

ABSTRACT
We have armed a tumor-selective oncolytic vaccinia virus (vvDD) with the chemokine (CK) CXCL11, in order to enhance its ability to attract CXCR3+ antitumor CTLs and possibly NK cells to the tumor microenvironment (TME) and improve its therapeutic efficacy. As expected, vvDD-CXCL11 attracted high numbers of tumor-specific T cells to the TME in a murine AB12 mesothelioma model. Intratumoral virus-directed CXCL11 expression enhanced local numbers of CD8+ CTLs and levels of granzyme B, while reducing expression of several suppressive molecules, TGF-β, COX2, and CCL22 in the TME. Unexpectedly, we observed that vvDD-CXCL11, but not parental vvDD, induced a systemic increase in tumor-specific IFNγ-producing CD8+ T cells in the spleen and other lymph organs, indicating the induction of systemic antitumor immunity. This effect was associated with enhanced therapeutic efficacy and a survival benefit in tumor-bearing mice treated with vvDD-CXCL11, mediated by CD8+ T cells and IFNγ, but not CD4+ T cells. These results demonstrate that intratumoral expression of CXCL11, in addition to promoting local trafficking of T cells and to a lesser extent NK cells, has a novel function as a factor eliciting systemic immunity to cancer-associated antigens. Our data provide a rationale for expressing CXCL11 to enhance the therapeutic efficacy of oncolytic viruses (OVs) and cancer vaccines.
KEYWORDS: Antitumor immunity, CD8+ T cells, chemokine, IFNγ, oncolytic virus, therapeutic efficacy, tumor microenvironment
Abbreviations
- Ab
antibody
- APC
antigen-presenting cell
- CIK
cytokine-induced killer cells
- CK
chemokine
- CM
culture medium
- COX-2
cyclooxygenase-2
- CTL
cytotoxic T lymphocyte
- DC
dendritic cells
- DsRed
the Discosoma red fluorescent protein
- FBS
fetal bovine serum
- HPRT
hypoxanthine guanine phosphoribosyl transferase
- i.p.
intraperitoneal
- IFNγ
interferon gamma
- MDSC
myeloid-derived suppressor cells
- MMC
mitomycin C
- OV
oncolytic virus
- PBMC
peripheral blood mononuclear cells
- PD-1
Programmed cell death protein 1
- RQ
relative quantity
- TAA
tumor-associated antigen
- TME
tumor microenvironment
- VV
vaccinia virus
- vvDD
a tumor-selective (tk and vgf) double deleted vaccinia virus
- WR
Western Reserve strain (of vaccinia virus).
Introduction
Cancer immunotherapy has become a promising therapeutic modality in the last few years.1-4 Tumor-specific T cell responses can be successfully induced by cancer vaccines including those involving viral vectors, immunostimulatory agents,5-7 immune checkpoint blockade4 or by adoptive T cell transfer.1 Yet, despite all the efforts and rational approaches of immunotherapy, only a minority of human patients benefit from these treatments in clinical trials. Studies have shown that tumor infiltration by T cells, especially Th1 and CD8+ T cells, is positively associated with overall patient survival in many types of cancer.8-11 However, the TME is highly immunosuppressive, which not only restricts the effects of tumor-specific T cells on tumor destruction, but also impacts the infiltration efficiency of the tumor-specific T cells.12,13 Recent studies have suggested that OVs may favorably modulate the TME. OVs can selectively infect, replicate, and kill cancer cells and associated stromal cells while sparing normal tissues. Viral mediated cell death results in the release of potent danger signals (DAMPs and PAMPs) and cross presentation of tumor-associated antigens to dendritic cells (DCs) and other antigen presenting cells, resulting in antitumor innate and adaptive immunity.14-17 Thus, viruses may function not only as oncolytic agents, but also potent modulators of the immune TME.
CKs are immune cell attractants facilitating inter-cellular communication and are involved in the induction and effector phases of immunity against infections and cancer.18 The induction of immune responses in the draining lymph nodes has been shown to critically depend on CCL19 and CCL21 (ligands for CCR7 expressed on mature DCs, and naive and memory T cells), and the CCR5-CCL5 interaction, mediating intranodal communication between activated lymphocytes and DCs.19,20 CXCR3 ligands, including CXCL9, CXCL10 and CXCL11 (I-TAC), have potent antitumor activity through attraction of cytotoxic T lymphocytes.21-24 We reasoned that efficient, intratumoral expression of a CXCR3 ligand, using a tumor-selective replicating vaccinia virus (VV), may function to enhance the antitumor response. While CXCL11 is generally recognized as an important factor promoting the entry of type-1 effector cells (CTLs, Th1 and NK cells) into inflamed or tumor tissues,24-27 no studies so far have addressed its potential role in combination with an oncolytic virus. We have previously reported on interesting findings with VV expressing CCL5 in which CCL5 was used only as a chemoattractant.28 It is important to point out that CXCL11 may attract only activated T (Th1) and NK cells, while CCL5 could attract both naive and activated immune cells including T, NK, monocytes, and immature DCs. From point-of-view of immunotherapy, we believe that CXCL11 is a superior CK when compared to CCL5.
