

ABSTRACT
Missing self recognition makes cancer sensitive to natural killer cell (NKc) reactivity. However, this model disregards the NKc licensing effect, which highly increases NKc reactivity through interactions of inhibitory killer cell immunoglobulin-like receptors (iKIR) with their cognate HLA-I ligands. The influence of iKIR/HLA-ligand (HLA-C1/C2) licensing interactions on the susceptibility to and progression of plasma cell (PC) dyscrasias was evaluated in 164 Caucasian patients and 286 controls. Compared to controls, myeloma accumulates KIR2DL1−L2+L3− genotypes (2.8% vs. 13.2%, p < 0.01, OR = 5.29) and less diverse peripheral repertoires of NKc clones. Less diverse and weaker-affinity repertoires of iKIR2D/HLA-C licensing interactions increased myeloma susceptibility. Thus, the complete absence of conventional iKIR2D/HLA-C licensing interactions (KIR2DL1−L2+L3−/C2C2, 2.56% vs. 0.35%; p < 0.05; OR = 15.014), single-KIR2DL3+/C1+ (20.51% vs. 10.84%; p < 0.05; OR = 2.795) and single-KIR2DL2+/C1+ (12.82% vs. 4.9%; p < 0.01; OR = 5.18) interactions were over-represented in myeloma, compared to controls. Additionally, KIR2DL1−L2+L3− (20% vs. 83%, p < 0.00001) as well as KIR3DL1− (23% vs. 82%, p < 0.00001) genotypes had a dramatic negative impact on the 3-y progression-free survival (PFS), particularly in patients with low-tumor burden. Remarkably, myeloma-PCs, compared to K562 and other hematological cancers, showed substantial over-expression of HLA-I (“increasing-self” instead of missing-self), including HLA-C, and mild expression of ligands for NKc activating receptors (aRec) CD112, CD155, ULBP-1 and MICA/B, which apparently renders myeloma-PCs susceptible to lysis mainly by licensed NKc. KIR2DL1−L2+L3−/C2C2 patients (with no conventional iKIR2D/HLA-C licensing interactions) lyse K562 but barely lyse myeloma-PCs (4% vs. 15%; p < 0.05, compared to controls). These results support a model where immunosurveillance of no-missing-self cancers, e.g., myeloma, mainly depends on NKc licensing.
KEYWORDS: HLA-C, HLA-I, KIR, KIR ligands, missing self, myeloma, NK cell activating receptors, NK cell licensing
Abbreviations
- aKIR
activating KIR
- aRec
activating receptor
- BM
Bone marrow
- BMPC
bone marrow plasma cell
- CFSE
carboxyfluorescein diacetate succinimidyl ester
- FISH
fluorescent in situ hybridization
- HLA
human leukocyte antigen
- iKIR
inhibitory KIRs
- ISS
International scoring system
- KIR:
killer cell immunoglobulin-like receptors
- MGUS
monoclonal gammopathy of uncertain significance
- MM
multiple myeloma
- NKc
natural killer cells
- PB
peripheral blood
- PC
plasma cell
- PCD
plasma cell dyscrasia
- PFS
progression-free survival
- SCT
autologous stem cell transplantation
- SMM
smoldering myeloma
Introduction
Consistent with the “missing self” hypothesis, NK cells sensing the loss of self-HLA-class-I (HLA-I) molecules trigger cytolysis of “stressed” cells (e.g., tumor or virus-infected), while sparing healthy tissues.1 The missing self hypothesis, however, disregards the effect of NK cell licensing, which promotes terminal maturation and functional competence of NK cells after signaling through activating receptors (aRec).2,3 NK cell licensing is dependent on the avid engagement of inhibitory killer cell immunoglobulin-like receptors (KIR) on NK cells with their cognate HLA-I ligands on cell tissues.3,4 KIR receptors may transmit inhibitory (iKIRs) or activating (aKIRs) signals.5 Within the iKIRs, KIR2DL1 recognizes the HLA-C2 allotype (Lys80), and KIR2DL2/3 the HLA-C1 allotype (Asn80),6 although KIR2DL2 may also interact with the HLA-C2 allotype.7 KIR2DL4 binds HLA-G, KIR3DL1 interacts with the Bw4 epitope of some HLA-A and HLA-B molecules,8 and KIR3DL2 interacts with HLA-A3 and A11 alleles.9 Binding of aKIRs to HLA-C-ligands has only been documented for KIR2DS1 and HLA-C2,10 as well as for KIR2DS4 and HLA-C2 and a few C1 allotypes.11 The ligands for other relevant activating (KIR2DS3, KIR2DS5 and KIR3DS1) and inhibitory (KIR2DL5 and KIR3DL3) KIR receptors are currently uncertain. Certainly, the NK cell cytotoxic function depends on the signals delivered through interaction of aRec with their ligands. Concerning ligands for “natural cytotoxicity receptors,” NKp30 interacts with B7-H6,12 NKp44 with NKp44L,13 but the non-viral ligands for NKp46 remain unknown. With regard to other aRecs, NKG2D recognizes the stress-inducible MICA/B.14 and ULBPs proteins.15 whereas DNAM-1 (CD226) specifically recognizes poliovirus receptor (CD155) and Nectin-2 (CD112).16
Opposing signals from inhibitory and activating receptors interacting with their corresponding ligands showed to control NK cell functionality. Therefore, not only the diverse receptors expressed on the NK cell, but also the inhibitory and activating ligands expressed on the target cells will determine NK cell response, susceptibility to autoimmunity,17 infection,18 and cancer,19 and the outcome of allogeneic stem cell transplantation (SCT),20 or liver transplantation.21 Malignant cells usually over-express ligands for activating receptors,16,22 and reduce the expression of inhibitory ones (HLA-I).23,24 With respect to this, it has been described that myeloma cells express ligands for NK cell activating receptors,25,26 but only scarce and contradictory results concerning the expression of inhibitory ligands (HLA-I).27,26,28 are available.
Plasma cell dyscrasias (PCDs) are a group of diseases characterized by the proliferation of clonal PCs and the presence of monoclonal gammopathy. PCDs include benign forms such as monoclonal gammopathy of undetermined significance (MGUS), and malignant forms such as plasmacytoma, smoldering multiple myeloma (SMM), multiple myeloma (MM) and PC leukemia. Although there have been important advances in the last decade,29 malignant forms of PCD remain largely incurable and their incidence is increasing, accounting for approximately 1% of cancers and 13% of hematologic cancers in Western countries.30 The role of KIR/HLA interactions in the pathogenesis, prognosis and progression of PCDs has not been extensively studied. However, it has been shown that in patients with MM, interaction between KIR3DL1/S1 and HLA-Bw4 can predict PFS after autologous SCT.31 The present study investigates the influence of KIR/HLA-I interactions involved in NK cell licensing on the susceptibility to and the progression of PCDs. Additionally, explores whether the expression of KIR receptors in NK cells or that of their ligands in malignant PCs, could determine NK cell functionality and the outcome of the disease.
Results
Clinical and biological endpoints of patients
This study included 86 MGUS and 78 myelomas (consisting of 25 SMM and 53 MM). Nonetheless, following the new criteria of the myeloma working group, 20 out of the 25 SMM patients could be currently classified as MM.32 Table 1 shows the demographic and clinic-biological characteristics of patients. No significant differences in sex or age were found between patient groups. Most MGUS patients showed a low risk profile, 50.6% of patients had no adverse risk factors, 49.4% had one and no patient had two. In contrast, 60% of SMM patients had two adverse risk factors, 28% had one and only 12% had none. MM patients mostly showed a high-risk profile, with 33.9% showing ISS (international score system)-III, 43.4% ISS-II and only 22.6% ISS-I. Increasing values of serum monoclonal component (1.1 ± 0.1, 1.6 ± 0.2 and 2.4 ± 0.3 g/dL), frequency of Bence Jones protein (13.9%, 47.8% and 73.9%), immunoparesis (23.3%, 84% and 90.6%), histological bone marrow plasma cell (BMPC, 3.96% ± 0.3, 16.5% ± 2.6 and 33.9% ± 2.9) and ratio of aberrant/normal BMPCs (73.1% ± 2.3, 89.9% ± 4.01 and 96.0% ± 1.1) were found in MGUS, SMM and MM patients, respectively. Similarly, an increased incidence of numeric abnormalities in chromosomes 5, 9 and 15, break at IgH (14q32) and deletions of 17p13.1 (p53) and 13q14 was observed, with at least one of those alterations being present in 21.6%, 61.9% and 70.8% of MGUS, SMM and MM patients, respectively.(see Table 1 for details)
Table 1.
Demographic data and clinic-biological characteristics of patients at diagnosis.
| Demographic data and clinical characteristics | MGUS (n = 86 ) | SMM1 (n = 25 ) | p < 2 | MM (n = 53 ) | p < 3 | |
|---|---|---|---|---|---|---|
| Sex: Female/Male (% Females) | 42/44 (48,8%) | 17/8 (68.0%) | 27/26 (50.9%) | |||
| Age: years (Mean ± SEM ) | 67.0 ± 1.4 | 65.6 ± 2.9 | 68.4 ± 1.7 | |||
| Risk and ISS stratification (n) 4 | 42 / 41 / 0 | 3 / 7 / 15 | 12 / 23 / 18 | |||
| Monoclonal component | ||||||
| Monoclonal component, g/dL (Mean ± SEM ) | 1.1 ± 0.1 | 1.6 ± 0.2 | 0.05 | 2.4 ± 0.3 | 0.001* | |
| Bence Jones protein, no Yes/No (% Yes) | 10/62 (13.9%) | 11/12 (47.8%) | 0.001 | 34/12 (73.9%) | 0.001 | |
| Immunoparesis, no Yes/No (% Yes) | 20/66 (23.3%) | 21/4 (84%) | 0.001 | 48/5 (90.6%) | 0.001 | |
| Plasma cells (histology and immunophenotype) | ||||||
| Histology-BMPC, % (Mean ± SEM ) | 3.96 ± 0.3 | 16.5 ± 2.6 | 0.001 | 33.9 ± 2.9 | 0.001* | |
| Ratio aberrant/total-BMPC (Mean ± SEM ) | 73.1 ± 2.3 | 89.9 ± 4.1 | 0.001 | 96.0 ± 1.1 | 0.001 | |
| Fluorescent in situ hybridization (FISH) | ||||||
| p53 deletion, no Yes/No (% Yes) | 1/73 (1.3%) | 2/20 (9.1%) | 4/46 (8.0%) | |||
| 13q deletion, no Yes/No (%Yes) | 3/70 (4.1%) | 2/20 (9.1%) | 12/37 (24.5%) | 0.001 | ||
| Break of IgH, no Yes/No (%Yes) | 2/73 (2.7%) | 4/18 (18.2%) | 0.05 | 9/38 (19.1%) | 0.01 | |
| Aneuploidy, no Yes/No (% Yes) | 11/61 (15.3%) | 6/14 (30.0%) | 22/24 (57.8%) | 0.001 | ||
| Any FISH alteration, no Yes/No (%Yes) | 15/61 (21.6%) | 13/8 (61.9%) | 0.001 | 34/14 (70.8%) | 0.001 | |
Chr, Chromosome; SEM indicates standard error of mean;
Following new criteria of the Myeloma Working Group,32 20 out of 25 SMM should be included in the MM group.
