

ABSTRACT
The presence of colony stimulating factor-1 (CSF1)/CSF1 receptor (CSF1R)-driven tumor-infiltrating macrophages and myeloid-derived suppressor cells (MDSCs) is shown to promote targeted therapy resistance. In this study, we demonstrate the superior effect of a combination of CSF1R inhibitor, PLX3397 and BRAF inhibitor, PLX4720, in suppressing primary and metastatic mouse BRAFV600E melanoma. Using flow cytometry to assess SM1WT1 melanoma-infiltrating leukocytes immediately post therapy, we found that PLX3397 reduced the recruitment of CD11b+ Gr1lo and CD11b+ Gr1int M2-like macrophages, but this was accompanied by an accumulation of CD11b+ Gr1hi cells. PDL1 expression on remaining myeloid cells potentially dampened the antitumor efficacy of PLX3397 and PLX4720 in combination, since PD1/PDL1 axis blockade improved outcome. We also reveal a role for PLX3397 in reducing tumor-infiltrating lymphocytes, and interestingly, this feature was rescued by the co-administration of PLX4720. Our findings, from three different mouse models of BRAF-mutated melanoma, support clinical approaches that co-target BRAF oncogene and CSF1R.
KEYWORDS: BRAFV600E, BRAF inhibitor, CSF1R, melanoma, M2 macrophage, PLX4720, PLX3397, tumor
Introduction
BRAF inhibitors (PLX4032; vemurafenib or GSK2118436; dabrafenib) have been shown to effectively suppress BRAFV600E-mutated melanoma, prolonging patient overall survival. However, clinical responses to these oncogene-targeted therapies are generally followed by patient therapy resistance and tumor recurrence.1-4 Substantial evidence showed that resistance to BRAF inhibition is not solely driven by tumor cell autochthonous mechanisms,5-9 but also via the induction of an immunosuppressive tumor microenvironment.10-12 In light of these findings, manipulating intratumoral immunity represents a promising strategy to overcome resistance and improve the ability of BRAF inhibitors to treat melanoma patients. While immunotherapies have been shown to enhance the anti-melanoma activity of BRAF inhibitors, these immunotherapeutic agents were mainly restricted to T cell checkpoint inhibitors, T cell co-stimulation agonists and adoptive cellular therapy.10,13-15 In contrast, approaches to modulate intratumoral macrophages and myeloid cells for overcoming BRAF inhibition resistance have not been widely explored.
Driven by tumor-derived soluble factors like CSF1, immunosuppressive M2 macrophages and MDSCs that express CSF1 receptor (CSF1R; CD115) are actively polarized and recruited to the tumor microenvironment.16,17 The presence of M2 macrophages and MDSCs has not only been shown to suppress cytotoxicity and/or promote exhaustion of antitumor T cells, but also result in therapy resistance.16,18,19 Administration of CSF1/CSF1R inhibitors (monoclonal antibodies or small molecule inhibitors) to inhibit M2 macrophages and MDSCs was shown not only to be effective as a cancer monotherapy, but also as an adjuvant therapy to overcome resistance to therapeutic approaches like vaccination, chemotherapy, adoptive cellular therapy, radiotherapy and T cell checkpoint blockade.20-28 Among the CSF1R inhibitors, PLX3397 is an effective small molecule receptor tyrosine kinase inhibitor for KIT/CSF1R/FLT3 that is currently being trialled as a single agent or in combination therapy for patients with glioblastoma, breast cancer, melanoma, and other cancers.
Here, we demonstrate superior anti-melanoma responses following a combination of PLX4720 (BRAF inhibitor; an analog of PLX4032) with PLX3397, in a series of mouse BRAFV600E-mutated melanomas with differential sensitivity to BRAF inhibition. We show that PLX3397 reduces the recruitment of CSF1R+ CD11b+ Gr1lo and CSF1R+ CD11b+ Gr1int M2-like tumor-associated macrophages (TAMs), accompanied by an accumulation of CD11b+ Gr1hi MDSC-like cells in the SM1WT1 BRAFV600E melanomas. The expression of PDL1 on remaining myeloid cells potentially dampened the antitumor efficacy of PLX3397 and PLX4720 in combination, since PD1/PDL1 axis blockade improved outcome. We also uncover a role for PLX3397 in reducing tumor-infiltrating NK cells, CD8+ T cells, CD4+ T cells, and CD4+ Foxp3+ Tregs, and this feature of PLX3397 is rescued by the co-administration of PLX4720.
Results
Combination of PLX4720 and PLX3397 enhances antitumor response against BRAFV600E melanomas
We first assessed the antitumor effect of PLX3397 alone or in combination with PLX4720 in three mouse models of BRAFV600E melanoma, namely the Tyr::CreERT2 BrafCA Ptenfl/fl (TBP) strain of melanoma prone mice and the tumor transplants, SM1WT1 and SM1WT1-LWT1. Consistent with published findings,14,29,30 PLX4720 suppressed the growth of de novo TBP melanomas, prolonging the survival of tumor-bearing mice (Fig. 1A and Fig. S1A). We observed that the survival of TBP-bearing mice treated with PLX3397 was prolonged, albeit its effect was inferior to PLX4720 (Fig. 1A and Fig. S1A). Notably, we found that the co-administration of PLX4720 and PLX3397 resulted in a significantly superior antitumor effect in comparison to either treatment alone with a 88% survival rate at 250 days for the combination relative to median survivals of 235 days for PLX4720, 120 days for PLX3397, and 70 days for control chow cohorts (Fig. 1A). This superior antitumor effect of PLX4720 in combination with PLX3397 was further confirmed in the transplantable SM1WT1 melanoma model (Fig. 1B). Next, in the experimental SM1WT1-LWT1 lung metastases model, we found that the combination therapeutic effect was also marginally increased, in comparison to PLX4720 treatment alone (Fig. S1B-C). Similar to the TBP model results, PLX3397 was found to be less effective than PLX4720 in suppressing the growth of SM1WT1 and experimental metastasis of SM1WT1-LWT1 melanomas (Fig. 1B and Fig. S1B-C).
Figure 1.
