion, the current study exhibited that HIF-1α was directly bound to pSTAT3 in EML4-ALK-translocated pADC cell lines under hypoxia. Taken together, the data suggest that EML4-ALK increases HIF-1α expression, which upregulates PD-L1 expression by binding to the PD-L1 promoter and cooperating with STAT3 in pADC under hypoxia, whereas EML4-ALK increases PD-L1 expression via STAT3 activation under normoxia (Fig. 6C). However, it remains unclear whether this binding occurs at the PD-L1 promoter.
With respect to the mechanism for ALK-mediated activation of HIF-1α, ALK-mediated signals increased HIF-1α at the transcriptional level in NPM-ALK-translocated ALCL cells via STAT3 rather than via posttranslational HIF-1α protein stabilization.28,33 In the present study, EML4-ALK transfection in lung cancer cells highly increased the transcription of HIF-1α under hypoxia, but minimally under normoxia, suggesting that EML4-ALK might upregulate HIF-1α transcription under hypoxia, but very little under normoxia. Moreover, the ubiquitination of HIF-1α was attenuated in EML4-ALK-transfected pADC cell lines under both normoxia and hypoxia, and, consistently, direct interaction between HIF-1α and VHL proteins was also found to be decreased in these cells. HIF-1α degradation was consequently delayed in EML4-ALK-transfected cell lines compared with control cells. These data suggest that EML4-ALK might also be involved in HIF-1α ubiquitination and stabilization in lung cancer cells, a finding that was inconsistent with ALCL. Several studies have demonstrated that ALK strongly activates STAT3 in cancer cells. Moreover, constitutive activation of STAT3 interfered with the interactions between HIF-1α and VHL, thereby inhibiting HIF-1α ubiquitination.1,34 These findings suggest that ALK might be involved in HIF-1α unbiquitination in cancer cells via STAT3, a finding that is consistent with pADCs, but not with ALCL. Combined, our experiments suggest that EML4-ALK might enhance HIF-1α activity at both the transcriptional and posttranslational levels in lung cancer cells under hypoxia. Furthermore, the levels of phospho-ALK (pALK) expression were markedly increased by CoCl2 (Fig. 4B) and were suppressed by a HIF-1α inhibitor (Fig. 4F), indicating that HIF-1α might also be able to regulate ALK phosphorylation in pADC cell lines under hypoxia. ALK activity is increased by its phosphorylation.2 Therefore, it is likely that HIF-1α might increase ALK activity under hypoxia in lung cancer cells, a finding that was consistent with an observation in a previous study.28 Based on these findings, it is feasible that crosstalk between ALK and HIF-1α establishes a fine regulatory feedback network for PD-L1 expression in ALK-translocated pADC under hypoxia.(Fig. 6C).
We also investigated the expression of cytokines including IL-6, IL-10 and IFNγ in EML4-ALK pADC cells, because these cytokines are known to activated STAT3 or upregulate PD-L1.35,36 The expression of IL-6 and IL-10 was not significantly increase in EML4-ALK transfected H23 cells (Fig. S3). In contrast, the expression of IFNγ in EML4-ALK transfected H23 cells significantly increased upto 2.5 to 3 times compared to control cells. However, the Ct value of IFNγ was measured between 30 and 32, suggesting relatively low level of IFNγ expression in these cells (Fig. S3). Thus, it is reasonable that these cytokines might have little effect, if any, on the upregulation of PD-L1 expression in ALK-translocated pADC.