Significant difference between MGUS and SMM (Pearson's χ2, Fisher's exact or Mann–Whitney tests).
Significant difference between MGUS and MM (Pearson's χ2, Fisher's exact or Mann–Whitney tests).
Risk factors (0/1/2 score) in MGUS and SMM, and international scoring system (ISS, I/II/III score) in MM. 33,34
Differences lower than p < 0.05 between SMM and MM.
Genotypes missing iKIR are associated with myeloma
The gene frequency of KIR receptors is shown in Table 2. KIR2DL1 gene was present in 97.2% of controls and 96.5% of MGUS, but only in 92.0% of SMM, 86.8% of MM (p < 0.01; Pc < 0.05; OR = 0.19) and 82.9% of MM with ISS > I (p < 0.001; Pc < 0.01; OR = 0.14). KIR2DL1 was also underrepresented in MM (p < 0.05; Pc > 0.05; OR = 0.24) and in MM with ISS > I (p < 0.01; Pc < 0.05; OR = 0.18) compared to MGUS. KIR2DL3 gene was present in 87.8% of controls and in 84.0% of SMM, but only in 75.5% of MM (p < 0.05; Pc > 0.05; OR = 0.41) and in 68.3% of MM with ISS > I (p < 0.01; Pc < 0.05; OR = 0.29). The frequency of KIR2DL3 was also lower in MM with ISS >I (p < 0.05; Pc < 0.05; OR = 0.35) than in MGUS (86.0%). KIR2DL2 was underrepresented in MGUS (55.8%), SMM (44.0%) and MM (54.7%) compared to controls (62.9%), but these differences were not statistically significant. Remarkably, dissociated expression of KIR2DL2 and KIR2DS2 (KIR2DL2 −S2 + genotype) was detected in two patients from the MM group with ISS >I (Table 2), but not in any MGUS or SMM patient or control. Additionally, similar lower frequencies of KIR3DL1 and KIR2DS4 were observed in MM (88.7%, p < 0.05; Pc < 0.05; OR = 0.31) and in MM with ISS >I (87.8%, p < 0.05; Pc < 0.05; OR = 0.29) than in controls (98.8%). Multivariate logistic regression analysis confirmed that both KIR2DL1 (p < 0.01; OR = 0.185) and KIR2DL2 (p < 0.05; OR = 0.557) were independent protective factors for myeloma (Table 3) .
Table 2.
KIR and KIR ligands in controls and patients with plasma cell dyscrasia.
| Controls (n = 286 ) | MGUS (n = 86 ) | SMM (n = 25 ) | MM (n = 53 ) | MM with ISS>I (n = 41 ) | |
|---|---|---|---|---|---|
| KIR gene frequency1 | n (%) | n (%) | n (%) | n (%) | n (%) |
| 2DL1 | 278 (97.2%) | 83 (96.5%) | 23 (92.0%) | 46 (86.8%)2 | 34 (82.9%)2 |
| 2DL2 | 180 (62.9%) | 48 (55.8%) | 11 (44.0%) | 29 (54.7%) | 25 (61.0%) |
| 2DL3 | 251 (87.8%) | 74 (86.0%) | 21 (84.0%) | 40 (75.5%)3 | 28 (68.3%)3 |
| 2DL5 | 146 (51.0%) | 51 (59.3%) | 11 (44.0%) | 28 (52.8%) | 23 (56.1%) |
| 3DL1 | 274 (98.8%) | 82 (95.3%) | 24 (96.0%) | 47 (88.7%)4 | 36 (87.8%)4 |
| 2DS1 | 104 (36.4%) | 37 (43.5%) | 7 (28.0%) | 20 (37.7%) | 16 (39.0%) |
| 2DS2 | 179 (62.6%) | 48 (55.8%) | 11 (44.0%) | 31 (58.5%)5 | 27 (65.9%)5 |
| 2DS3 | 94 (33.0%) | 32 (37.2%) | 8 (32.0%) | 16 (30.2%) | 14 (34.1%) |
| 2DS4 | 274 (98.8%) | 82 (95.3%) | 24 (96.0%) | 47 (88.7%)4 | 36 (87.8%)4 |
| 2DS5 | 76 (26.6%) | 31 (36.5%) | 5 (20.0%) | 16 (30.2%) | 13 (31.7%) |
| 3DS1 | 111 (38.8%) | 37 (43.0%) | 7 (28.0%) | 22 (41.5%) | 17 (41.5%) |
| iKIR2D genotypes | |||||
| 2DL1+L2+L3+ | 142 (49.7%) | 36 (41.9%) | 7 (28.0%)6 | 16 (30.2%)6 | 12 (29.3%)6 |
| 2DL1+L2+L3− | 26 (9.1%) | 9 (10.5%) | 2 (8.0%) | 6 (11.3%) | 6 (14.6%) |
| 2DL1+L2−L3+ | 110 (38.5%) | 38 (44.2%) | 14 (56.0%) | 24 (45.3%) | 16 (39.0%) |
| 2DL1−L2+L3− | 8 (2.8%) | 3 (3.5%) | 2 (8.0%) | 7 (13.2%)7 | 7 (17.1%)7 |
| KIR genotype | |||||
| AA | 76 (26.3%) | 16 (18.6%) | 9 (36.0%) | 13 (24.5%) | 8 (19.5%) |
| Bx | 211 (73.8%) | 70 (81.4%) | 16 (64.0%) | 40 (75.5%) | 33 (80.5%) |
| KIR ligands | |||||
| C1C1 | 95 (33.3%) | 25 (29.1%) | 8 (32.0%) | 21 (39.6%) | 15 (33.6%) |
| C1C2 | 136 (47.6%) | 40 (46.5%) | 14 (56.0%) | 24 (45.2%) | 22 (56.3%) |
| C2C2 | 55 (19.2%) | 21 (24.4%) | 3 (12.0%) | 8 (15.1%) | 4 (9.75%) |
| Bw4 | 230 (80.4%) | 62 (79.5%) | 20 (80.0%) | 42 (79.2%) | 32 (78.0%) |
| A3/A11 | 87 (32.4%) | 25 (32.1%) | 5 (20.0%) | 16 (30.2%) | 13 (31.7%) |
Framework genes are not considered (3DL2, 3DL3, 2DL4, 3DP1).
Control vs. MM (p < 0.01; Pc < 0.05; OR = 0.19); Control vs. MM with ISS > I (p < 0.001; Pc < 0.01; OR = 0.14); MGUS vs. MM (p < 0.05; Pc > 0.05; OR = 0.24) and MGUS vs. MM with ISS >I (p < 0.01; Pc > 0.05; OR = 0.18).
Control vs. MM (p < 0.05; Pc >0.05; OR = 0.41); Control vs. MM with (ISS >I p< 0.01; Pc< 0.05; OR = 0.29); MGUS vs. MM with ISS >I (p < 0.05; Pc >0.05; OR = 0.35).
Control vs. MM (p < 0.05; Pc >0.05; OR = 0.31) and Control vs. MM with ISS >I (p < 0.05; Pc >0.05; OR = 0.29).
KIR2DL2−S2+ dissociated genotype was found in two MM with ISS > I.
Control vs. SMM (p < 0.05; Pc >0.05; OR = 0.39); Control vs. MM (p < 0.01; Pc < 0.05; OR = 0.44) and Control vs. MM ISS > I (p < 0.05; Pc > 0.05; OR = 0.42).
Control vs. MM (p < 0.01; Pc < 0.05; OR = 5.23); Control vs. MM with ISS > I (p < 0.001; Pc < 0.01; OR = 7.15); MGUS vs. MM (p < 0.05; Pc > 0.05; OR = 4.2) and MGUS vs. MM with ISS>I (p < 0.05; Pc >0.05; OR = 5.88).
Table 3.
Multivariate logistic regression analysis comparing controls and myelomas (SMM+MM).
| Dependent variable | Co-variables1 | OR | 95%CI | p |
|---|---|---|---|---|
| iKIR genes | Sex | 1.138 | 0.672–1.926 | 0.630 |
| Age | 1.015 | 0.991–1.039 | 0.214 | |
| KIR2DL1 | 0.185 | 0.065–0.527 | 0.002 | |
| KIR2DL2 | 0.557 | 0.325–0.955 | 0.033 | |
| KIR3DL1/2DS4 | 0.470 | 0.166–1.333 | 0.156 | |
| iKIR2D genotype | KIR2DL1−L2+L3− | 6.743 | 2.348–19.359 | 0.000 |
| KIR2DL1+L2−L3+ | 2.055 | 1.152–3.668 | 0.015 | |
| KIR2DL1+L2+L3− | 1.862 | 0.750–4.623 | 0.180 | |
| KIR2DL1+L2+L3+2 | 1.0 | N.A.2 | 0.002 |
OR, odd ratio; 95%CI, 95% confidence interval.
Sex and age were included in all analyses and no significant results were found in all cases.
Reference category. N.A.: no applicable.
Consequently, KIR2DL1+L2+L3+ genotype was found to be underrepresented in SMM (28.0%; p < 0.05; Pc < 0.05; OR = 0.39) and MM (30.2%; p < 0.01; Pc < 0.05; OR = 0.44) compared to controls (49.7%), whereas the KIR2DL1 −L2+L3 − genotype was significantly associated to MM (13.2%; p < 0.01; Pc < 0.05; OR = 5.23) and MM with ISS >I (17.1%; p < 0.001; Pc < 0.01; OR = 7.15) compared to controls (2.8%) (Table 2). Multivariate logistic regression analysis confirmed that KIR2DL1−L2+L3− (p < 0.0001; OR = 6.74) and KIR2DL1+L2−L3+ (p < 0.05; OR = 2.055) genotypes compared to KIR2DL1+L2+L3+, were independently associated with the emergence of myeloma (Table 3). Moreover, compared to controls frequency of KIR3DL1− genotype was increased within KIR2DL1+L2+L3− myeloma patients (0.0% vs. 25.0%, p < 0.05; Pc < 0.05) and frequency of KIR2DL3− genotype was increased within KIR3DL1− myeloma patients (9.1% vs. 57.1%, p < 0.05; Pc < 0.05). Our data suggest that not only double negative KIR2DL1−L3−, but also KIR2DL3−3DL1− genotype might be involved in the susceptibility to myeloma, but since the significant association of KIR2DL3−3DL1− genotype with myeloma susceptibility was lost after Bonferroni correction, this association should be contrasted in larger series.