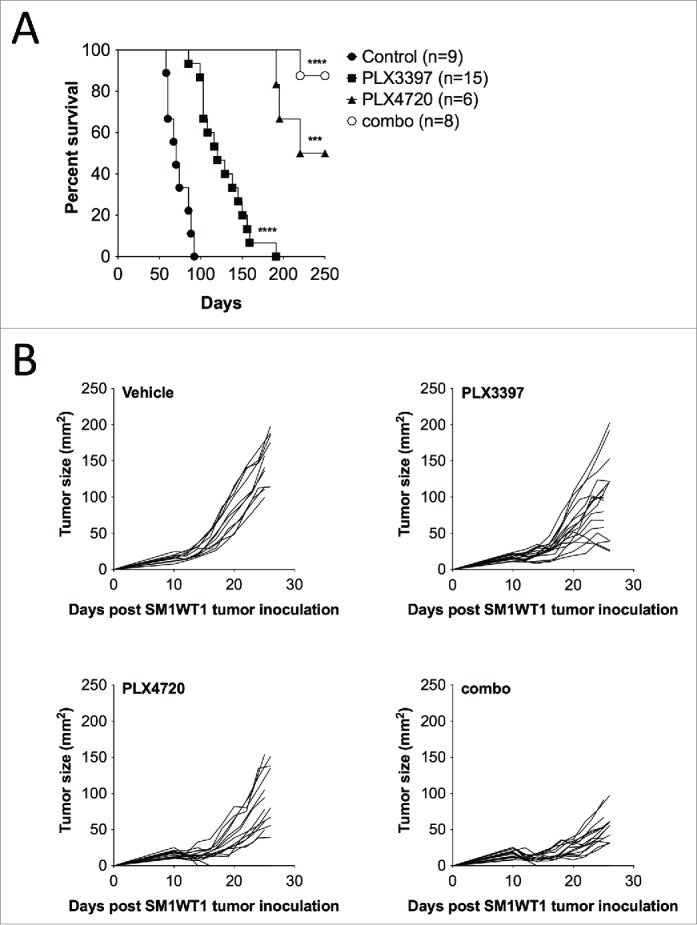
Combination of PLX4720 and PLX3397 suppresses primary BRAFV600E melanoma. (A) Groups of TBP mice were given topical 4-OHT at weaning (day 23) to initiate tumors. One week after tumor induction (day 30), mice were given control rodent diet, PLX3397 (511 mg/kg), PLX4720 (417 mg/kg), or PLX3397+PLX4720 (290 and 200 mg/kg, respectively) (combination). Mice were monitored weekly for tumor growth and measurements were taken with a digital caliper. Survival endpoint was reached at 1 cm3. No signs of toxicity were observed in the treatment cohorts. Kaplan–Meier plots were prepared and log-rank statistics were determined. (B) Groups of B6 mice (n = 7–8) were injected subcutaneously with SM1WT1 (1.0 × 106) on day 0. On day 10, tumor-bearing mice were treated with vehicle, PLX3397 (50 mg/kg), PLX4720 (20 mg/kg), PLX4720 (50 mg/kg) + PLX3397 (20 mg/kg) (combination) daily from days 10–20. Tumor growth was measured using a digital caliper, and tumor sizes are presented as individual tumor size. Data shown are pooled from two independent experiments, with each line representing an individual mouse.
Marker expression profile of intratumoral CD11b+ Gr1+ myeloid cells
Given the reported function of the CSF1/CSF1R axis in modulating macrophages and MDSCs, we then performed flow cytometry analyses on tumor-infiltrating leukocytes (TILs) isolated from established SM1WT1 tumors to identify and characterize these intratumoral CD11b+ myeloid cells. Our flow cytometry analyses of TILs showed 3 subsets of CD11b+ Gr1+ cells, which can be differentiated by their Gr1 expression level (Gr1hi, Gr1int and Gr1lo) (Fig. 2A). Consistent with their expression of CSF1R, CD11b+ Gr1int and CD11b+ Gr1lo cells were shown positive for F4/80 expression (Fig. 2B). These two subsets of cells were also shown to be Ly6G− CD11c+ and MHCII+, with Ly6Cint/lo/− for CD11b+ Gr1lo cells whereas Ly6Chi for CD11b+ Gr1int cells (Fig. 2B). In contrast, CD11b+ Gr1hi cells were CSF1R− Ly6G+ Ly6Clo/− F4/80−/lo CD11c−/lo and MHCII− (Fig. 2B). Our analyses also demonstrated that CD206, a classical M2 macrophages marker,31 was mainly found on CD11b+ Gr1int and CD11b+ Gr1lo cells.(Fig. 2B)
Figure 2.
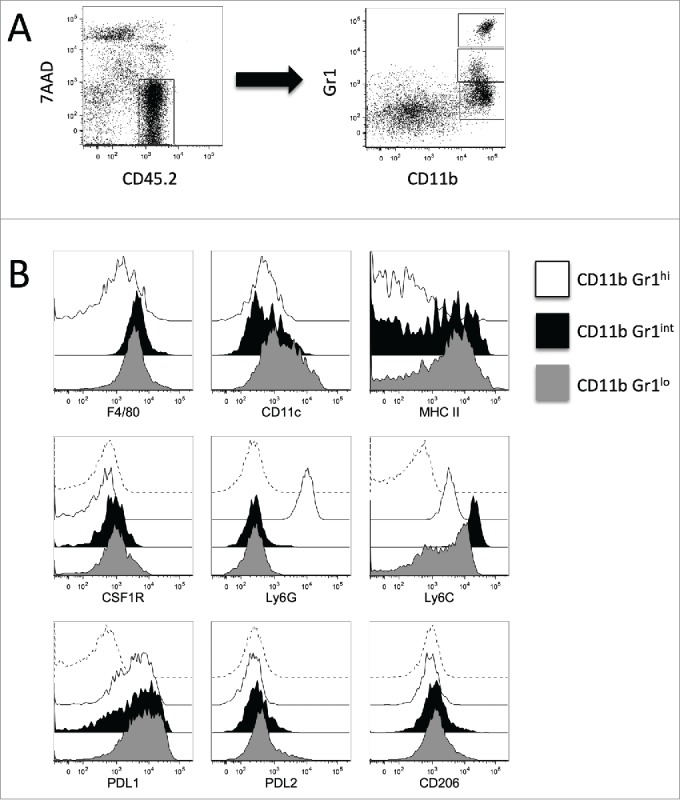
Surface markers of intratumor CD11b+ Gr1+ myeloid cells. Groups of B6 mice (n = 7–8) were injected subcutaneously with SM1WT1 (1.0 × 106) on day 0. On day 10 or 11, tumor-bearing mice were treated with vehicle for three consecutive days. Tumors were harvested 16 to 24 h after last treatment for flow cytometric analyses. (A) Representative FACS plot to demonstrate gating strategy to analyze tumor-infiltrating leukocytes. Tumor-infiltrating leukocytes were gated from CD45.2+ 7AAD− cells (left panel) and further subset into CD11b+ Gr1+ cells (right panel). (B) Representative overlaid histogram plots of total CD11b+ Gr1hi (white histogram), CD11b+ Gr1int (black histogram), and CD11b+ Gr1lo (gray histogram) for surface markers are as indicated. Isotype Ab stained tumor samples (dashed histogram), gated on total CD11b+ cells, are shown for CSF1R, Ly6G, Ly6C, PDL1, PDL2 and CD206 marker analyses.