In our experiments, PD-L1 expression was positively correlated with the number of PD-1+ TILs in pADCs from patients, suggesting that PD-1/PD-L1 interaction in tumors may contribute to immune evasion in patients with ALK-translocated lung cancer. Consistent with this suggestion, our study demonstrated that strong PD-L1 expression was significantly associated with an unfavorable clinical outcome in terms of PFS and OS of patients. However, PD-L1 expression in tumors had no influence on the responsiveness to crizotinib in patients with ALK-translocated lung cancer, an observation that might be attributed to partial responses to crizotinib in most patients (89.4%) (data not shown). In contrast to ALK-translocated lung cancer, PD-L1 expression was associated with better clinical outcome in lung cancer patients treated with EGFR TKI.19 These observations suggest that PD-L1 expression might have different clinical implications in patient outcome according to the types of molecular target therapy, tumor histology, and genetic alterations. Meanwhile, melanoma cells resistant to a BRAF inhibitor upregulated PD-L1 expression via MAPK signaling,32 suggesting that molecular target therapy might affect PD-L1 expression in tumor cells. Consistently, PD-L1 expression was retained or increased in tumor tissues after acquiring resistance to crizotinib compared with tumor tissues before crizotinib treatment in six of seven patients (Fig. S2). There are several possible explanations for this PD-L1 expression pattern in these patients. First, ALK activity might be restored and/or persistent in tumors due to failed inhibition of ALK signaling in pADCs, thereby upregulating PD-L1 expression even after crizotinib treatment, a finding that is consistent with the increased expression levels of pALK in pADCs from patients after acquiring resistance to crizotinib treatment (Fig. S2). Second, PD-1/PD-L1 pathway-mediated immune escape in ALK-translocated pADCs might affect the clinical outcome for patients treated with crizotinib, partly accounting for the crizotinib resistance mechanism in patients with pADCs. Although explanations for this issue remain unclear, persistent expression of PD-L1 in crizotinib-resistant pADCs is intriguing in that alternative or combined therapies should be considered for these patients. Recently, it was demonstrated that PD-1/PD-L1-targeted therapy might have significant therapeutic efficacy in EGFR-mutated lung cancer even after acquiring resistance to EGFR TKI.17 Thus, it seems reasonable that PD-1/PD-L1-targeted immunotherapy may be an alternative therapeutic strategy for patients with acquired resistance to crizotinib besides second generation ALK inhibitors.
In conclusion, our study first demonstrates that ALK translocation upregulates PD-L1 expression in pADC via HIF-1α and STAT3, providing invaluable information for the development of therapeutic reagents and clinical implications in PD-1/PD-L1-targeted immunotherapy for patients with ALK-translocated pADCs.
Materials and methods
Patients and samples
A cohort of pADC patients (n = 532), including those harboring ALK translocation (n = 58, 11%), who underwent surgery without prior chemotherapy at Seoul National University Hospital (SNUH, Seoul, Republic of Korea) were evaluated. Tissue microarrays with 2-mm core diameters were constructed from formalin-fixed paraffin-embedded (FFPE) tumor tissues. Clinicopathological data were obtained from medical records. EGFR and KRAS mutation and ALK translocation status were evaluated as described previously.37,38
Another cohort of 214 patients who had recurrence or metastatic pADC with ALK translocation and were treated with the ALK TKI crizotinib at SNUH was collected. Among them, FFPE tissues before ALK TKI treatment were available in 90 patients for the evaluation of PD-L1 expression. In addition, to estimate PD-L1 expression in tumors from patients before crizotinib treatment and after acquiring resistance, we established a cohort of seven patients with paired biopsies. Crizotinib was administered orally at a dose of 250 mg twice daily as part of a clinical trial (A8081007 [NCT00932893], A8081014 [NCT01154140]), until tumor progression, death, significant uncontrolled toxicity or patient refusal. Chest computed tomography was performed per protocol to confirm patient response and assess disease progression. Treatment response was evaluated using RECIST 1.0.39 PFS was measured from the first day of inhibitor treatment until the first objective sign of disease progression or death. OS was measured from the date of diagnosis until death from any cause. This study followed the World Medical Association Declaration of Helsinki recommendations and was approved by the institutional review board of SNUH (H-1404-100-572).
Cell lines and reagents
NSCLC cell lines, H23 (EGFR wild-type [WT], ALK WT; ATCC® CRL-5800) and H2228 (EML4-ALK variant 3; ATCC® CRL-5935) cells, were purchased from the American Type Culture Collection. Cell lines were authenticated by morphology and growth characteristics, frozen and cultured. Cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin. Crizotinib (PF-02341066, an ALK inhibitor) was purchased from Selleckchem (catalog no. S1068). S3I-201 (a STAT3-specific inhibitor; catalog no. SML0330), CoCl2 (a chemical inducer of HIF-1α; catalog no. 232696), PX-12 (a HIF-1α-specific inhibitor; catalog no. M5324) and cycloheximide (a translation blocker; catalog no. 01810) were purchased from Sigma-Aldrich, and MG132 (a proteasome inhibitor) was purchased from Calbiochem (catalog no. 474790).