Finally, no differences in the frequency of KIR genotypes (AA or Bx), KIR ligands (HLA-C1C1, -C1C2 or -C2C2, Bw4-epitope and HLA-A3/A11 alleles) or HLA-A, HLA-B or HLA-C alleles (data not shown) were observed between patients and controls (Table 2).
Repertoires of NK cells expressing lower diversity of KIR2D receptors in myeloma patients
There were no differences in the absolute number of total NK cells or NK cells expressing KIR2D receptors (KIR2D+) or not (KIR2D−) in peripheral blood (PB) between MGUS, Myeloma or control groups. However, patients with the KIR2DL1−L2+L3− genotype replenished the pool of KIR2D+ circulating NK cells exclusively with KIR2DL2/S2+ clones, while patients with the KIR2DL1+L2+L3+ genotype exhibited a much more diverse repertoire of clones showing all possible KIR2DL combinations (Fig. 1) .
Figure 1.
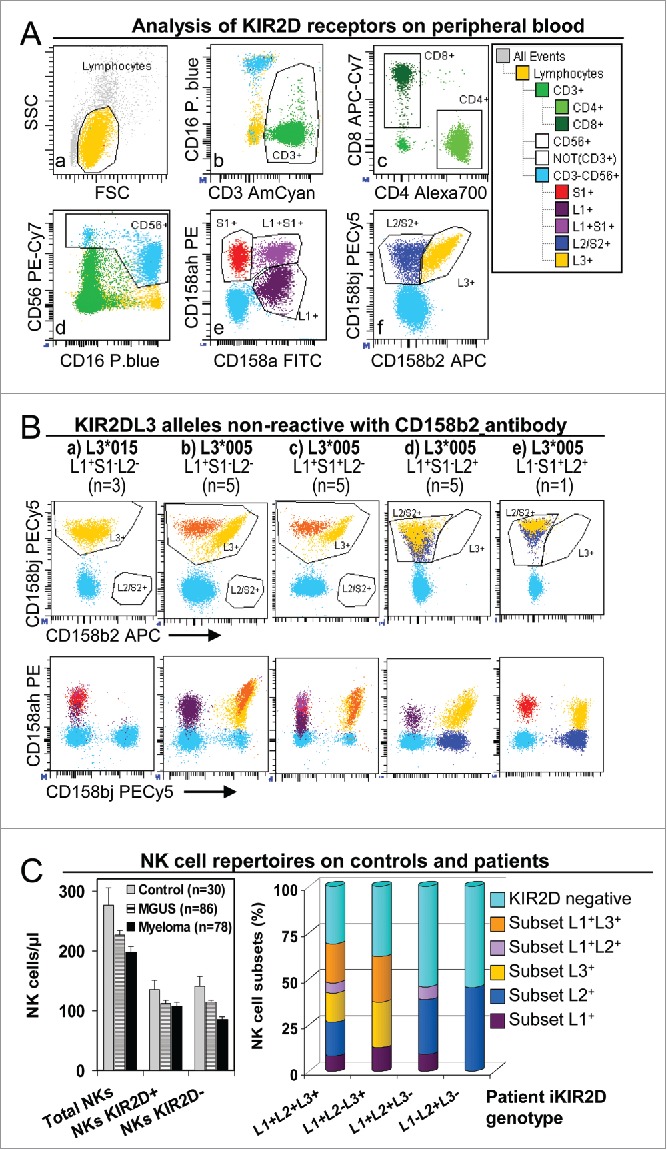
NK cell repertoires in peripheral blood of controls and patients. (A) Analysis of KIR2D expression in peripheral blood NK and T cells. Gates to identify total lymphocyte (a), as well as CD3+ (b), CD4+ and CD8+ (c), CD16+CD56+ (d), KIR2DS1+, L1+ and L1+S1+ (e), and KIR2DL2/S2+ and L3+ (f) subsets were set in appropriate dotplots.35 These gates were logically combined to assigned KIR2D expression to CD3+CD4+, CD3+CD8+ and CD3−CD16+CD56+ (NK) lymphocytes in a representative patient with the KIR2DL1+S1+L2/S2+L3+ genotype. (B) Special analyses were implemented for patients with KIR2DL3 alleles that were non-reactive with anti-KIR2DL3 antibody clone 180701 (presumably KIR2DL3*005 or *015 alleles).36 As described, KIR2DL3*005 was cross-reactive with anti-KIR2DL1/S1 antibody clone EB6 but not with KIR2DL1 antibody clone 143211. In these patients, the possibility of three genetic backgrounds required cytometric analysis to be adapted: (1) for KIR2DL2− individuals (a, b and c; n = 13) all cells stained with CD158b/j (clone GL183) were assigned as L3+ (yellow and orange), disregarding alleged-alleles *005 and/or *015 were homozygous (a, n = 3 ), or heterozygous with others KIR2DL3 alleles (orange) which are reactive with the CD158b antibody (b, n = 5 and c, n = 5); (2) for KIR2DL2+ individuals (d and e; n = 6) cells co-reacting with anti-CD158b/j and CD158a/h antibodies were assigned as L3+ (yellow), and cells that were non-reactive with CD158a/h antibody as L2+ (dark blue); (3) for KIR2DS1+/KIR2DL3*005 individuals (c and e; n = 6 ), S1+ cells were considered only as those that reacted with CD158a/h but not with CD158a nor CD158bj antibodies (violet and red, in c and e, respectively). (C) NK cell repertoires in peripheral blood of controls and patients. Left, no differences in the number (cells/μL) of total NK cells (CD3−CD16+CD56+) or NK cells expressing KIR2D receptors (KIR2D+) or not (KIR2D−) between controls and MGUS or myeloma (SMM+MM) patients were detected by using CD158a/h+CD158bj antibodies. Right, distribution of peripheral blood NK cell subsets (percent of total NK cells) expressing KIR2DL1, and/or KIR2DL2/S2 and/or KIR2DL3, in total patients (MGUS+SMM+MM) with different KIR2D genotypes. KIR2DL1−L2+L3− showed only KIR2DL− or KIR2DL2+ clones.
iKIR2D/HLA-C licensing interactions with lower diversity and weaker affinity are associated with myeloma
The frequency of iKIR2D/HLA-C licensing interactions in controls and patients was analyzed with regard to the previously described (conventional) KIR-ligand affinity hierarchy: KIR2DL1/C2 > KIR2DL2/C1 > KIR2DL3/C1 (Table 4).37 Global significant differences in the repertoire of iKIR2D/HLA-C licensing interactions were found between controls and myeloma patients (p < 0.01, Fisher test; Pc < 0.05), but not between controls and MGUS patients.
Table 4.
Diversity of iKIR2D/HLA-C licensing interactions in controls and myeloma patients.
| Frequency, n (%) |
Multivariate logistic regression |
||||
|---|---|---|---|---|---|
| Combinations of conventional iKIR2D/HLA-C licensing interactions1 | Controls (n=286 ) | SMM+MM (n=78 ) | OR | Analysis 95%CI | p |
| No iKIR2D/HLA-C interactions2 | 1 (0.35%) | 2 (2.56%) | 15.014 | 1.171–195.55 | 0.037 |
| Single-KIR2DL3+/C1+ | 31 (10.84%) | 16 (20.51%) | 2.795 | 1.183–6.603 | 0.019 |
| Single-KIR2DL2+/C1+ | 14 (4.9%) | 10 (12.82%) | 5.182 | 1.795–14.960 | 0.002 |
| Single-KIR2DL1+/C2+ | 53 (18.5%) | 9 (11.54%) | 1.089 | 0.426–2.728 | 0.858 |
| Double-KIR2DL2+L3+/C1+ | 51 (17.83%) | 6 (7.69%) | 0.727 | 0.255–2.068 | 0.550 |
| Double-KIR2DL1+L3+/C2+C1+ | 53 (18.53%) | 18 (23.08%) | 1.936 | 0.855–4.382 | 0.113 |
| Double-KIR2DL1+L2+/C2+C1+ | 13 (4.56%) | 5 (6.41%) | 2.335 | 0.703–7.758 | 0.166 |
| Triple-KIR2DL1+L2+L3+/C2+C1+3 | 70 (24.48%) | 12 (15.38%) | 1.0 | N.A. | 0.004 |
OR, odd ratio; 95%CI, 95% confidence interval.
Ordered following previously described (conventional) KIR2D-ligand affinity hierarchy: KIR2DL3/C1 < KIR2DL2/C1 < KIR2DL1/C2.37
Patients with no conventional iKIR2D/HLA-C licensing interactions (but with the non-conventional KIR2DL2/C2 interaction).
Reference category. N.A.: no applicable.