PLX3397 reduces intratumoral CD11b+ Gr1int and CD11b+ Gr1lo cells but increases CD11b+ Gr1hi cells
To determine the changes in intratumoral CD11b+ myeloid cells in mice treated with PLX4720, PLX3397, or the combination, we performed our flow cytometry analyses on TILs overnight after 3 days of therapy. Our analyses showed a substantial reduction in TILs (CD45.2+ 7AAD−) (Fig. 2A and Fig. 3A) in PLX3397- or PLX4720+PLX3397 (hereafter termed combination)-treated mice, in comparison to vehicle- or PLX4720-treated mice. In concert with CSF1R expression levels, we found that CD11b+ Gr1int and CD11b+ Gr1lo cells were significantly reduced in the tumors of PLX3397- or combination-treated mice, in comparison to vehicle-treated mice (Fig. 3B). Interestingly, the tumors of the PLX3397- and combination-treated mice demonstrated a significant increase in CD11b+ Gr1hi cells (Fig. 3B). While PLX4720 was shown to marginally reduce CD11b+ Gr1int cells, we did not detect any changes in the frequencies or numbers of CD11b+ Gr1hi and CD11b+ Gr1lo cells in these tumors.(Fig. 3B)
Figure 3.

PLX3397 reduces CD11b+ Gr1int, and CD11b+ Gr1lo cells but increases CD11b+ Gr1hi cells. Groups of B6 mice (n = 7–8) were injected subcutaneously with SM1WT1 (1.0 × 106) on day 0. On day 10 or 11, tumor-bearing mice were treated with indicated treatments for three consecutive days. Tumors were harvested 16 to 24 h after last treatment for flow cytometric analyses. (A and B) Frequencies and (C and D) number of cells per gram (g) of tissue of (A and C) live CD45.2+ (CD45.2+ 7AAD−) cells and (B and D) CD11b+ Gr1hi, CD11b+ Gr1int, and CD11b+ Gr1lo cells (gated from live CD45.2+ cells) between vehicle, PLX3397 (50 mg/kg), PLX4720 (20 mg/kg), PLX4720+PLX3397 (combination) are shown. Data are presented as the mean ± SD with individual symbols representing individual mice. Statistical differences in frequencies and number of cells per gram of tissue of live CD45.2+ cells, CD11b+ Gr1hi, CD11b+ Gr1int and CD11b+ Gr1lo cells between vehicle, PLX3397, PLX4720 and combo were determined by an unpaired t-test (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). Data shown are representative of (A and C) 3 and (B and D) 2 independent experiments.
PLX4720 partially rescues lymphocyte-depleting effect of PLX3397
Given the modulation of CD11b+ myeloid cells in the tumors of PLX3397- and combination-treated mice, we next examined whether these changes would impact on tumor-infiltrating NK cells (NK1.1+ TCRβ−), total CD8+ T cells (CD8a+ TCRβ+), total CD4+ T cells (CD4+ TCRβ+) and Tregs (CD4+ Foxp3+) (Fig. 4A). While no significant lymphocyte number changes were detected between vehicle- and PLX4720-treaed mice, to our surprise, PLX3397 greatly reduced the number of tumor-infiltrating NK and T cells (Fig. 4C). These number changes in the immune subsets analyzed (except Tregs), were not detected using frequency parameter (Fig. 4B). Our flow cytometry analyses demonstrated that while intratumoral T cells were CSF1R− c-Kit− FLT3−, a small proportion of the tumor-infiltrating NK cells was expressing c-Kit (Fig. S2). Of note, numbers of NK cells and total CD4+ T cells were also reduced in the tumors of combination-treated mice, compared to vehicle-treated mice (Fig. 4C). Interestingly, we observed substantial increases in NK cells, total CD4+ T and CD8+ T cell numbers, but not Tregs, in combination-treated mice compared with mice treated with PLX3397 alone (Fig. 4C), indicating a lymphocyte-rescue effect of PLX4720. We next examined the cytokine production of these intratumoral T cells. In concert with the frequencies of CD4+ Foxp3+ cells between PLX4720- and combination-treated mice (Fig. 4B), the IL-10 production of CD4+ T cells was reduced (Fig. 5A). The reduction of Foxp3 frequency among CD4+ T cells isolated from tumors of combination-treated mice was accompanied by an increase in IFNγ+ CD4+ T cells, when compared to vehicle-treated mice (Fig. 5A). The increase of IFNγ-production in CD4+ T cells was not observed in the CD8+ T cell population, possibly due to the incomplete rescue following PLX3397 co-treatment (Fig. 5B). No significant changes in the frequencies of IL-2- or TNF-producing CD4+ and CD8+ T cells were noted between vehicle-treated and PLX-treated mice (Fig. 5A-B). Thus overall, the suppression of SM1WT1 melanomas was associated with reduced numbers of CD11b+ Gr1int and CD11b+ Gr1lo M2-like TAMs, and CD4+ Foxp3+ Tregs; but increases in CD11b+ Gr1hi cells, T cells, and NK cells.
Figure 4.