We hypothesized that oncolytic VV armed with certain CKs could enhance the modulation of the TME directly, promote the trafficking of antitumor T cells into tumor tissues, and elicit antitumor effects. In this report, we have explored a CXCL11-armed oncolytic poxvirus to treat tumor models of mesothelioma as well as colorectal cancer. We observed that in addition to its ability to promote the effector phase of anticancer immunity, CXCL11-armed virus also uniquely promotes the induction phase of immunity, acting as a potent therapeutic vaccine.
Results
CXCL11 expression did not impact virus infection and replication in vitro
We constructed a new oncolytic VV expressing murine CXCL11, in the backbone with deletion of viral genes encoding thymidine kinase and vaccinia growth factor for tumor selectivity, called vvDD-CXCL11 (Fig. S1A). To confirm that the virus displays normal functions as an oncolytic virus and that it expresses the CK CXCL11, we infected two murine cancer cell lines, mesothelioma AB12-luc cells and MC38-luc colon cancer cells, with vvDD-CXCL11 or parental virus (vvDD) at an MOI of 1, and then measured the replication efficiency by both plaque assays and viral marker gene expression (A34R). We measured CXCL11 expression by both RT-qPCR and ELISA assays. Replication efficiency of the two viruses were similar as determined by plaque assay of total infectious viruses (Fig. 1A) and quantity of A34R mRNA expression in infected cancer cells (Fig. 1B; p = 0.79). CXCL11-armed virus express much more CXCL11 mRNA (Fig. 1C, p < 0.0001) in the infected AB12-luc cells, and secreted 29-fold more CXCL11 in the supernatant of the infected AB12-luc cancer cells (Fig. 1D). Similar results were observed in MC38-luc colon cancer cells (data not shown). The new vvDD-CXCL11 virus and parental virus also had very similar cytotoxicity in AB12-luc cells (Fig. S1B), and in MC38 colon cancer cells (data not shown). In summary, these data demonstrate that inclusion of the CXCL11 gene did not affect the functionality of the virus as an oncolytic virus and secreted CXCL11 CK in vitro.
Figure 1.
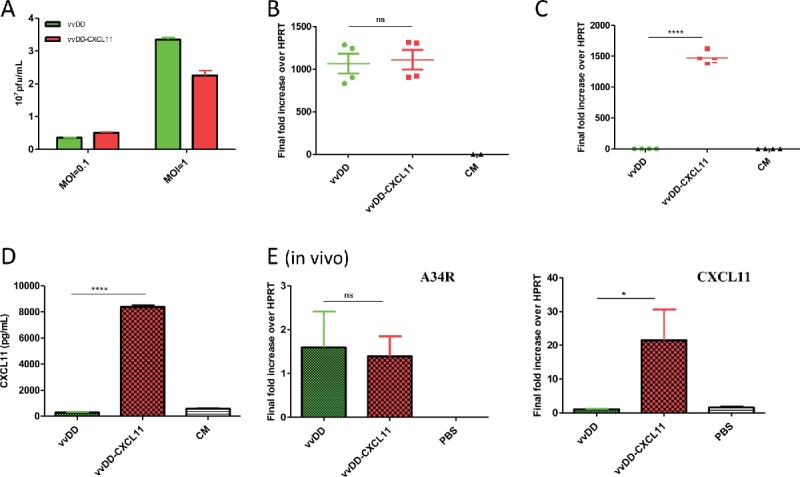
Viral replication and CXCL11 expression from the virus both in vitro and in vivo. AB12-luc cells were infected with vvDD-CXCL11 or vvDD. The infected cells were harvested 24 h post infection for RNA purification. The production of virus progeny from infected cancer cells at 48 h post infection was determined by plaque assay (A). Purified total RNAs were subject to RT-qPCR for quantitative detection of A34R mRNA (viral gene) (B) or CXCL11 (C). The quantity of secreted CXCL11 was measured by ELISA assay (D). For in vivo analyses (E), 4 × 105 AB12-luc cells were inoculated i.p. into naive BalB/c mice at day 0 and then injected i.p. with vvDD-CXCL11 or control virus vvDD (1 × 108 pfu/mouse) or PBS on day 5. Tumor nodules were harvested on day 9 and RNA isolated for qPCR to determine viral replication (via A34R expression, left panel) and CXCL11 expression (right panel). Symbols: * stands for p < 0.05; ** p < 0.01; *** p < 0.001; and NS: not significant. Abbreviations: CM, culture medium; HPRT, hypoxanthine guanine phosphoribosyl transferase, a house-keeping gene.