The multivariate logistic regression analysis showed that compared to the combination with the most diverse and highest affinity iKIR2D/HLA-C licensing interactions (reference category, triple-KIR2DL1+L2+L3+/C1+C2+; 15.38% vs. 24.48% in patients and controls, respectively), less diverse and weaker-affinity iKIR2D/HLA-C licensing interactions independently contributed to increase myeloma susceptibility. Thus, the complete absence of conventional iKIR2D/HLA-C licensing interactions (but with the non-conventional KIR2DL2/C2 one) was detected only in 1 out of 286 controls (0.35%), but in 1 out of 86 MGUS (1.16%) and in 2 out of 78 myeloma patients (2.56%; p < 0.05; OR = 15.014; controls vs. Myeloma patients). This unusual KIR2D/HLA-C combination always occurred in KIR2DL1-negative, C2-homozygous individuals. Additionally, the following weakest iKIR2D/HLA-C licensing interactions, single-KIR2DL3+/C1+ (20.51% in myeloma vs. 10.84% in controls; p < 0.05; OR = 2.795) and single-KIR2DL2+/C1+ (12.82% in myeloma vs. 4.9% in controls; p < 0.01; OR = 5.18) also contributed independently to myeloma susceptibility. Combinations of iKIR2D/HLA-C licensing interactions with higher affinity in myeloma compared controls, such as single-KIR2DL1+/C2+ (11.54% vs. 18.5%), double-KIR2DL2+L3+/C1+ (7.69% vs. 17.83%), double-KIR2DL1+L3+/C2+C1+ (23.08% vs. 18.53%), double-KIR2DL1+L2+/C2+C1+ (6.41% vs. 4.56%) did not show significant differences.(Table 4)
KIR2DL1−L2+L3− and KIR3DL1− genotypes negatively impacted in the clinic-biological variables and patient outcome
We explored the impact of different iKIR2D genotypes on the clinic-biological variables and patient outcome (Table 5 and Fig. 4). Although no significant differences were found in the mean age of patients with different KIR2DL genotypes, significantly higher frequency of patients under 47 (including 2 patients at 35 and 37) was found within KIR2DL1−L2+L3− genotype compared with the remaining genotypes (25% vs. 2%, p < 0.05; Pc < 0.05; OR= 5.99). Remarkably, the two younger patients had the KIR2DL1−L2+L3− genotype in the HLA-C2/C2 background, and consequently lacked any conventional iKIR2D/HLA-C licensing interactions. Additionally, patients with the KIR2DL1−L2+L3− genotype showed higher numbers of histology-BMPC (30.3% vs. 14.5%; p < 0.01; Pc < 0.05), monoclonal component (2.9 vs. 1.75 g/dL; p < 0.05), ratio of aberrant/total-BMPC (94.3% vs. 81.9%; p < 0.05; Pc < 0.05) and FISH alterations (90.0% vs. 39.1%; p < 0.001; Pc < 0.005; OR, 22.27), compared to the remaining patients. Moreover, patients with KIR2DL1−L2+L3− genotype showed a higher number of histology-BMPCs (30.3% vs. 11.3%; p < 0.01; Pc < 0.05) and ratio of aberrant/total-BMPC (94.3% vs. 81.2%; p < 0.05; Pc < 0.05) than patients with KIR2DL1+L2+L3+ genotype.
Table 5.
Impact of iKIR2D genotype on clinic-biological variables and the outcome of total patients (MGUS+SMM+MM).
| |
2DL1+L2+L3+ (n=59 ) |
2DL1+L2−L3+ (n=76 ) |
2DL1+L2+L3− (n=18 ) |
2DL1−L2+L3− (n=12 ) |
|---|---|---|---|---|
| Age | ||||
| Years (Mean ± SEM ) | 65.4 ± 1.5 | 67.6 ± 1.5 | 64.7 ± 2.4 | 65.3 ± 5.24 |
| ≤47 years / >47 y (%) | 4/55 (6.8%) | 3/73 (3.9) | 1/16 (5.9%) | 3/9 (25.0%)1 |
| Plasma cell biology | ||||
| Histology-BMPC, % (Mean ± SEM ) | 11.3 ± 1.9 | 15.8 ± 2.2 | 19.9 ± 5.9 | 30.0 ± 7.2* |
| Monoclonal comp., g/dL (Mean ± SEM ) | 1.69 ± 0.18 | 1.77 ± 0.21 | 1.9 ± 0.5 | 2.9 ± 1.0 |
| Ratio aberrant/total-BMPC (Mean ± SEM ) | 81.2 ± 2.6 | 81.9 ± 2.5 | 87.8 ± 5.4 | 94.3 ± 2.4** |
| FISH alteration, no Yes/No (%)2 | 10/48 (42.6%) | 24/41 (36.9%) | 6/10 (37.5%) | 9/1 (90.0%)3 |
2DL1−L2+L3− vs. the other groups (p < 0.05; Pc > 0.05; OR = 5.99).
2DL1−L2+L3− vs. the other groups (p < 0.01; Pc < 0.05); 2DL1−L2+L3− vs. 2DL1+L2+L3+ (p < 0.01; Pc < 0.05).
2DL1−L2+L3− vs. the other groups (p < 0.05; Pc > 0.05); 2DL1−L2+L3− vs. 2DL1+L2+L3+ (p < 0.05; Pc > 0.05).
Any alteration of p53, 13q deletions, break of IgH or aneuploidy of Chr-5, -9 or -15.
2DL1−L2+L3− vs. the other groups (p < 0.001; Pc < 0.005; OR = 22.27).
Figure 4.
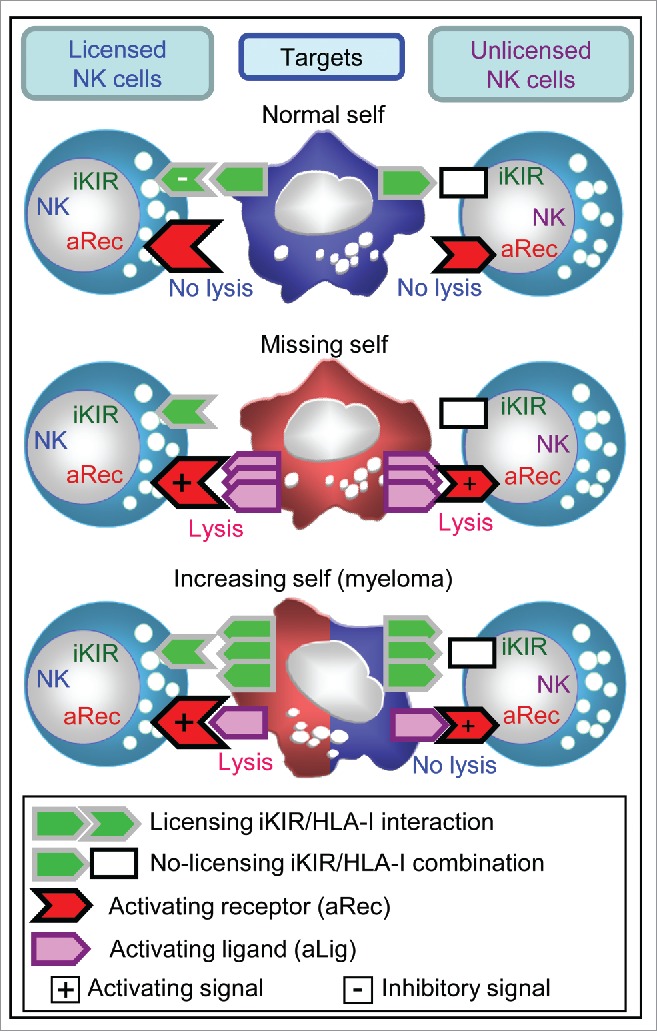
Proposed model to explain cytolytic activity of licensed and unlicensed NK cells against normal-self (top), missing-self (medium) and increasing-self (bottom) targets. Our hypothesis sustains that licensed NK cells (left) responding through activating receptors (aRec) can kill missing-self and increasing-self tumors expressing ligands for aRec (aLig), but not normal cells. By contrast, unlicensed NK cells (right) would kill missing-self tumors,41 but not normal tissue or increasing-self tumors with a mild expression of aLig such as myeloma.
Importantly, not only KIR2DL1− (33.3% vs. 77.8%, p < 0.01; Pc < 0.05) but also KIR3DL1− (35.5% vs. 75.5%, p < 0.001; Pc < 0.01) genotypes showed significantly lower three-year PFS than the remaining genotypes (Fig. 2). Interestingly, this difference in the PFS was more marked in patients with low-tumor burden (aberrant BMPCs < 95%, which was equivalent to 6.4 ± 0.8% of PCs in the bone marrow (BM)) both for KIR2DL1− (20% vs. 83%, p < 0.00001; Pc < 0.0001) and KIR3DL1− (23% vs. 82%, p < 0.00001; Pc < 0.0001), compared with the remaining genotypes. In contrast, in patients with high-tumor burden (aberrant BMPCs > 95%, which was equivalent to 29.3% ± 2.7 of PCs in the BM) KIR2DL1− (50% vs. 55%, p < 0.05) and KIR3DL1− (33.6% vs. 56.7%, p < 0.05), showed no significant differences in the PFS, compared with the rest of genotypes. Taken in consideration that only 3 out of 86 MGUS patients (3.5%) progressed on their diseases in the 3-y follow-up (data not shown), it is noteworthy that the only MGUS patient showing no conventional iKIR2D/HLA-C licensing interactions (KIR2DL1−L2+L3−/C2C2), a 37 y old woman, progressed to MM during the third follow-up year.
Figure 2.
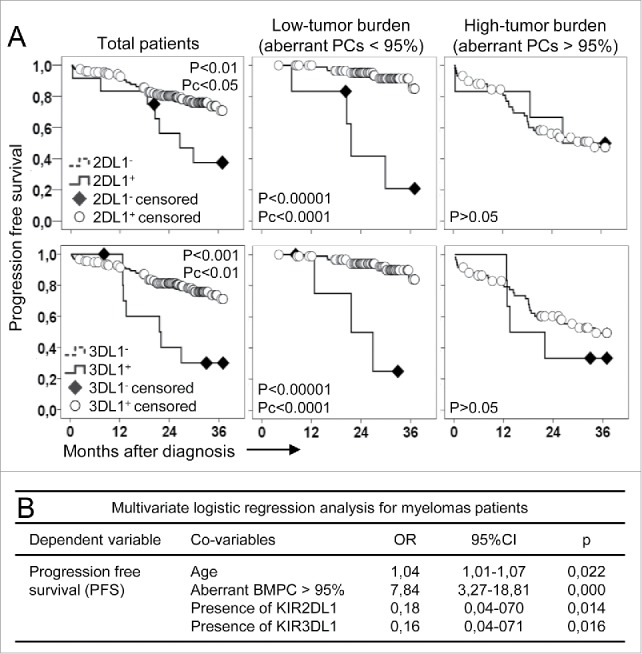
The impact of KIR genotype on myeloma progression free survival depends on tumor burden. (A) Patients with KIR2DL1− (KIR2DL1−L2+L3−, p < 0.01; Pc p < 0.05; top, left graphs) or KIR3DL1− (p < 0.001; Pc < 0.01; bottom, left graphs) genotypes showed significant lower progression free survival than the remaining patients. These differences were more intense and significant (p < 0.00001; Pc p < 0.0001 for both genotypes, middle graphs) in patients with low-tumor burden (aberrant plasma cells -PCs- <95%, equivalent to 6.4 ± 0.8% of PCs in the bone marrow). No differences in the progression free survival were observed in patients with high-tumor burden (aberrant PCs > 95%, equivalent to 29.3% ± 2.7% of PCs in the bone marrow; right graphs). (B) Multivariate logistic regression analysis confirmed that age (OR, 1.04; p = 0.022) and high-tumor burden (OR, 7.84; p = 0.000) significantly and independently increased the risk of myeloma progression. However, presence of any KIR2DL1 (OR, 0.178; p = 0.014) or KIR3DL1 (OR, 0.164; p = 0.016) significantly and independently protected from disease progression.