PLX3397 reduces intratumor NK cells and T cells. Groups of B6 mice (n = 7–8) were injected subcutaneously with SM1WT1 (1.0 × 106) on day 0. On day 10 or 11, tumor-bearing mice were treated with indicated treatments for three consecutive days. Tumors were harvested 16 to 24 h after last treatment for flow cytometric analyses. (A) Representative FACS plots for indicated immune subsets, gated from live CD45.2+ cells. (B) Frequencies and (C) number of cells per gram (g) of tissue of indicated immune subsets between vehicle, PLX3397 (50 mg/kg), PLX4720 (20 mg/kg), PLX4720+PLX3397 (combination) are shown. Data are presented as the mean ± SD with individual symbols representing individual mice. Statistical differences in frequencies and number of cells per gram of tissue of indicated immune subsets between vehicle, PLX3397, PLX4720 and combination were determined by an unpaired t-test (*p < 0.05; **p < 0.01; ****p < 0.0001). Data shown are representative of three (NK cells, CD8+ T cells, and CD4+ T cells) or one (Foxp3+ Tregs) independent experiment(s).
Figure 5.

Intracellular cytokine profile of intratumor T cells. Groups of B6 mice (n = 7–8) were injected subcutaneously with SM1WT1 (1.0 ×106on day 0. On day 11, tumor-bearing mice were treated with indicated treatments for three consecutive days. Tumors were harvested 16 to 24 h after last treatment for flow cytometric analyses. Frequencies of IL-10-, IFNγ-, IL-2-, or TNF-expressing of (A) CD4+ T cells and (B) CD8+ T cells between vehicle, PLX3397 (50 mg/kg), PLX4720 (20 mg/kg), PLX4720+PLX3397 (combination) are shown. Data are presented as the mean ± SD with individual symbols representing individual mice. Statistical differences in frequencies and number of cells per gram of tissue of indicated immune subsets between vehicle, PLX3397, PLX4720, and combination were determined by an unpaired t-test (*p < 0.05; **p < 0.01).
PD1/PDL1 blockade enhances therapeutic effect of PLX4720 and PLX3397
Despite the significant reduction of tumor immune suppressor cells (M2 macrophages and Tregs), we observed that combination-treated mice eventually relapsed following the cessation of therapy. We reasoned that the incomplete tumor clearance in combination-treated mice was driven by the expansion of CD11b+ Gr1hi cells, and remnants of CD11b+ Gr1int and CD11b+ Gr1lo cells. Indeed, we found that a co-administration of anti-Ly6G mAb (clone 1A8) further enhanced the antitumor effect of PLX4720 and PLX3397 therapy (Fig. 6), indicating a role of the expanded Ly6G+ CD11b+ Gr1hi cells in suppressing PLX3397-mediated antitumor immunity. While the frequencies of PD1-expressing CD4+ and CD8+ T cells were not modulated by PLX4720 and/or PLX3397 (Fig. 7B), we found striking increases in expression of PDL1 and PDL2 on all intratumoral CD11b+ Gr1+ cells isolated from the combination-treated mice (Figs. 2B and 7A). These findings prompted us to assess whether the PD1/PDL1 pathway might be targeted along with PLX4720 and PLX3397 combination therapy. Consistent with our speculation, by using PD1 and PDL1 blocking antibodies, we showed that the SM1WT1 tumor in combination-treated mice was further suppressed when the PD1/PDL1 signaling axis was blocked.(Fig. 7C)
Figure 6.
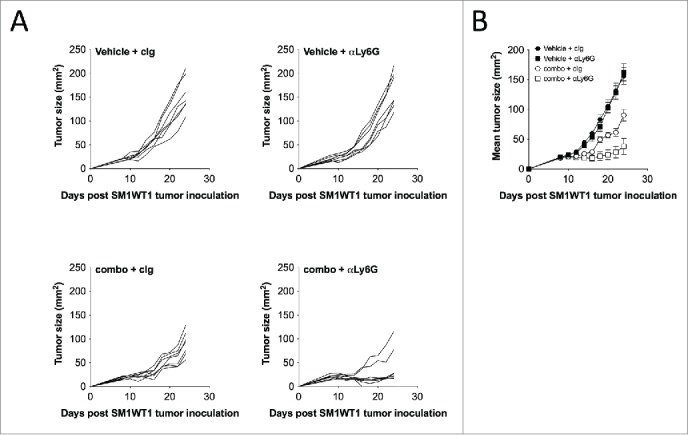
Depletion of Ly6G+ cells enhances PLX4720 and PLX3397 combination therapy effect. Groups of B6 mice (n = 7–8) were injected subcutaneously with SM1WT1 (1.0 × 106) on day 0. On day 11, tumor-bearing mice were treated with vehicle or PLX4720 (20 mg/kg) +PLX3397 (50 mg/kg) (combination) daily from days 11–21. Vehicle- or combination-treated mice were then treated with 500 μg of control Ig or anti-Ly6G on day 14, 17 and 20. Tumor growth was measured using a digital caliper, and tumor sizes are (A) presented as individual tumor size or (B) presented as mean ± SEM. Statistical differences in tumor sizes between treated mice were determined by an unpaired t-test (Day 18; combination + cIg vs. combination + anti-Ly6G p = 0.0081) (Day 20; combination + cIg vs. combination + anti-Ly6G p = 0.0049) (Day 22; combination + cIg vs. combination + anti-Ly6G p = 0.0171) (Day 24; combination + cIg vs. combination + anti-Ly6G p = 0.0091).
Figure 7.