Transfection using an EML4-ALK construct and ALK short interfering RNA (siRNA)
The cells were plated in six-well plates (5 × 105/well) in opti-MEM media (Gibco, catalog no. 31985‐070) and were transfected with pcDNA, EML4-ALK v1, EML4-ALK v3, ALK specific siRNA or scramble (sc) siRNA using Lipofectamine 2000 (Invitrogen, catalog no. 11668-019). The transfected cells were cultured for 6 h, and the culture medium was then replaced with fresh complete medium. The EML4-ALK v1 and v3 constructs were kindly provided by Hiroyuki Mano (Jichi Medical University, Tochigi, Japan). ALK siRNA 1, 2 and 3 and sc siRNA were synthesized by Bioneer as reported previously (Table S1).22
Protein-DNA binding assay
Nuclear extracts were prepared from pcDNA-, EML4-ALK v1- or EML4-ALK v3-transfected H23 cells and subjected to a protein-DNA binding assay to detect the binding of STAT3 to the PD-L1 promoter in vitro. To this end, the EpiQuik™ General Protein-DNA Binding Assay Kit (Colorimetric) (Epigentek, catalog no. P-2004-96) was used. The DNA probe corresponding to the STAT3-binding sequence in the PD-L1 promoter was constructed using the following sequence: 5′-CGATTTCACCGAAGGTC -3′ (underline, putative pSTAT3 binding sequence).
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed using the ChIP-IT Express Enzymatic Kit (Active Motif, catalog no. 53009). Briefly, chromatin from cells was cross-linked with 1% formaldehyde, sheared to an average size of ˜500 bp and then immunoprecipitated using an anti-HIF-1α antibody (Santa Cruz Biotechnology, catalog no. sc-13515). Two putative binding sites of HIF-1α containing canonical hypoxia response elements (HRE; TACGTG) within the PD-L1 promoter region were identified by TFSEARCH and included HRE1 (located between exons 1 and 2) and HRE2 (located between exons 4 and 5). The ChIP-qPCR primers (Table S1) were designed to amplify the regions containing HRE1 and HRE2. Real-time quantitative PCR (qPCR) was performed using these primers and SYBR Green master mix (Takara Bio, catalog no. RR82LR), or the amplified product was visualized on agarose gels after electrophoresis.
Immunoprecipitation and ubiquitination assay
H23 cells were transfected with pcDNA, EML4-ALK v1 or EML4-ALK v3 for 24 h and incubated with CoCl2 (100 μM). Three hours after incubation, cells were lysed in 10 mM HEPES at pH 7.9, 10 mM KCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT buffer and 40 µg/mL Complete Protease Inhibitor mix (Roche, catalog no. 118735800001). Immunoprecipitates were prepared using anti-HIF-1α antibody, immobilized on protein A/G agarose beads (Santa Cruz Biotechnology, catalog no. sc-2003), and then subjected to Western blotting using anti-ubiquitin antibody (Santa Cruz Biotechnology, catalog no. sc-166553) or anti-VHL antibody (BD PharMingen, catalog no. 556347).
Quantitative reverse transcription PCR (qRT-PCR)
To evaluate the expression levels of PD-L1, ALK, HIF-1α, IL-6, IL-10 and IFNγ, total RNA was extracted from the cells using TRIzol (Life Technologies, catalog no. 15596-018), and subjected to reverse transcription using the PrimeScript™ 1st Strand cDNA Synthesis Kit (Takara Bio, catalog no. 6110A). qRT-PCR analysis was performed using the SYBR® qRT-PCR Kit (Clontech Laboratories, catalog no. 638313) and Step One Plus thermocycler (Applied Biosystems). GAPDH was used as the internal control. The primer sequences used for qRT-PCR are shown in Table S1. Each measurement was performed at least three times in triplicate. The relative expression level [2(-ddCt)] of each molecule was calculated as follows: dCt = Ct(molecule) − Ct(GAPDH); ddCt = dCt(experiment or target) − dCt(control or reference).
Western blotting
Total cellular protein was extracted using lysis buffer, and 50 μg protein were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The resolved proteins were transferred to polyvinylidene difluoride membranes (Millipore, catalog no. IPVH00010), which were then incubated with antibodies against PD-L1 (clone E1L3N, #13684), ALK (clone D5F3, #3633), pALK (Tyr1604, #3341), STAT3 (clone 124H6, #9139), pSTAT3 (Tyr705) (clone D3A7, #9145) (Cell Signaling), HIF-1α (BD Bioscience, catalog no. 610958), and β-actin (Santa Cruz Biotechnology, catalog no. sc-47778). The immunoblots were visualized using an enhanced chemiluminescence detection system (Amersham Pharmacia Biotech, catalog no. RPN 2109).