Increased expression of total-HLA-I and HLA-C (“Increasing self”) and mild expression of aRec-ligands in BMPCs determines NK cell cytolytic function
In order to explain why iKIR/HLA-I licensing interactions might provide protection against myeloma, the expression of both iKIR-ligands (total-HLA-I and HLA-C) and aRec-ligands (CD112, CD155, ULBP-1 and MICA/B) were analyzed on normal-BMPCs in 10 non-dyscrasia donors and on aberrant-BMPCs in 15 consecutive MM patients at diagnosis as well as on their normal-residual lymphocytes (Fig. 3). Remarkably, as opposed to K562 or other hematological tumors (ALL-T or AML) that reduced (“missing-self”) or preserved (“normal-self”) the expression of total-HLA-I and HLA-C, aberrant-BMPCs showed increased expression of total-HLA-I and HLA-C (“increasing-self”) compared to normal-BMPCs and lymphocytes (p < 0.01). Indeed, not a single patient showing a reduction of total-HLA-I or HLA-C expression was found. Besides, compared to normal-BMPCs and lymphocytes, aberrant BMPCs showed increased expression of aRec-ligands CD112 and CD155 (p < 0.01, any case) and conserved expression of ULBP-1 and MICA/B, but much lower expression of aRec-ligands than K562.
Figure 3.
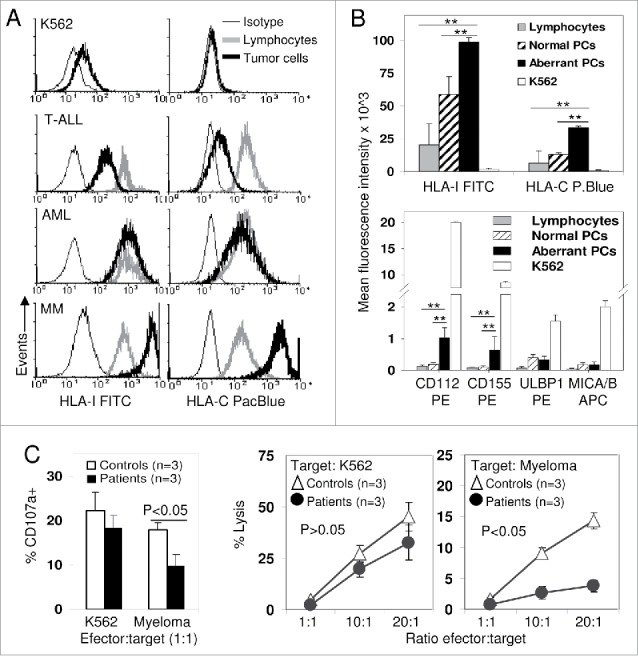
Increased expression of HLA-I (“Increasing self”) determines that myeloma plasma cells lysis mainly depends on NK cell licensing. (A) Expression of total-HLA-I (W6/32 antibody, left column) and HLA-C (DT9 antibody, right column) was evaluated in K562 cell line and in residual normal bone marrow lymphocytes (gray line) and tumor cells (bold black line) from T acute lymphoblastic leukemia (T-ALL), acute myeloblastic leukemia (AML) and MM patients. Cells were also stained with isotype control (thin line). K562 and T-ALL blasts completely or partially lost expression of total-HLA-I and HLA-C; AML blasts conserved normal expression of both molecules, but myeloma PCs showed an increased expression of both total-HLA-I and HLA-C compared to normal lymphocytes. (B) Plasma cells from myeloma patients (black bars, n = 15) showed higher expression (Mean fluorescence intensity) of total-HLA-I and HLA-C as well as ligands for activating receptors CD112 and CD155 than PCs from normal donors (striped bars, n = 10) and normal residual lymphocytes (gray bars, n = 15). K562 (white bars) showed much lower expression of HLA-I and HLA-C, and much higher expression of activating ligands than myeloma PCs. **, indicates p < 0.01 in the DMS post-hoc analysis from the ANOVA comparing both normal PCs and residual lymphocytes with myeloma PCs. (C) Ex vivo, un-stimulated purified NK cells from three individuals with variable number of iKIR2D/HLA-C licensing interactions (Controls, white bars and symbols) and three patients with no conventional iKIR2D/HLA-C licensing interactions (KIR2DL1−L2+L3−/C2C2, black bars and symbols) showed similar degranulation and cytotoxic capacity against K562 cell line. However, patients with no conventional iKIR2D/HLA-C licensing interactions showed lower degranulation (p < 0.05) and cytotoxicity (p < 0.05) activity against myeloma PCs (n = 3), cryopreserved from two C2C2 and one C1C1 MM patients. Data represent mean ± SEM of three independent experiments performed for every patient or control against every myeloma PC target. NK cells from controls showed at least one iKIR2DL licensing interaction either for C1C1 or C2C2 myeloma targets.
Finally, we investigated the role of iKIR2D/HLA-C licensing interactions on the responsiveness of NK cell against “increasing-self” tumors. So that CD107a degranulation and cytotoxic capacity was evaluated in three patients from our series with no conventional iKIR2D/HLA-C licensing interactions (KIR2DL1−L2+L3− genotype in the HLA-C2/C2 background) and compared with three individuals showing a variable number of iKIR2D/HLA-C licensing interactions (controls). To preserve the original licensing status of NK cells in each individual, directly ex-vivo un-stimulated NK cells were assayed. Both CD107a degranulation and cytotoxicity assays demonstrated that NK cells from patients with total absence of conventional iKIR2D/HLA-C licensing interactions had very similar response than controls against K562 (“missing-self” target rich in aRec-ligands). However, when target consisted in ex-vivo purified BMPCs from MM patients (“increasing-self” target with moderate expression of aRec-ligands), NK cells from patients with no conventional iKIR2D/HLA-C licensing interactions exhibited lower degranulation ( p < 0.05) and cytotoxic capacity (p < 0.05) than NK cells from controls.
Discussion
Since the late 80′s, the “missing self” hypothesis has provided a reasonable explanation for cancer immunosurveillance: malignant cells reduce the expression of HLA molecules to avoid the attack of cytotoxic T cells, thereby enabling NK cells to sense this loss through iKIRs.1 Although HLA loss is a widespread phenomenon in cancer,23,24 it is not certain whether this is a general event. Thus, differential expression of HLA among cancers could have contributed to the variety of models proposed to explain NK allo-reactivity in haploidentical SCT.38 Certainly, our data and others.27 demonstrate that myeloma-PCs, compared to normal lymphocytes and other hematological cancers, showed increased expression of HLA-I (“increasing self”), including HLA-C, and mild expression of ligands for NK cell aRec (CD112, CD155, ULBP-1 and MICA/B),25 which apparently renders myeloma-PCs susceptible to lysis mainly by licensed NK cells. Our data would support a new model where NK cell response to myeloma, and hypothetically to other non-missing-self tumors, would be highly dependent on NK cell licensing, as licensed NK cells would respond more efficiently against no-missing-self targets expressing low or moderate amounts of ligands for NK cell aRec (Fig. 4). This might have important implications for the design of future NK cell-based therapies and for the selection of donors for haploidentical SCT.
Our proposition for a role of NK cell licensing in the immunosurveillance of myeloma is based on the higher frequency of genotypes missing iKIRs, particularly KIR2DL1, KIR2DL2, KIR2DL3 and KIR3DL1 found in these patients, probably reflecting increased homozygosity.39 and the presence of repertoires of iKIR2D/HLA licensing interactions with lower diversity and weaker affinity. Indeed, less diverse and weaker affinity iKIR2D/HLA interactions,37,40 such as those showing complete absence of conventional iKIR2D/HLA-C licensing interactions (KIR2DL1−L2+L3−/C2C2), single-KIR2DL3+/C1+ and single-KIR2DL2+/C1+ were accumulated in myeloma patients, while stronger licensing combinations KIR2DL1+/C2+, double-KIR2DL2+L3+/C1+ and triple-KIR2DL1+L2+L3+/C1+C2+ were diminished, leading to repertoires of licensed NK cell with lower diversity and potentially decreased cytotoxic capacity.41 To demonstrate whether or not iKIR/HLA-I licensing interactions can provide protection against increasing-self tumors, CD107a degranulation and cytotoxic capacity was analyzed in three patients from our series with no conventional iKIR2D/HLA-C licensing interactions (KIR2DL1−L2+L3− genotype in the HLA-C2/C2 background). As recently described, unlicensed NK cells maintained cytolytic capacity against missing-self tumor (e.g., K562),42 but barely lyse “increasing-self” tumors such as myeloma. In contrast, NK cells from individuals with a variable number of iKIR2D/HLA-C licensing interactions lyse normally myeloma cells, as previously described.43 As mentioned above, to preserve the original licensing status of NK cells in each individual, NK cells were assayed un-stimulated, directly ex-vivo, and in absence of exogenous cytokines; consequently, in vitro responses against myeloma targets were modest but sufficient to demonstrate that iKIR2D/HLA-C licensing interactions improves the ability of NK cells to provide better responses against increasing-self targets with a mild expression of ligands for NK cell aRec.
Current knowledge cannot easily explain why licensed NK cells, as opposed to unlicensed ones, are able to lyse increasing-self targets such us myeloma. Although further research on the activating and inhibitory signal transduction pathways in licensed and un-licensed NK cells should be conducted, a reasonable explanation can be found in recent results describing that NK cell education is strongly linked to a dynamic and coordinated expression of DNAM-1 and to conformational changes in LFA-1.44 The authors described that the expression of the adhesion/activation DNAM-1 molecule on NK cell membrane positively correlates with the quantity and quality of iKIR2D/HLA-C licensing interactions as well as with the magnitude of NK cell functional responses. Thus, upon target cell recognition, a higher expression of DNAM-1 in educated NK cells would favor changes in LFA-1 conformational state and a rapid colocalization of both active LFA-1 and DNAM-1 at the immune synapse, which might promote the formation of more stable target cell conjugates,45,46 consequently increasing the chances to lyse targets less sensitive to NK cell killing such as myeloma tumor cells.
During the maturation process, PCs undergo several episodes of gene recombination which make them genetically unstable. In this sense, FISH analysis determined that 68.1% of myelomas in our series accumulated at least one out of the six studied alterations. Obviously, such variety and high frequency of genetic alterations demand diverse immunosurveillance mechanisms that permanently recognize and eliminate transformed cells, with NK cells playing essential roles.25 Accordingly, the reduction of effective (“licensed”) NK cell repertoire would seriously impair immunosurveillance, favoring not only the emergence, but also the development of more aggressive PCDs. Indeed, diversity loss is particularly obvious in KIR2DL1−L2+L3− patients, in whom an earlier onset of MM and PCDs with worse biological features was observed. Remarkably, progressive PCD was diagnosed in 2 out of the 3 patients showing complete absence of conventional iKIR2D/HLA-C licensing interactions at the early ages of 35 and 37. In accordance with our hypothesis, not only KIR2DL1−L2+L3− patients but also KIR3DL1− (57.1% of them KIR3DL1−KIR2DL3−) showed much poorer outcome, particularly in patients with low-tumor burden, where the activity of NK cells may still prevail.