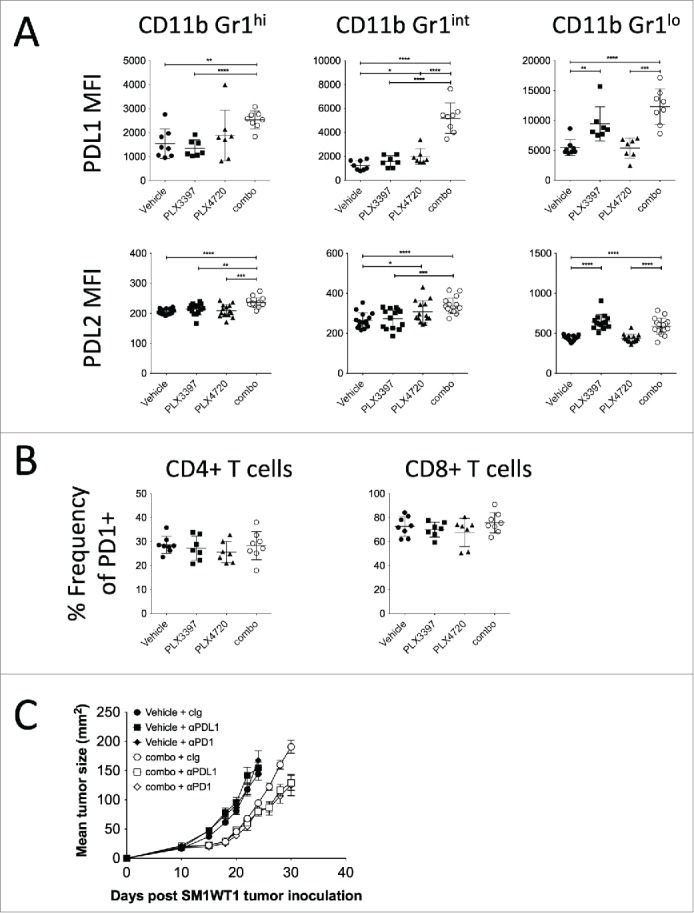
PD1/PDL1 axis blockade enhances PLX4720 and PLX3397 combination therapy effect. (A and B) Groups of B6 mice (n = 7–8) were injected subcutaneously with SM1WT1 (1.0 ×106) on day 0. On day 10 or 11, tumor-bearing mice were treated with indicated treatments for three consecutive days. Tumors were harvested 16 to 24 h after last treatment for flow cytometric analyses. (A) PDL1 MFI and PDL2 MFI of CD11b+ Gr1hi, CD11b+ Gr1int, and CD11b+ Gr1lo cells between vehicle, PLX3397 (50 mg/kg), PLX4720 (20 mg/kg), PLX4720+PLX3397 (combination) are shown. Data are presented as the mean ± SD with individual symbols representing individual mice. Statistical differences in PDL1 MFI and PDL2 MFI of indicated immune subsets between vehicle, PLX3397, PLX4720 and combo were determined by an unpaired t-test (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). Data shown are representative of one (PDL1 MFI) or two (PDL2 MFI) independent experiments. (B) Frequencies of PD1-expressing CD4+ and CD8+ T cells between vehicle, PLX3397 (50 mg/kg), PLX4720 (20 mg/kg), PLX4720+PLX3397 (combination) are shown. Data are presented as the mean ± SD with individual symbols representing individual mice. Data shown are representative of one independent experiment. (C) Groups of B6 mice (n = 6–8) were injected subcutaneously with SM1WT1 (1.0 × 106) on day 0. On day 11, tumor-bearing mice were treated with vehicle or PLX4720 (20 mg/kg) +PLX3397 (50 mg/kg) (combination) daily from days 11–16. Vehicle-treated mice were then treated with 250 μg of control Ig (cIg), anti-PD1, or anti-PDL1 on day 16 and 20. Combination-treated mice were then treated with 250 μg of control Ig, anti-PD1, or anti-PDL1 on day 16, 20, 24 and 28. Tumor growth was measured using a digital caliper, and tumor sizes are presented as mean ± SEM. Statistical differences in tumor sizes between treated mice were determined by an unpaired t test (Day 26; combination + cIg vs. combination + anti-PDL1 p = 0.0032) (Day 26; combination + cIg vs. combination + anti-PD1 p = 0.0102) (Day 28; combination + cIg vs. combination + anti-PDL1 p = 0.0035) (Day 28; combination + cIg vs. combination + anti-PD1 p = 0.0021) (Day 30; combination + cIg vs. combination + anti-PDL1 p = 0.0029) (Day 30; combination + cIg vs. combination + anti-PD1 p = 0.0069).
Discussion
Development of resistance following oncogene inhibition is common in the clinic, where tumor cells acquire or develop pro-survival mechanisms to maintain their growth.5-9 While BRAF inhibitors have displayed a rapid and dramatic response rate in BRAF-mutated melanoma patients, most of these patients eventually relapse.1-4 In contrast, immunotherapy has been shown to induce long-term response in patients.32,33 In light of these findings, a combination of an immunotherapeutic agent and a BRAF inhibitor has great potential to overcome oncogene inhibition resistance. Indeed, we and others have previously demonstrated superior antitumor effects of a BRAF inhibitor in combination with T cell-based immunotherapy agents in suppressing the growth mouse BRAFV600E melanomas.13,10,14,15 Here, we demonstrated an enhanced anti-melanoma effect by a combination of PLX4720 and CSF1R inhibitor, PLX3397, providing pre-clinical evidence for a similar therapeutic combination in a Phase I trial for BRAF-mutated advanced stage melanoma (NCT01826448).
Similar to published studies,20-22,27,28 PLX3397 was shown to reduce SM1WT1 intratumoral CSF1R-expressing CD11b+ Gr1int and CD11b+ Gr1lo M2-like TAMs, and this was accompanied a large expansion of CD11b+ Gr1hi MDSC-like cells. However, we also observed a sizeable reduction in intratumor NK cells, T cells, and Tregs in PLX3397-treated mice. While c-Kit is known to be involved in regulating thymic T cell development,34 its expression and function in intratumor T cells has not been well characterized. Alternatively, the presence of c-Kit+ intratumor NK cells in B16F10 melanoma has been previously reported.35 While the reduction of intratumoral NK cell number seen in PLX3397-treated mice could be explained by NK cell c-Kit expression, the mechanism of PLX3397 in reducing intratumoral T cells remains to be discerned. Given the absence of CSF1R/c-Kit/FLT3 expression in intratumoral T cells, it is possible that this T cell-depletion is mediated via the decreased myeloid component in the SM1WT1 tumors. The co-depletion of effector lymphocytes and M2-like TAMs, with an expansion of CD11b+ Gr1hi MDSC-like cells, might result in the relatively minor antitumor effect of PLX3397-treated mice compared to vehicle-treated mice.