vvDD-CXCL11 is a superior therapeutic agent against established tumors
We tested the virus replication, CXCL11 expression and later the therapeutic efficacy in two intraperitoneal (i.p.) tumor models: AB12 mesothelioma and MC38 colon cancer. Tumor-bearing mice were treated with i.p. vaccinia as described in Materials and Methods. Cells isolated from the tumor 4 d after viral injection were used to isolate RNA for qPCR analysis to determine in vivo viral replication and CXCL11 expression. The results showed that the viral gene A34R expression (as a marker gene) was similar in the tumor in mice treated with either virus (Fig. 1E. Left; p = 0.83), indicating similar viral genome copies and viral gene expression. In contrast, about 20-fold more CXCL11 expression was found in the tumor from mice treated with vvDD-CXCL11 (Fig. 1E right; p = 0.03).
We then examined the therapeutic efficacy of vvDD-CXCL11 relative to the parental virus vvDD in established models of peritoneal carcinomatosis from mesothelioma as well as colorectal cancer. AB12-luc cancer cells were injected i.p. into naive BALB/c mice, and the tumor-bearing mice were randomized into groups and were treated i.p. with PBS, vvDD or vvDD-CXCL11 at 1 × 108 pfu/mouse 5 d post tumor cell inoculation. Tumor burden was measured by in vivo bioluminescence imaging on the day right before viral injection (days 5) and day 18 post tumor cell inoculation (Fig. 2A). It is clear that three groups of mice had similar tumor burden right before viral treatment (day 5). On day 18, in PBS-treated group three mice had large tumor masses and the other one died of tumor burden on day 17. Tumors in the vvDD-treated group also progressed. However, vvDD-CXCL11-treated mice displayed much less tumor burden. The survival analysis demonstrated vvDD-CXCL11-treated animals survived longer than vvDD or PBS treated animals, in tumor models of either AB12-luc (Fig. 2B, p = 0.0016 or p = 0.0023, respectively). Similar pattern has also been shown for MC38-luc colon cancer model even though the effects seemed to be smaller ( Fig. S2. p = 0.002 or p < 0.0001, respectively). These data support the conclusion that vvDD-CXCL11 is superior as a therapeutic agent in two tumor models. Immunotherapy for mesothelioma is a highly promising but also very challenging approach in human patients.29 For these reasons, we chose to conduct further experiments in the AB12 mesothelioma model.
Figure 2.

vvDD-CXCL11 treatment elicited antitumor effects in AB12-luc tumor model. AB12-luc cells (4 × 105) were inoculated i.p. into BALB/c mice and injected i.p. with PBS, vvDD or vvDD-CXCL11 (1 × 108 pfu/mouse) 5 d post tumor cell inoculation. (A). Tumor burden was measured by in vivo bioluminescence imaging on days 5 (D5) and 18 (D18) post tumor cell inoculation (n = 10 per group; only five mice per group are shown). One mouse (#4) in the PBS treated group has died of tumor burden on day 17, thus no image on D18. (B). Animal survival in AB12-luc tumor-bearing mice is presented using Kaplan–Meier survival curves. The curves represented data pooled from three independent experiments (n = 28 or more as indicated). p values are presented in the context of Results.
vvDD-CXCL11 skewed the TME into a more favorable one for antitumor immunity
To explore the mechanisms of vvDD-CXCL11-elicited antitumor effects, we determined the number of infiltrated CD8+ T cells and NK cells in the tumor mass. Both the percentage of CD8+ T cells in tumor infiltrating leukocytes and the number of CD8+ T cells per gram of tumor mass were higher in tumors from mice treated with vvDD-CXCL11, compared with vvDD (Fig. 3A and B, p = 0.011 and 0.035, respectively) or PBS treated mice. However, vvDD-CXCL11 increased NK cell infiltrate in a small number of mice (2 out of 9 mice) compared to vvDD, even though both viruses enhanced trafficking of NK cells into tumors when compared to PBS ( Fig. S3. p ≥ 0.05 between vvDD and vvDD-CXCL11-treated groups). Among the immunosuppressive cell types, we have examined both MDSC and CD4+CD25+FoxP3+ cells, and found no difference in the tumor tissues between PBS, vvDD or vvDD-CXCL11-treated groups (Fig. S4). Interestingly, no difference in infiltrated CD4+ T cells was observed either (Fig. S5).