Flow cytometry
Surface staining was performed using FITC-conjugated mouse anti-human PD-L1 (clone MIH2) (LSBio, catalog no. LS-C188353) or FITC-conjugated mouse IgG (isotype controls) (LSBio, catalog no. LS-C188355), followed by flow cytometry analysis (Epics XL; Coulter).
Immunohistochemistry (IHC)
IHC was performed using rabbit anti-PD-L1 (E1L3N) XP® monoclonal antibody (mAb) (Cell Signaling, #13684), rabbit anti-pSTAT3 mAb (Tyr705; Cell Signaling, #9145), mouse anti-HIF-1α mAb (ESEE122; Novus Biologicals, catalog no. NB100-131) and anti-pALK antibody (Tyr1507; Abcam, catalog no. ab73996) and the Benchmark XT autostainer (Ventana Medical Systems). PD-L1 IHC was evaluated based on the intensity and proportion of membranous staining in tumor cells and was scored as follows: 0, negative; 1, weak or moderate in ≤5% of tumor cells; 2, moderate in ≥5% of tumor cells; 3, strong in ≥5% of tumor cells. Cases with scores of 2 or 3 were deemed positive for PD-L1 expression. The nuclear expression of pSTAT3 and HIF-1α and membranous and/or cytoplasmic pALK expression were evaluated based on the immunoreactivity (graded 0, 1, 2 and 3) and proportion, and the results (H score) are recorded by multiplying the percentage of positive cells by the intensity. CD8+ and PD-1+ TILs were immunostained and automatically enumerated as described previously.21
Statistical analysis
All statistical analyses were performed using SPSS software (version 21; IBM Corp.). Comparisons between variables were performed using the χ2 test or Student's t-test. Survival analysis was performed using the Kaplan–Meier method with the log-rank test. Two-sided p values less than 0.05 were considered statistically significant in all analyses.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Prof. Jong-Wan Park (Department of Pharmacology, Seoul National University College of Medicine, Seoul, Republic of Korea) for helpful discussions.
Funding
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant no.: HI14C0069).
Reference
- 1.Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer 2013; 13:685-700; PMID:24060861; http://dx.doi.org/ 10.1038/nrc3580 [DOI] [PubMed] [Google Scholar]
- 2.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H et al.. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007; 448:561-6; PMID:17625570; http://dx.doi.org/ 10.1038/nature05945 [DOI] [PubMed] [Google Scholar]
- 3.Lindeman NI, Cagle PT, Beasley MB, Chitale DA, Dacic S, Giaccone G, Jenkins RB, Kwiatkowski DJ, Saldivar JS, Squire J et al.. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. Arch Pathol Lab Med 2013; 137:828-60; PMID:23551194; http://dx.doi.org/ 10.5858/arpa.2012-0720-OA [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee JK, Park HS, Kim DW, Kulig K, Kim TM, Lee SH, Jeon YK, Chung DH, Heo DS, Kim WH et al.. Comparative analyses of overall survival in patients with anaplastic lymphoma kinase-positive and matched wild-type advanced nonsmall cell lung cancer. Cancer 2012; 118:3579-86; PMID:22086654; http://dx.doi.org/ 10.1002/cncr.26668 [DOI] [PubMed] [Google Scholar]
- 5.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Janne PA, Costa DB et al.. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010; 363:1693-703; PMID:20979469; http://dx.doi.org/ 10.1056/NEJMoa1006448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F et al.. Crizotinib vs. chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013; 368:2385-94; PMID:23724913; http://dx.doi.org/ 10.1056/NEJMoa1214886 [DOI] [PubMed] [Google Scholar]
- 7.Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T et al.. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014; 371:2167-77; PMID:25470694; http://dx.doi.org/ 10.1056/NEJMoa1408440 [DOI] [PubMed] [Google Scholar]
- 8.Kim S, Kim TM, Kim DW, Go H, Keam B, Lee SH, Ku JL, Chung DH, Heo DS. Heterogeneity of genetic changes associated with acquired crizotinib resistance in ALK-rearranged lung cancer. J Thorac Oncol 2013; 8:415-22; PMID:23344087; http://dx.doi.org/ 10.1097/JTO.0b013e318283dcc0 [DOI] [PubMed] [Google Scholar]