We and others showed that BRAF inhibitors possess immunostimulating activities on T cells and NK cells (BRAF wildtype), mediated by the paradoxical activation of MAPK pathway.36,37 During completion of our study, Mok and colleagues reported a combination of PLX4032 and PLX3397 that demonstrated superior antitumor effect in suppressing the growth of SM1 melanoma.38 Of note, SM1WT1 was derived from SM1 melanoma, but is directly more easily transplantable.14 Both SM1- and SM1WT1-bearing mice eventually relapse upon termination of combination therapy.38 Our data showed that the relapse of SM1WT1 melanomas was partly driven by the PD1/PDL1 axis. This observation concurred with the expansion of PDL1/PDL2-expressing CD11b+ Gr1hi MDSC-like cells and presence of the remaining PDL1/PDL2-expressing CD11b+ Gr1int and CD11b+ Gr1lo TAMs. Although the presence of CD11b+ Gr1hi cells was not being assessed in Mok et al. PLX4032 and PLX3397 combination therapy study,38 they have previously reported that PLX3397 therapy alone increases the frequency of intratumor CD11b+ Gr1hi Ly6G+ cells22 and they recently concluded that the direct effect of TAMs or pro-survival cytokines produced by TAMs (e.g., TNF) did not confer resistance to Braf inhibitor.38 Together with our study, we reason that the expansion of these intratumor CD11b+ Gr1hi Ly6G+ MDSC-like cells is a driver of tumor relapse. While we found that intratumor NK cells and T cells were depleted in PLX3397-treated mice, the numbers of intratumor NK cells and effector T cells, but not Tregs, were rescued by co-administering PLX4720. Lymphocyte-depleting activity of PLX3397 was not reported in Mok et al., but rather the contrary. This may be due to the basally lower T cell infiltrates found in SM1 tumors, compared with SM1WT1 tumors, or the greater effect of combination therapy [including a high dose of PLX4032 (100 mg/kg)] observed at the time of flow analysis.38 Therefore, the superior antitumor effect of this combination therapy is likely driven by the depletion of immune suppressor cells (M2 macrophages and Tregs) caused by PLX3397, together with the recovery of NK cells and effector T cells enabled by PLX4720. We also demonstrated for the first time the combination activity in the de novo melanoma prone BRAFV600E TBP mutant strain of mice. This is important because here the melanomas develop from normal tissue in the host. Our study has provided a more complete understanding of combining BRAF and CSF1R inhibitors in pre-clinical mouse models of melanoma and in treating melanoma patients with lymphocyte infiltrates.39,40
Materials and methods
Mice
C57BL/6 wild-type (WT) male mice were purchased from the ARC Animal Resources Center. C57BL/6J Tyr::CreERT2 BrafCA Ptenfl/fl (TBP) were maintained as previously described.41 Groups of 4 to 15 (6 to 12 weeks) mice per experiment were used for experimental tumor assays, to ensure adequate power to detect biological differences. All experiments were approved by the QIMR Berghofer Medical Research Institute Animal Ethics Committee and the Yale Institutional Animal Care and Use Committee.
Tumor cell lines
The C57BL/6 SM1WT1 and SM1WT1-LWT1 were maintained as previously described.37,14 For in vivo experiments, the indicated cell numbers were subcutaneously or intravenously injected into mice in a 100 or 200 μL volume, respectively.
Antibodies and reagents
PLX4720 (a BRAF inhibitor) and PLX3397 (a small molecule receptor tyrosine kinase inhibitor for KIT/CSF1R/FLT3) were obtained from Plexxikon Inc.. For in vivo studies of SM1WT1 and SM1WT1-LWT1, PLX4720 was dissolved in DMSO (Calbiochem), followed by PBS (a final volume of 50 μL), which was then injected daily i.p. into mice at 20 mg/kg. An equal volume of DMSO and PBS mixture was used as Vehicle. PLX3397 was dissolved in DMSO, and then further diluted in an aqueous mixture of 0.5 % hydroxypropyl methyl cellulose (HPMC) (Sigma) and 1 % polysorbate 80 (PS80) (Fluka). The PLX3397 drug suspension was administered by daily oral gavage into mice at 50 mg/kg. An equal volume of DMSO and aqueous mixture was used as Vehicle. Treatment of mice in the TBP model was performed by feeding mice chow ad libitum either lacking drug (control) or compounded to contain PLX4720, PLX3397, or both. PLX4720 is a sister compound to vemurafenib with preferred properties for mouse studies.42 The doses of drug were previously determined to achieve serum levels similar Cmax seen in human clinical trials.3,43 Purified anti-mouse PD1 mAb (RMP1-14; BioXCell), anti-mouse PDL1 (10F.9G2; BioXCell), anti-mouse Ly6G (1A8; BioXCell) and control Ig (2A3; BioXCell or 1-1; Leinco Technologies) were used in the schedule and at the doses indicated.
In vivo treatments
1 × 106 SM1WT1 tumor cells were subcutaneously injected into mice in a 100 μL volume (day 0) and treatments given as indicated in the figure legends. Tumor growth was measured using a digital caliper, and tumor sizes are presented as mean tumor growth or individual tumor growth profile. 5.0 × 105 or 7.5 × 105 SM1WT1-LWT1 tumor cells were intravenously injected into mice in a 200 μL volume (day 0) and treatments given as indicated in the figure legends. On day 14, mice were harvested and lung metastases were quantified as previously described.37 For flow cytometry analyses of TILs, mice with established SM1WT1 tumor (day 10 or 11) were treated with the indicated reagents daily for three consecutive days and immune cells were isolated 16 to 24 h post last treatment (day 13 or 14). For the TBP model, localized tumors were induced in mice immediately on postnatal day 23 as previously described44 and treatment was initiated one week after tumor induction.
Flow cytometry analysis
Tumors were harvested from mice that had been treated with Vehicle, PLX4720, and/or PLX3397, and processed for flow cytometry analysis as previously described.45 For surface staining, TIL suspensions were stained with eFluor780 anti-CD45.2 (104; eBioscience), eFluor450 or BV605 anti-CD4 (RM4-5; eBioscience and Biolegend), PE-Cy7 or BV421 anti-CD8a (53-6.7; eBioscience or Biolegend), FITC- or PE-anti-TCRβ (H57-597; eBioscience), APC- or PE-Cy7 anti-NK1.1 (PK136; eBioscience), PE-Cy7 anti-CD11b (M1/70; eBioscience), eFluor450 anti-Gr1 (RB6-8C5; eBioscience), APC-anti-CD115 (AFS98; eBioscience), FITC-anti-Ly6G (1A8; BD Pharmingen), PE-Cy7 anti-Ly6C (AL-21; BD Pharmingen), PE-anti-F4/80 (BM8; eBioscience), FITC-anti-CD11c (N418, eBioscience), PE-anti-MHC II (M5/114.15.2; eBioscience), APC-anti-PDL1 (10F.9G2; Biolegend), FITC-anti-PDL2 (122; eBioscience), Alexa Fluor 467-anti-CD206 (C068C2; Biolegend), FITC anti-PD1 (J43; eBioscience), PE-anti-CD135 (A2F10; Biolegend), APC-anti-CD117 (2B8; Biolegend), and respective isotype antibodies in the presence of anti-CD16/32 (2.4G2). BD Liquid Counting Beads (Cat. No. 335925) were added to sample for cell number analyses. 7AAD (Biolegend) was used to exclude dead cells. For intracellular transcription factor staining, surface-stained cells were fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience), according to the manufacturer's protocol, and stained using FITC-anti-Foxp3 (FJK-16s, eBioscience). For intracellular staining of IFNγ, IL-10, IL-2 and TNF, cells were stimulated in vitro with Cell Stimulation Cocktail (plus protein transport inhibitors) (500X) (eBioscience; Cat No. 00-4975-93) for 4 h, and then surface stained as aforementioned. Surface-stained cells were then fixed and permeabilized using BD Cytofix/Cytoperm (BD Biosciences) according to the manufacturer's protocol, and stained with PE-anti-IFNγ (XMG1.2; eBioscience), FITC-anti-IL-10 (JES5-16E3; eBioscience), APC-anti-IL-2 (JES6-5H4; eBioscience), and BV605-anti-TNF (MP6-XT22; Biolegend), and respective isotype antibodies. Cells were acquired on the BD FACSCANTO II (BD Biosciences) and analysis was carried out using FlowJo (Tree Star).