In consonance with our results, a protective role of KIR3DL1 against myeloma progression has been reported in a series of 182 patients after autologous SCT.31 Indeed, shorter PFS was described not only for KIR3DS1+ homozygous patients (KIR3DL1−, as these 2 genes are alleles) but also particularly for KIR3DL1+KIR3DS1+ heterozygous patients with low-tumor burden (having complete or partial remission) who lacked the KIR3DL1/Bw4 licensing interaction and, therefore, functionally these patients could also be considered KIR3DL1−. Gabriel's study did not describe any impact on PFS for KIR2DL1; however, authors showed that KIR2DL1 frequency was 94%, slightly higher than ours (86.8%), but lower than that described for British Caucasians (97.0 to 97.2%).47,48 Differences in KIR2DL1 frequency between both series, in view of the rapid progressive disease observed for KIR2DL1− patients in our series, could be due to earlier exitus or to the exclusion of patients undergoing tandem auto or auto-allo SCT in Gabriel's series. Although no further studies analyzing KIR genes in myeloma have been reported, it is noteworthy that Australian Caucasians show one of the lowest KIR2DL1 frequencies (84%).49 and one of the highest rates of myeloma (http://www.aihw.gov.au/WorkArea/DownloadAsset.aspx?id= 60129546402).
In Western countries, the frequency of myeloma is likely to increase as the population ages, so the availability of new therapeutic strategies will be necessary. Our results provide information for understanding immunosurveillance of a kind of tumor that highly express HLA-I and, therefore, cannot be explained by the missing self theory. Consequently, this information might be useful for the design of future NK cell-based therapies for myeloma or hypothetically for other non-missing self tumors and for a better selection of haploidentical SCT donors.
Patients and methods
Patients and controls
BM and PB were obtained at diagnosis from 164 Caucasian patients suffering from PCDs, recruited consecutively between 2010 and 2012 from Hospital Clínico Universitario Virgen de la Arrixaca, Hospital Rafael Mendez and Hospital General Universitario Santa Lucía, Murcia, Spain (Table 1). Differential diagnosis of myeloma from MGUS was based on the presence of at least 10% clonal BMPCs and gammopathy in serum or urine. Histological BMPCs quantification was done following standard procedures in May-Grünwald-Giemsa stained aspirates. Myeloma was classified as SMM or MM, depending on the absence or presence of myeloma-related organ or tissue dysfunction, including hypercalcemia, renal insufficiency, anemia and bone disease. The study included 86 MGUS, 25 SMM and 53 MM patients. Importantly, following new criteria of the International Myeloma Working Group, 20 out of the 25 SMM patients should be currently classified as MM.32 Risk factors for MGUS and SMM.33 and International Staging Systems (ISS) for MM.34 were estimated as described elsewhere.
HLA-I (HLA-A, -B and -C) and KIR genotypes were studied in patients and in 286 ethnic-, sex- and age-matched, unrelated healthy controls. KIR2D receptor expression on PB lymphocytes was analyzed in all patients and in 30 controls. Expression of iKIR-ligands (total-HLA-I and HLA-C) as well as aRec-ligands (CD112, CD155, ULBP-1 and MICA/B) was evaluated on normal-BMPCs in 10 non-dyscrasia donors and on aberrant-BMPCs in 15 consecutive MM patients at diagnosis as well as on their normal-residual lymphocytes.
The study was approved by the respective Research Ethics Committee at each hospital, and informed consent was obtained from all patients and controls in accordance with the Declaration of Helsinki.
HLA-A, -B and -C and KIR genotyping
HLA-A and B genotyping was performed using PCR-SSOP and luminex technology (LabType, OneLambda, Canoga Park, CA), in order to identify patients and controls carrying A3/A11 alleles or HLA-Bw4 epitopes. HLA-C and KIR typing was performed as previously described.21 HLA-C genotyping allowed distinction between HLA-CAsn80 (group-C1) and HLA-CLys80 (group-C2) alleles. HLA-C genotypes were classified into C1C1, C1C2 and C2C2 groups. KIR genes encoding iKIRs (2DL1-L3/2DL5 and 3DL1-L3) and aKIRs (2DS1-S5 and 3DS1),37,50 as well as KIR2DL4, which has both inhibitory and activating potential, were detected.51 Distinction between KIR2DL5A and KIR2DL5B was not possible. KIR genotypes were classified into two groups, A and B.52
Flow cytometry
Immunophenotyping for discrimination between normal and aberrant BMPCs was performed as described,53 using 7-color staining with CD19, CD20, CD27, CD38, CD45, CD56 and CD138 monoclonal antibodies (mAbs). KIR2D expression on CD3−CD16+CD56+ NK cells as well as on CD3+CD4+ and CD3+CD8+ T cells was analyzed using methods summarized in Fig. 1A, B, using 9-color staining with CD3, CD4+, CD8+, CD16, CD56, CD158a,h (clone EB6, which recognized both KIR2DL1 and 2DS1), CD158b1/b2,j (clone GL183, KIR2DL2, 2DL3 and 2DS2), CD158a (clone 143211, KIR2DL1) and CD158b2 (clone 180701, KIR2DL3) mAbs. Total-HLA-I and HLA-C expression on normal and tumor cells was evaluated using W6/32 (Serotec, Oxford, England) and DT9 (kindly provided by Dr. Simon Brackenridge, University of Oxford) mAbs, respectively. Expression of ligands for NK cell aRec on tumor cells was analyzed using CD112, CD155, ULBP-1 and MICA/B mAbs. Reagents were obtained from Becton Dickinson (BD, San Jose, CA), except for CD20 (Miltenyi Biotec, Bergisch Gladbach, Germany), CD138 (IQP, Groningen, The Netherlands), CD158a/h and CD158b1/b2/j (Beckman Coulter, Fullerton, CA), CD158a, CD158b2 and ULBP (R&D systems, Minneapolis, USA), and CD112, CD155 and MICA/B (Biolegend, San Diego, CA). All studies were done on FACSCanto-II and LSR-II using DIVASoftware (BD).
Fluorescent in situ hybridization (FISH)
BMPCs were purified using RosetteSep® Human Multiple-Myeloma-Cell Enrichment Cocktail (StemCell Technologies, Inc., Grenoble, France), fixed with Carnoy's solution and stored at −20°C. Numeric abnormalities in chromosomes 5, 9 and 15, break at IgH (14q32) and deletions of 17p13.1 (p53) and 13q14 were studied by FISH using DNA-probes (Vysis Inc., Downers Grove, IL). Sufficient purified BMPCs to complete FISH study were obtained from 145 out of 164 patients (88.4%).
NK cell cytotoxicity and CD107a-degranulation assays
NK cells were purified with RosetteSep™ Human-NK-Cell Enrichment Cocktail (StemCell technologies, Grenoble, France) using 20mL of PB from three individuals with a variable number of iKIR2D/HLA-C licensing interactions (Controls) and from three KIR2DL1−L2+L3−/C2C2 patients with no conventional iKIR2D/HLA-C licensing interactions (but with the non-conventional KIR2DL2/C2 interaction). Myeloma PCs were purified from 3 MM patients (one C1C1 and 2 C2C2) using RosetteSep™ and cryopreserved.
Cytotoxic activity of freshly purified, un-stimulated NK cells was evaluated using K562 cell line or cryopreserved myeloma PCs as targets, following previously described methods.54 Briefly, targets were stained with 0.25 µM 5- (and 6-) carboxyfluorescein diacetate succinimidyl ester (CFSE, Molecular probes, Leiden, The Netherlands) for 10 min at 37°C, extensively washed to remove any rest of CFSE and resuspended in complete medium. NK cell effectors were washed in complete medium and viability of both target and effector cells examined with 0.5% trypan-blue. Effector cells were seeded with a constant number of 10,000 CFSE-labeled target cells at different effector/target ratios 1:1, 10:1, and 20:1 in triplicate, in absence of stimulatory cytokines. In parallel, target cells were incubated alone to measure basal cell death. Cells were incubated in a V-bottom 96-well microplates in a total volume of 150 μL of complete medium for 4 h in a 5% CO2 atmosphere at 37°C. Cell mixtures were then washed in PBS–1% BSA and incubated in the same buffer containing 20µg/mL 7-amino actinomycin D (7-AAD, Sigma, France) for 10 min at 4°C in the dark. Cells were then washed and acquired right afterwards on a LSR-II flow cytometer. Mean value of triplicates was used to calculate the percentage of lysis as follows: experimental – spontaneous apoptotic target cells.
CD107a degranulation assay was performed with the same target and effector cells. Control sample without target cells was included to detect spontaneous degranulation. An effector/target ratio of 1:1 (100.000 cells each in 150 µL) in triplicate was used. Vioblue anti-CD107a (10 µL, Miltenyi Biotec) was added in each well before incubation. Effectors and targets were then co-incubated at 37°C for 4 h; after the first hour, monensin (BD) at a final concentration of 2 mM, was added to inhibit cell secretion. After 4 h, cells were washed and stained with a mixture of mAbs to analyze the surface expression of CD107a on NK cells.
Statistical analysis
Data was collected in a database (Excel2003; Microsoft Corporation, Seattle, WA) and analyzed using SPSS-15.0 (SPSS Inc.., Chicago, IL). Pearson's χ2 and 2-tailed Fisher's exact tests were used to compare categorized variables. Numerical variables were compared using Mann Whitney or ANOVA and the DMS post-hoc tests. The Kaplan–Meier method and Log-rank test were used to compare differences in the PFS. Multivariate logistic regression analysis was applied to confirm positive associations. The strength of association was estimated by odds ratio (OR) and 95% confidence interval (95%CI). p values < 0.05 were considered significant. The Bonferroni method was used to correct for the number of comparisons (Pc) when necessary.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank B. Las Heras, S. Soriano and F. Villar for technical help, as well as the hematologist from the hematology services from Clinic University Hospital Virgen de la Arrixaca (Murcia, Spain), Rafael Mendez Hospital (Lorca, Spain) and University Hospital Santa Lucía (Cartagena, Spain) for their kind collaboration in enrolling patients. We are also grateful to Dr. Simon Brackenridge, University of Oxford, who kindly provided DT-9 antibody.