- 9.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252-64; PMID:22437870; http://dx.doi.org/ 10.1038/nrc3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol 2007; 19:813-24; PMID:17606980; http://dx.doi.org/ 10.1093/intimm/dxm057 [DOI] [PubMed] [Google Scholar]
- 11.Sanmamed MF, Chen L. Inducible expression of B7-H1 (PD-L1) and its selective role in tumor site immune modulation. Cancer J 2014; 20:256-61; PMID:25098285; http://dx.doi.org/ 10.1097/PPO.0000000000000061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A 2002; 99:12293-7; PMID:12218188; http://dx.doi.org/ 10.1073/pnas.192461099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K et al.. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N Eng J Med 2012; 366:2455-65; PMID:22658128; http://dx.doi.org/22658127 10.1056/NEJMoa1200694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al.. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443-54; PMID:22658127; http://dx.doi.org/ 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, Anders RA. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 2014; 20:5064-74; PMID:24714771; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN et al.. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014; 515:563-7; PMID:25428504; http://dx.doi.org/ 10.1038/nature14011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ et al.. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov 2013; 3:1355-63; PMID:24078774; http://dx.doi.org/ 10.1158/2159-8290.CD-13-0310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Azuma K, Ota K, Kawahara A, Hattori S, Iwama E, Harada T, Matsumoto K, Takayama K, Takamori S, Kage M et al.. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol 2014; 25:1935-40; PMID:25009014; http://dx.doi.org/ 10.1093/annonc/mdu242 [DOI] [PubMed] [Google Scholar]
- 19.D'Incecco A, Andreozzi M, Ludovini V, Rossi E, Capodanno A, Landi L, Tibaldi C, Minuti G, Salvini J, Coppi E et al.. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br J Cancer 2015; 112:95-102; PMID:25349974; http://dx.doi.org/ 10.1038/bjc.2014.555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang CY, Lin MW, Chang YL, Wu CT, Yang PC. Programmed cell death-ligand 1 expression in surgically resected stage I pulmonary adenocarcinoma and its correlation with driver mutations and clinical outcomes. Eur J Cancer 2014; 50:1361-9; PMID:24548766; http://dx.doi.org/ 10.1016/j.ejca.2014.01.018 [DOI] [PubMed] [Google Scholar]
- 21.Kim MY, Koh J, Kim S, Go H, Jeon YK, Chung DH. Clinicopathological analysis of PD-L1 and PD-L2 expression in pulmonary squamous cell carcinoma: Comparison with tumor-infiltrating T cells and the status of oncogenic drivers. Lung Cancer 2015; 88:24-33; PMID:25662388; http://dx.doi.org/ 10.1016/j.lungcan.2015.01.016 [DOI] [PubMed] [Google Scholar]
- 22.Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, Wang HY, Wysocka M, Cheng M, Ruggeri BA et al.. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A 2008; 105:20852-7; PMID:19088198; http://dx.doi.org/ 10.1073/pnas.0810958105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ait-Tahar K, Cerundolo V, Banham AH, Hatton C, Blanchard T, Kusec R, Becker M, Smith GL, Pulford K. B and CTL responses to the ALK protein in patients with ALK-positive ALCL. Int J Cancer 2006; 118:688-95; PMID:16114011; http://dx.doi.org/ 10.1002/ijc.21410 [DOI] [PubMed] [Google Scholar]
- 24.Passoni L, Gallo B, Biganzoli E, Stefanoni R, Massimino M, Di Nicola M, Gianni AM, Gambacorti-Passerini C. In vivo T-cell immune response against anaplastic lymphoma kinase in patients with anaplastic large cell lymphomas. Haematologica 2006; 91:48-55; PMID:16434370 [PubMed] [Google Scholar]
- 25.Ait-Tahar K, Damm-Welk C, Burkhardt B, Zimmermann M, Klapper W, Reiter A, Pulford K, Woessmann W. Correlation of the autoantibody response to the ALK oncoantigen in pediatric anaplastic lymphoma kinase-positive anaplastic large cell lymphoma with tumor dissemination and relapse risk. Blood 2010; 115:3314-9; PMID:20185586; http://dx.doi.org/ 10.1182/blood-2009-11-251892 [DOI] [PubMed] [Google Scholar]