Statistics
Statistical analyses were carried out using Graph Pad Prism software. Significant differences in SM1WT1 tumor growth and SM1WT1-LWT1 lung metastases were determined by an unpaired t-test. Significant differences in mouse survival in the TBP cohorts were determined using Kaplan–Meier plots and log-rank statistics. Based on ethical considerations related to maximum tumor size in mice, an endpoint of 1 cm3 tumor volume was used to determine survival in these TBP cohorts. Significant differences in cell subsets were determined by an unpaired t-test. Values of p < 0.05 were considered significant.
Supplementary Material
Disclosure of potential conflicts of interest
Mark Smyth declares a scientific research agreement grant with Bristol Myers Squibb. Mark Smyth is a consultant for Kymab, F-star, and AMGEN. Gideon Bollag of is an employee and stockholder of Plexxikon, Inc.. The remaining authors of this manuscript have declared that no conflict of interest exists.
Acknowledgments
The authors wish to thank Liam Town, Kate Elder and Joanne Sutton for breeding, genotyping and maintenance and care of the mice used in this study.
Funding
S.N. and M.J.S were supported by a National Health and Medical Research Council of Australia (NH&MRC) Australia Fellowship (628623) and Senior Principal Research Fellowship (1078671), and a QIMR Berghofer Ride to Conquer Cancer Grant.
References
- 1.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M et al.. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Eng J Med 2011; 364:2507; PMID:21639808; http://dx.doi.org/22608338 10.1056/NEJMoa1103782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, Infante JR, Millward M, Pavlick AC et al.. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet 2012; 379:1893; PMID:22608338; http://dx.doi.org/ 10.1016/S0140-6736(12)60398-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O'Dwyer PJ, Lee RJ, Grippo JF, Nolop K et al.. Inhibition of mutated, activated BRAF in metastatic melanoma. N Eng J Med 2010; 363:809; PMID:20818844; http://dx.doi.org/24108405 10.1056/NEJMoa1002011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT et al.. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Eng J Med 2012; 366:707; PMID:22356324; http://dx.doi.org/24108405 10.1056/NEJMoa1112302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartsough E, Shao Y, Aplin AE. Resistance to RAF inhibitors revisited. J Invest Dermatol 2014; 134:319; PMID:24108405; http://dx.doi.org/ 10.1038/jid.2013.358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nature Reviews. Cancer 2014; 14:455; PMID:24957944; http://dx.doi.org/ 10.1038/nrc3760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med 2013; 19:1401; PMID:24202393; http://dx.doi.org/ 10.1038/nm.3392 [DOI] [PubMed] [Google Scholar]
- 8.Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, Kong X, Bosenberg MC, Wiesner T, Rosen N et al.. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015; 520:368; PMID:25807485; http://dx.doi.org/ 10.1038/nature14336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM et al.. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014; 508:118; PMID:24670642; http://dx.doi.org/ 10.1038/nature13121 [DOI] [PubMed] [Google Scholar]
- 10.Cooper ZA, Juneja VR, Sage PT, Frederick DT, Piris A, Mitra D, Lo JA, Hodi FS, Freeman GJ, Bosenberg MW et al.. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol Res 2014; 2:643; PMID:24903021; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C et al.. BRAF Inhibition Is Associated with Enhanced Melanoma Antigen Expression and a More Favorable Tumor Microenvironment in Patients with Metastatic Melanoma. Clin Cancer Res 2013; 19:1225; PMID:23307859; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith MP, Sanchez-Laorden B, O'Brien K, Brunton H, Ferguson J, Young H, Dhomen N, Flaherty KT, Frederick DT, Cooper ZA et al.. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFalpha. Cancer Discov 2014; 4:1214; PMID:25256614; http://dx.doi.org/ 10.1158/2159-8290.CD-13-1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Acquavella N, Clever D, Yu Z, Roelke-Parker M, Palmer DC, Xi L, Pflicke H, Ji Y, Gros A, Hanada K et al.. Type I cytokines synergize with oncogene inhibition to induce tumor growth arrest. Cancer Immunol Res 2015; 3:37; PMID:25358764; http://dx.doi.org/ 10.1158/2326-6066.CIR-14-0122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, Haynes NM, Kinross K, Yagita H, Koya RC et al.. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J Clin Invest 2013; 123:1371; PMID:23454771; http://dx.doi.org/ 10.1172/JCI66236 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, Minasyan A, Graham NA, Graeber TG, Chodon T et al.. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res 2012; 72:3928; PMID:22693252; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162; PMID:19197294; http://dx.doi.org/ 10.1038/nri2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harbor Perspect Biol 2014; 6:a021857; PMID:24890514; http://dx.doi.org/25753580 10.1101/cshperspect.a021857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biswas SK, Allavena P, Mantovani A. Tumor-associated macrophages: functional diversity, clinical significance, and open questions. Seminars Immunopathol 2013; 35:585; PMID:23657835; http://dx.doi.org/25753580 10.1007/s00281-013-0367-7 [DOI] [PubMed] [Google Scholar]
- 19.Mantovani A, Allavena P. The interaction of anticancer therapies with tumor-associated macrophages. J Exp Med 2015; 212:435; PMID:25753580; http://dx.doi.org/ 10.1084/jem.20150295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA et al.. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 2011; 1:54; PMID:22039576; http://dx.doi.org/ 10.1158/2159-8274.CD-10-0028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D et al.. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 2013; 73:1128; PMID:23221383; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, Graeber TG, West BL, Bollag G, Ribas A. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res 2014; 74:153; PMID:24247719; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V et al.. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 2013; 19:1264; PMID:24056773; http://dx.doi.org/ 10.1038/nm.3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I et al.. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014; 25:846; PMID:24898549; http://dx.doi.org/ 10.1016/j.ccr.2014.05.016 [DOI] [PubMed] [Google Scholar]