Funding
This work was supported by excellence project of Seneca (GERM/06/2008), CajaMurcia Foundations, Instituto de Salud Carlos III (ISCiii) Project FISPI13/02297 (co-financed with FEDER funds) and Association Pablo Ugarte (APU). Isabel Legaz was financed by the Sara Borrell Program from the Fondo de Investigación Sanitaria del ISCiii, Ministerio de Economía y Competitividad, Spain. Lourdes Gimeno was supported by ISCiii (CA09/00099). M. Rocío López-Álvarez was supported by Seneca Foundation (04087/GERM/06 Project), Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBEREHD) and Programa Nacional de Movilidad de Recursos Humanos, Plan Nacional de I-D+i 2008–2011, Ministerio de Educación. María V. Martínez-Sánchez was supported by APU. A.M. designed research, analyzed data and wrote the paper; M.R.A-L. guided the interpretation of the immunological data. A.P. coordinated collection of clinical data and helped with clinical interpretation of data; M.V.M-S., I.L., L.G., N.R.M. and A.Mrowiec performed experimental analysis; J.A.C., M.V.B., M.R.L-A., A.M.G-A. and M.M. contributed to the immunological interpretation of data; C.G., M.C.G-G, V.C-P. J.L.F. and J.M.M. coordinated the enrolment of patients and controls; J.M.B. helped with statistical analyses.
References
- 1.Kärre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986; 319:675-788; PMID:3951539; http://dx.doi.org/16079848 10.1038/319675a0 [DOI] [PubMed] [Google Scholar]
- 2.Kim S, Poursine-Laurent J, Truscott SM, Lybarger L, Song YJ, Yang L, French AR, Sunwoo JB, Lemieux S, Hansen TH et al.. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 2005; 436:709-13; PMID:16079848; http://dx.doi.org/ 10.1038/nature03847 [DOI] [PubMed] [Google Scholar]
- 3.Anfossi N, André P, Guia S, Falk CS, Roetynck S, Stewart CA, Breso V, Frassati C, Reviron D, Middleton D et al.. Human NK cell education by inhibitory receptors for MHC class I. Immunity 2006; 25:331-42; PMID:16901727; http://dx.doi.org/ 10.1016/j.immuni.2006.06.013 [DOI] [PubMed] [Google Scholar]
- 4.Thomas LM, Peterson ME, Long EO. Cutting edge: NK cell licensing modulates adhesion to target cells. J Immunol 2013; 191:3981-5; PMID:24038086; http://dx.doi.org/ 10.4049/jimmunol.1301159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moretta A, Sivori S, Vitale M, Pende D, Morelli L, Augugliaro R, Bottino C, Moretta L. Existence of both inhibitory (p58) and activatory (p50) receptors for HLA-C molecules in human natural killer cells. J Exp Med 1995; 182:875-84; PMID:7650491; http://dx.doi.org/ 10.1084/jem.182.3.875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mandelboim O, Reyburn HT, Valés-Gómez M, Pazmany L, Colonna M, Borsellino G, Strominger JL. Protection from lysis by Natural killer cells of group 1 and 2 specificity is mediated by residue 80 in human histocompatibility leukocyte antigen C alleles and also occurs with empty major hiscompatibility complex molecules. J Exp Med 1996; 184:913-22; PMID:9064351; http://dx.doi.org/ 10.1084/jem.184.3.913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schönberg K, Sribar M, Enczmann J, Fischer JC, Uhrberg M. Analyses of HLA-C-specific KIR repertoires in donors with group A and B haplotypes suggest a ligand-instructed model of NK cell receptor acquisition. Blood 2011; 117:98-107; PMID:20935255; http://dx.doi.org/ 10.1182/blood-2010-03-273656 [DOI] [PubMed] [Google Scholar]
- 8.Foley BA, De Santis D, Van Beelen E, Lathbury LJ, Christiansen FT, Witt CS. The reactivity of Bw4+ HLA-B and HLA-A alleles with KIR3DL1: implications for patient and donor suitability for haploidentical stem cell transplantations. Blood 2008; 112:435-43; PMID:18385451; http://dx.doi.org/ 10.1182/blood-2008-01-132902 [DOI] [PubMed] [Google Scholar]
- 9.Hansasuta P, Dong T, Thananchai H, Willberg C, Aldemir H, Rowland-Jones S, Braud VM. Recognition of HLA-A3 and HLA-11 by KIR3DL2 is peptide-specific. Eur J Immunol 2004; 34:1673-9; PMID:15162437; http://dx.doi.org/ 10.1002/eji.200425089 [DOI] [PubMed] [Google Scholar]
- 10.Chewning JH, Gudme CN, Hsu KC, Selvakumar A, Dupont B. KIR2DS1-positive NK cells mediate alloresponse against the C2 HLA-KIR ligand group in vitro. J Immunol 2007; 179:854-68; PMID:17617576; http://dx.doi.org/ 10.4049/jimmunol.179.2.854 [DOI] [PubMed] [Google Scholar]
- 11.Katz G, Markel G, Mizrahi S, Arnon TI, Mandelboim O. Recognition of HLA-Cw4 and HLA-Cw6 by the NK cell killer cell Ig-like receptors NK cell receptor two domain short tail number 4. J Immunol 2001; 166:7260-7; PMID:11390475; http://dx.doi.org/ 10.4049/jimmunol.166.12.7260 [DOI] [PubMed] [Google Scholar]
- 12.Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC, Biassoni R, Moretta L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol 2001; 19:197-223; PMID:11244035; http://dx.doi.org/ 10.1146/annurev.immunol.19.1.197 [DOI] [PubMed] [Google Scholar]
- 13.Baychelier F, Sennepin A, Ermonval M, Dorgham K, Debré P, Vieillard V. Identification of a cellular ligand for the natural cytotoxicity receptor NKp44. Blood 2013; 122:2935-42; PMID:23958951; http://dx.doi.org/ 10.1182/blood-2013-03-489054 [DOI] [PubMed] [Google Scholar]
- 14.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999; 285:727-9; PMID:10426993; http://dx.doi.org/ 10.1126/science.285.5428.727 [DOI] [PubMed] [Google Scholar]
- 15.Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, Kubin M, Chalupny NJ. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001; 14:123-33; PMID:11239445; http://dx.doi.org/ 10.1016/S1074-7613(01)00095-4 [DOI] [PubMed] [Google Scholar]
- 16.Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, Cantoni C, Grassi J, Marcenaro S, Reymond N et al.. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J Exp Med 2003; 198:557-67; PMID:12913096; http://dx.doi.org/ 10.1084/jem.20030788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Díaz-Peña R, Blanco-Gelaz MA, López-Larrea C. KIR genes and their role in spondyloarthropathies. Adv Exp Med Biol 2009; 649:286-99; PMID:19731638; http://dx.doi.org/18246204 10.1007/978-1-4419-0298-6_22 [DOI] [PubMed] [Google Scholar]
- 18.Ahlenstiel G, Martin MP, Gao X, Carrington M, Rehermann B. Distinct KIR/HLA compound genotypes affect the kinetics of human antiviral natural killer cell responses. J Clin Invest 2008; 118:1017-26; PMID:18246204; http://dx.doi.org/ 10.1172/JCI32400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purdy AK, Campbell KS. Natural killer cells and cancer: regulation by the killer cell Ig-likereceptors (KIR). Cancer Biol Ther 2009; 8:2211-20; PMID:19923897; http://dx.doi.org/ 10.4161/cbt.8.23.10455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kröger N, Zabelina T, Berger J, Duske H, Klyuchnikov E, Binder T, Stübig T, Hilde-brandt Y, Atanackovic D, Alchalby H et al.. Donor KIR haplotype B improves progression-free and overall survival after allogeneic hematopoietic stem cell transplantation for multiple myeloma. Leukemia 2011; 25:1657-61; PMID:21647155; http://dx.doi.org/ 10.1038/leu.2011.138 [DOI] [PubMed] [Google Scholar]
- 21.Legaz I, López-Álvarez MR, Campillo JA, Moya-Quiles MR, Bolarín JM, de la Peña J, Salgado G, Gimeno L, García-Alonso AM, Muro M, Miras M et al.. KIR gene mismatching and KIR/C ligands in liver transplantation: consequences for short-term liver allograft injury. Transplantation 2013; 95:1037-44; PMID:23478359; http://dx.doi.org/ 10.1097/TP.0b013e318286486c [DOI] [PubMed] [Google Scholar]
- 22.Chan CJ, Andrews DM, Smyth MJ. Receptors that interact with nectin and nectin-like proteins in the immunosurveillance and immunotherapy of cancer. Curr Opin Immunol 2012; 24:246-51; PMID:22285893; http://dx.doi.org/ 10.1016/j.coi.2012.01.009 [DOI] [PubMed] [Google Scholar]
- 23.Garrido F, Cabrera T, Concha A, Glew S, Ruiz-Cabello F, Stern PL. Natural history of HLA expression during tumour development. Immunol Today 1993; 14:491-9; PMID:8274189; http://dx.doi.org/ 10.1016/0167-5699(93)90264-L [DOI] [PubMed] [Google Scholar]
- 24.Garrido F, Cabrera T, Lopez-Nevot MA, Ruiz-Cabello F. HLA class I antigens in human tumors. Adv Cancer Res 1995; 67:155-95; PMID:8571814; http://dx.doi.org/ 10.1016/S0065-230X(08)60713-7 [DOI] [PubMed] [Google Scholar]
- 25.El-Sherbiny YM, Meade JL, Holmes TD, McGonagle D, Mackie SL, Morgan AW, Cook G, Feyler S, Richards SJ, Davies FE et al.. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res 2007; 67:8444-9; PMID:17875681; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-4230 [DOI] [PubMed] [Google Scholar]
- 26.Carbone E, Neri P, Mesuraca M, Fulciniti MT, Otsuki T, Pende D, Groh V, Spies T, Pollio G, Cosman D et al.. HLAclass I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood 2005; 105:251-8; PMID:15328155; http://dx.doi.org/ 10.1182/blood-2004-04-1422 [DOI] [PubMed] [Google Scholar]