- 26.Koh J, Go H, Keam B, Kim MY, Nam SJ, Kim TM, Lee SH, Min HS, Kim YT, Kim DW et al Clinicopathologic analysis of programmed cell death-1 and programmed cell death-ligand 1 and 2 expressions in pulmonary adenocarcinoma: comparison with histology and driver oncogenic alteration status. Mod Pathol 2015; 24:1154-66; PMID:26183759; http://dx.doi.org/24778419 10.1038/modpathol.2015.63 [DOI] [PubMed] [Google Scholar]
- 27.Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med 2014; 211:781-90; PMID:24778419; http://dx.doi.org/ 10.1084/jem.20131916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinengo C, Poggio T, Menotti M, Scalzo MS, Mastini C, Ambrogio C, Pellegrino E, Riera L, Piva R, Ribatti D et al.. ALK-dependent control of hypoxia-inducible factors mediates tumor growth and metastasis. Cancer Res 2014; 74:6094-106; PMID:25193384; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-0268 [DOI] [PubMed] [Google Scholar]
- 29.Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer 2012; 12:9-22; PMID:22169972; http://dx.doi.org/23972814 10.1038/nrc3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reck M, Heigener DF, Mok T, Soria JC, Rabe KF. Management of non-small-cell lung cancer: recent developments. Lancet 2013; 382:709-19; PMID:23972814; http://dx.doi.org/ 10.1016/S0140-6736(13)61502-0 [DOI] [PubMed] [Google Scholar]
- 31.Xu C, Fillmore CM, Koyama S, Wu H, Zhao Y, Chen Z, Herter-Sprie GS, Akbay EA, Tchaicha JH, Altabef A et al.. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell 2014; 25:590-604; PMID:24794706; http://dx.doi.org/ 10.1016/j.ccr.2014.03.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res 2013; 19:598-609; PMID:23095323; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-2731 [DOI] [PubMed] [Google Scholar]
- 33.Marzec M, Liu X, Wong W, Yang Y, Pasha T, Kantekure K, Zhang P, Woetmann A, Cheng M, Odum N et al.. Oncogenic kinase NPM/ALK induces expression of HIF1alpha mRNA. Oncogene 2011; 30:1372-8; PMID:21102525; http://dx.doi.org/ 10.1038/onc.2010.505 [DOI] [PubMed] [Google Scholar]
- 34.Jung JE, Kim HS, Lee CS, Shin YJ, Kim YN, Kang GH, Kim TY, Juhnn YS, Kim SJ, Park JW et al.. STAT3 inhibits the degradation of HIF-1alpha by pVHL-mediated ubiquitination. Exp Mol Med 2008; 40:479-85; PMID:18985005; http://dx.doi.org/ 10.3858/emm.2008.40.5.479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kasprzycka M, Marzec M, Liu X, Zhang Q, Wasik MA. Nucleophosmin/anaplastic lymphoma kinase (NPM/ALK) oncoprotein induces the T regulatory cell phenotype by activating STAT3. Proc Natl Acad Sci U S A 2006; 103:9964-9; PMID:16766651; http://dx.doi.org/ 10.1073/pnas.0603507103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abiko K, Matsumura N, Hamanishi J, Horikawa N, Murakami R, Yamaguchi K, Yoshioka Y, Baba T, Konishi I, Mandai M. IFN-gamma from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br J Cancer 2015; 112:1501-9; PMID:25867264; http://dx.doi.org/ 10.1038/bjc.2015.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim YT, Seong YW, Jung YJ, Jeon YK, Park IK, Kang CH, Kim JH. The presence of mutations in epidermal growth factor receptor gene is not a prognostic factor for long-term outcome after surgical resection of non-small-cell lung cancer. J Thorac Oncol 2013; 8:171-8; PMID:23287850; http://dx.doi.org/ 10.1097/JTO.0b013e318277a3bb [DOI] [PubMed] [Google Scholar]
- 38.Park HS, Lee JK, Kim DW, Kulig K, Kim TM, Lee SH, Jeon YK, Chung DH, Heo DS. Immunohistochemical screening for anaplastic lymphoma kinase (ALK) rearrangement in advanced non-small cell lung cancer patients. Lung Cancer 2012; 77:288-92; PMID:22465695; http://dx.doi.org/ 10.1016/j.lungcan.2012.03.004 [DOI] [PubMed] [Google Scholar]
- 39.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC et al.. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000; 92:205-16; PMID:10655437; http://dx.doi.org/ 10.1093/jnci/92.3.205 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.