- 25.Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, Daniel D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8 T cells. Oncoimmunology 2013; 2:e26968; PMID:24498562; http://dx.doi.org/ 10.4161/onci.26968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Sluis TC, Sluijter M, van Duikeren S, West BL, Melief CJ, Arens R, van der Burg SH, van Hall T. Therapeutic peptide vaccine-induced CD8 T cells strongly modulate intratumoral macrophages required for tumor regression. Cancer Immunol Res 2015; 3(9):1042-51; PMID:25888578; http://dx.doi.org/ 10.1158/2326-6066. [DOI] [PubMed] [Google Scholar]
- 27.Xu J, Escamilla J, Mok S, David J, Priceman S, West B, Bollag G, McBride W, Wu L. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res 2013; 73:2782; PMID:23418320; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-3981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 2014; 74:5057; PMID:25082815; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ho PC, Meeth KM, Tsui YC, Srivastava B, Bosenberg MW, Kaech SM. Immune-based antitumor effects of BRAF inhibitors rely on signaling by CD40L and IFNgamma. Cancer Res 2014; 74:3205; PMID:24736544; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-3461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steinberg SM, Zhang P, Malik BT, Boni A, Shabaneh TB, Byrne KT, Mullins DW, Brinckerhoff CE, Ernstoff MS, Bosenberg MW, Turk MJ. BRAF inhibition alleviates immune suppression in murine autochthonous melanoma. Cancer Immunol Res 2014; 2:1044; PMID:25183499; http://dx.doi.org/ 10.1158/2326-6066.CIR-14-0074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends in Immunol 2002; 23:549; PMID:12401408; http://dx.doi.org/15766667 10.1016/S1471-4906(02)02302-5 [DOI] [PubMed] [Google Scholar]
- 32.June CH, Riddell SR, Schumacher TN. Adoptive cellular therapy: a race to the finish line. Science Trans Med 2015; 7:280ps7; PMID:25810311; http://dx.doi.org/15766667 10.1126/scitranslmed.aaa3643 [DOI] [PubMed] [Google Scholar]
- 33.Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annual Rev Med 2014; 65:185; PMID:24188664; http://dx.doi.org/15766667 10.1146/annurev-med-092012-112807 [DOI] [PubMed] [Google Scholar]
- 34.Pelayo R, Welner R, Perry SS, Huang J, Baba Y, Yokota T, Kincade PW. Lymphoid progenitors and primary routes to becoming cells of the immune system. Curr Opin Immunol 2005; 17:100; PMID:15766667; http://dx.doi.org/ 10.1016/j.coi.2005.01.012 [DOI] [PubMed] [Google Scholar]
- 35.Terme M, Ullrich E, Aymeric L, Meinhardt K, Coudert JD, Desbois M, Ghiringhelli F, Viaud S, Ryffel B, Yagita H et al.. Cancer-induced immunosuppression: IL-18-elicited immunoablative NK cells. Cancer Res 2012; 72:2757; PMID:22427351; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3379 [DOI] [PubMed] [Google Scholar]
- 36.Callahan MK, Masters G, Pratilas CA, Ariyan C, Katz J, Kitano S, Russell V, Gordon RA, Vyas S, Yuan J et al.. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol Res 2014; 2:70; PMID:24416731; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferrari de Andrade L, Ngiow SF, Stannard K, Rusakiewicz S, Kalimutho M, Khanna KK, Tey SK, Takeda K, Zitvogel L, Martinet L et al.. Natural killer cells are essential for the ability of BRAF inhibitors to control BRAFV600E-mutant metastatic melanoma. Cancer Res 2014; 74:7298; PMID:25351955; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-1339 [DOI] [PubMed] [Google Scholar]
- 38.Mok S, Tsoi J, Koya RC, Hu-Lieskovan S, West BL, Bollag G, Graeber TG, Ribas A. Inhibition of colony stimulating factor-1 receptor improves antitumor efficacy of BRAF inhibition. BMC Cancer 2015; 15:356; PMID:25939769; http://dx.doi.org/ 10.1186/s12885-015-1377-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, Chen L. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Science Translational Med 2012; 4:127ra37; PMID:22461641; http://dx.doi.org/25977340 10.1126/scitranslmed.3003689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Research 2015; 75:2139; PMID:25977340; http://dx.doi.org/ 10.1158/0008-5472.CAN-15-0255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr, You MJ, DePinho RA, McMahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nature Genetics 2009; 41:544; PMID:19282848; http://dx.doi.org/ 10.1038/ng.356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass NK et al.. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA 2008; 105:3041; PMID:18287029; http://dx.doi.org/ 10.1073/pnas.0711741105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tap WD, Wainberg ZA, Anthony SP, Ibrahim PN, Zhang C, Healey JH, Chmielowski B, Staddon AP, Cohn AL, Shapiro GI et al.. Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. N Eng J Med 2015; 373:428; PMID:26222558; http://dx.doi.org/22172720 10.1056/NEJMoa1411366 [DOI] [PubMed] [Google Scholar]
- 44.Damsky WE, Curley DP, Santhanakrishnan M, Rosenbaum LE, Platt JT, Gould Rothberg BE, Taketo MM, Dankort D, Rimm DL, McMahon M, Bosenberg M. β-catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell 2011; 20:741; PMID:22172720; http://dx.doi.org/ 10.1016/j.ccr.2011.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MWL, Smyth MJ. Anti-TIM3 Antibody Promotes T Cell IFN-γ–Mediated Antitumor Immunity and Suppresses Established Tumors. Cancer Res 2011; 71:3540; PMID:21430066; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-0096 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.