- 27.Bernal M, Garrido P Jiménez P, Carretero R, Almagro M, López P, Navarro P, Garrido F, Ruiz-Cabello F. Changes in activatory and inhibitory natural killer (NK) receptors may induce progression to multiple myeloma: implications for tumor evasion of T and NKcells. Hum Immunol 2009; 70:854-7; PMID:19580833; http://dx.doi.org/ 10.1016/j.humimm.2009.07.004 [DOI] [PubMed] [Google Scholar]
- 28.Perez-Andres M, Almeida J, Martin-Ayuso M, De Las Heras N, Moro MJ, Martin-Nuñez G, Galende J, Cuello R, Abuín I, Moreno I et al.. Soluble and membrane levels of molecules involved in the interaction between clonal plasma cells and the immunological microenvironment in multiple myeloma and their association with the characteristics of the disease. Int J Cancer 2009; 124:367-75; PMID:19003959; http://dx.doi.org/ 10.1002/ijc.23941 [DOI] [PubMed] [Google Scholar]
- 29.Palumbo A, Attal M, Roussel M. Shifts in the therapeutic paradigm for patients newly diagnosed with multiple myeloma: maintenance therapy and overall survival. Clin Cancer Res 2011; 17:1253-63; PMID:21411441; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-1925 [DOI] [PubMed] [Google Scholar]
- 30.Palumbo A, Anderson K. Multiple Myeloma. N Engl J Med 2011; 364:1046-60; PMID:21410373; http://dx.doi.org/ 10.1056/NEJMra1011442 [DOI] [PubMed] [Google Scholar]
- 31.Gabriel IH, Sergeant R, Szydlo R, Apperley JF, DeLavallade H, Alsuliman A, Khoder A, Marin D, Kanfer E, Cooper N et al.. Interaction between KIR3DS1 and HLA-Bw4 predicts for progression-free survival after autologous stem cell transplantation in patients with multiple myeloma. Blood 2010; 116:2033-9; PMID:20562327; http://dx.doi.org/ 10.1182/blood-2010-03-273706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, Kumar S, Hillengass J, Kastritis E, Richardson P et al.. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014; 15:e538-48; PMID:25439696; http://dx.doi.org/ 10.1016/S1470-2045(14)70442-5 [DOI] [PubMed] [Google Scholar]
- 33.Perez-Persona E, Vidriales MB, Mateo G, García-Sanz R, Mateos MV, de Coca AG, Galende J, Martín-Nuñez G, Alonso JM, de Las Heras N et al.. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood 2007; 110:2586-92; PMID:17576818; http://dx.doi.org/ 10.1182/blood-2007-05-088443 [DOI] [PubMed] [Google Scholar]
- 34.Greipp PR, San Miguel J, Durie BG, Crowley JJ, Barlogie B, Bladé J, Boccadoro M, Child JA, Avet-Loiseau H, Kyle RA et al.. International staging system for multiple myeloma. J Clin Oncol 2005; 23:3412-20; PMID:15809451; http://dx.doi.org/ 10.1200/JCO.2005.04.242 [DOI] [PubMed] [Google Scholar]
- 35.Cognet C, Farnarier C, Gauthier L, Frassati C, André P, Magérus-Chatinet A, Anfossi N, Rieux-Laucat F, Vivier E, Schleinitz N. Expression of the HLA-C2-specific activating killer-cell Ig-like receptor KIR2DS1 on NK and T cells. Clin Immunol 2010; 135:26-32; PMID:20093094; http://dx.doi.org/ 10.1016/j.clim.2009.12.009 [DOI] [PubMed] [Google Scholar]
- 36.Falco M, Romeo E, Marcenaro S, Martini S, Vitale M, Bottino C, Mingari MC, Moretta L, Moretta A, Pende D. Combined genotypic and phenotypic killer cell Ig-like receptor analyses reveal KIR2DL3 alleles displaying unexpected monoclonal antibody reactivity: identification of the amino acid residues critical for staining. J Immunol 2010; 185:433-41; PMID:20525888; http://dx.doi.org/ 10.4049/jimmunol.0903632 [DOI] [PubMed] [Google Scholar]
- 37.Parham P. MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol 2005; 5:201-14; PMID:15719024; http://dx.doi.org/ 10.1038/nri1570 [DOI] [PubMed] [Google Scholar]
- 38.Leung W. Infusions of allogeneic natural killer cells as cancer therapy. Clin Cancer Res 2014; 20:3390-400; PMID:24987108; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-1766 [DOI] [PubMed] [Google Scholar]
- 39.Jiang W, Johnson C, Jayaraman J, Simecek N, Noble J, Moffatt MFC, Cookson WO, Trowsdale J, Traherne JA. Copy number variation leads to considerable diversity for B but not A haplotypes of the human KIR genes encoding NK cell receptors. Genome Res 2012; 22:1845-54; PMID:22948769; http://dx.doi.org/ 10.1101/gr.137976.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu J, Heller G, Chewning J, Kim S, Yokoyama WM, Hsu KC. Hierarchy of the human natural killer cell response is determined by class and quantity of inhibitory receptors for self-HLA-B and HLA-C ligands. J Immunol 2007; 179:5977-89; PMID:17947671; http://dx.doi.org/ 10.4049/jimmunol.179.9.5977 [DOI] [PubMed] [Google Scholar]
- 41.Brodin P, Lakshmikanth T, Johansson S, Kärre K, Höglund P. The strength of inhibitory input during education quantitatively tunes the functional responsiveness of individual natural killer cells. Blood 2009; 113:2434-41; PMID:18974374; http://dx.doi.org/ 10.1182/blood-2008-05-156836 [DOI] [PubMed] [Google Scholar]
- 42.Carrega P, Pezzino G, Queirolo P, Bonaccorsi I, Falco M, Vita G, Pende D, Misefari A, Moretta A, Mingari MC et al.. Susceptibility of human melanoma cells to autologous natural killer (NK) cell killing: HLA-related effector mechanisms and role of unlicensed NK cells. Plos One 2009; 4:e8132; PMID:19997637; http://dx.doi.org/ 10.1371/journal.pone.0008132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katodritou E, Terpos E, North J, Kottaridis P, Verrou E, Gastari V, Chadjiaggelidou C, Sivakumaran S, Jide-Banwo S, Tsirogianni M et al.. Tumor-primed natural killer cells from patients with multiple myeloma lyse autologous, NK-resistant, bone marrow-derived malignant plasma cells. Am J Hematol 2011; 86:967-73; PMID:21919039; http://dx.doi.org/ 10.1002/ajh.22163 [DOI] [PubMed] [Google Scholar]
- 44.Enqvist M, Ask EH, Forslund E, Carlsten M, Abrahamsen G, Béziat V, Andersson S, Schaffer M, Spurkland A, Bryceson Y et al.. Coordinated expression of DNAM-1 and LFA-1 in educated NK cells. J Immunol 2015; 194:4518-27; PMID:25825444; http://dx.doi.org/ 10.4049/jimmunol.1401972 [DOI] [PubMed] [Google Scholar]
- 45.Ramsbottom KM, Hawkins ED, Shimoni R, McGrath M, Chan CJ, Russell SM, Smyth MJ, Oliaro J. Cutting edge: DNAX accessory molecule 1-deficient CD8+ T cells display immunological synapse defects that impair antitumor immunity. J Immunol 2014; 192:553-7; PMID:24337740; http://dx.doi.org/ 10.4049/jimmunol.1302197 [DOI] [PubMed] [Google Scholar]
- 46.Hou S, Ge K, Zheng X, Wei H, Sun R, Tian Z. CD226 protein is involved in immune synapse formation and triggers natural killer (NK) cell activation via its first extracellular domain. J Biol Chem 2014; 289:6969-77; PMID:24451371; http://dx.doi.org/ 10.1074/jbc.M113.498253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harvey D, Pointon JJ, Sleator C, Meenagh A, Farrar C, Sun JY, Senitzer D, Middleton D, Brown MA, Wordsworth BP. Analysis of killer immunoglobulin-like receptor genes in ankylosing spondylitis. Ann Rheum Dis 2009; 68:595-8; PMID:19019897; http://dx.doi.org/ 10.1136/ard.2008.095927 [DOI] [PubMed] [Google Scholar]
- 48.Jones DC, Edgar RS, Ahmad T, Cummings JR, Jewell DP, Trowsdale J, Young NT. Killer Ig-like receptor (KIR) genotype and HLA ligand combinations in ulcerative colitis susceptibility. Genes Immun 2006; 7:576-82; PMID:16929347; http://dx.doi.org/ 10.1038/sj.gene.6364333 [DOI] [PubMed] [Google Scholar]
- 49.Witt CS, Dewing C, Sayer DC, Uhrberg M, Parham P, Christiansen FT. Population frequencies and putative haplotypes of the killer cell immunoglobulin-like receptor sequences and evidence for recombination. Transplantation 1999; 68:1784-9; PMID:10609957; http://dx.doi.org/ 10.1097/00007890-199912150-00024 [DOI] [PubMed] [Google Scholar]
- 50.Moretta L, Moretta A. Killer immunoglobulin-like receptors. Curr Opin Immunol 2004; 16:626-33; PMID:15342010; http://dx.doi.org/ 10.1016/j.coi.2004.07.010 [DOI] [PubMed] [Google Scholar]
- 51.Faure M, Long EO. KIR2DL4 (CD158d), an NK cell-activating receptor with inhibitory potential. J Immunol 2002; 168:6208-14; PMID:12055234; http://dx.doi.org/ 10.4049/jimmunol.168.12.6208 [DOI] [PubMed] [Google Scholar]
- 52.Uhrberg M, Valiante NM, Shum BP, Shilling HG, Lienert-Weidenbach K, Corliss B, Tyan D, Lanier LL, Parham P. Human diversity in killer cell inhibitory receptor genes. Immunity 1997; 7:753-63; PMID:9430221; http://dx.doi.org/ 10.1016/S1074-7613(00)80394-5 [DOI] [PubMed] [Google Scholar]
- 53.San Miguel JF, Almeida J, Mateo G, Bladé J, López-Berges C, Caballero D, Hernández J, Moro MJ, Fernández-Calvo J, Díaz-Mediavilla J et al.. Immunophenotypic evaluation of the plasma cell compartment in multiple myeloma: a tool for comparing the efficacy of different treatment strategies and predicting outcome. Blood 2002; 99:1853-6; PMID:11861305; http://dx.doi.org/ 10.1182/blood.V99.5.1853 [DOI] [PubMed] [Google Scholar]
- 54.López-Alvarez MR, Martínez-Sánchez MV, Salgado-Cecilia MG, Campillo JA, Heine-Suñer D, Villar-Permuy F, Fuster JL, Bas A, Gil-Herrera J, Muro M et al.. Association of monoclonal expansion of Epstein-Barr virus-negative CD158a+ NK cells secreting large amounts of gamma interferon with hemophagocytic lymphohistiocytosis. Clin Vaccine Immunol 2009; 16:142-5; PMID:19020108; http://dx.doi.org/ 10.1128/CVI.00358-08 [DOI] [PMC free article] [PubMed] [Google Scholar]