Figure 3.

vvDD-CXCL11 treatment skewed the TME into a more favorable one for antitumor immunity. Tumor-bearing mice were treated as described in Fig. 2. Tumors nodules were harvested at day 9 post tumor inoculation, and were enzyme-digested and stained with PerCP-cy5.5-α-CD45 Ab and APC-α-CD8+ Ab (A, B). RNA was isolated for RT-qPCR to determine the expression of TGF-β (C), COX2 (D), CCL22 (E) and Granzyme B (F) in the TME. Symbols: * stands for p < 0.05; ** p < 0.01; *** p < 0.001; and NS: not significant. Abbreviations: RQ, relative quantity.
We analyzed several important molecules in the TME (Fig. 3, C-F) and found that the immune-suppressive molecules TGF-β, COX-2 and CCL22 (CK for regulatory T cell recruitment) were decreased in tumors from mice treated with vvDD-CXCL11, compared with vvDD (Fig. 3C-E, p = 0.03, 0.02 and 0.001, respectively) or PBS treated mice. In contrast, the cell lytic marker granzyme B was increased in tumors from vvDD-CXCL11-treated mice compared with vvDD (Fig. 3F, p = 0.01) or PBS treated mice, consistent with an active, antitumor cellular immune response. These data indicated that vvDD-CXCL11 modulates the TME toward a more immune-active state.
Virally expressed CXCL11 promotes a systemic antitumor CD8+ T cell response
We further explored whether expression of CXCL11 in the tumor led to the development of a systemic antitumor response by analyzing the CD8+ T cells in both the spleen and mesenteric lymph nodes. First, we checked the CD8+ T cells in mesenteric lymph nodes from peritoneal tumor-bearing mice treated with PBS, vvDD or vvDD-CXCL11. Single cells from mesenteric lymph nodes were stained with APC-α-CD8+ Ab. At the primary phase – day 9 post tumor inoculation – numbers of CD8+ T cells in mesenteric lymph nodes were similar in both virus-treated experimental groups (data not shown). However, at the effector phase – day 20 post tumor inoculation – the percentage of CD8+ T cells in mesenteric lymph nodes from mice treated with vvDD-CXCL11 virus was about 20%, which was significantly higher than that of mice treated with vvDD (Fig. 4A, p = 0.002) or PBS (about 8%). Next, we examined the numbers of CD8+ T cells in the spleens by staining with APC-α-CD8+ Ab. We found that the percentage of splenic CD8+ T cells was not significantly different in the virus-treated groups at both 9 d and 20 d after infection (Fig. 4B, p = 0.60). However, at the effector phase (day 20), vvDD-CXCL11 treated mice had more absolute numbers of splenic CD8+ T cells compared to those from mice treated with vvDD (Fig. 4C, D, p = 0.007) or PBS (p = 0.005).
Figure 4.
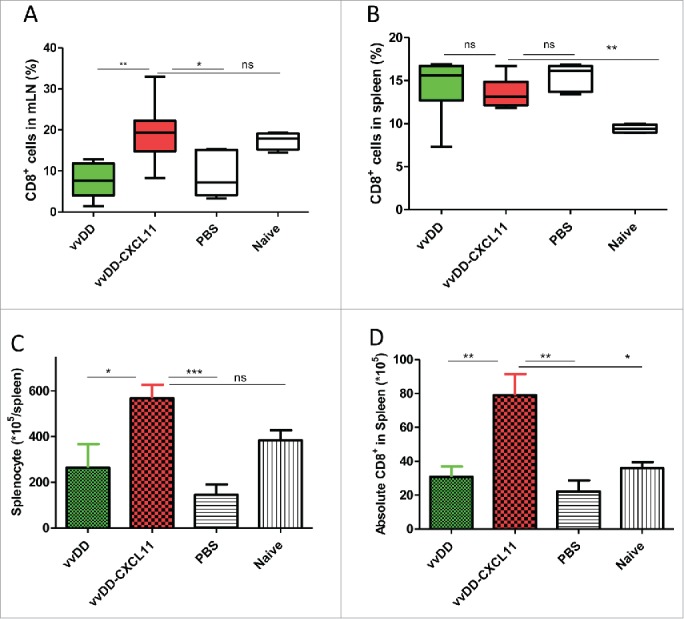
vvDD-CXCL11 treatment led to the accumulation of more CD8+ T cells in the spleen and mesenteric lymph nodes. Tumor-bearing mice were treated as described in Fig. 2. Mesenteric lymph nodes and spleens were harvested and single cells were stained with APC-α-CD8+ Ab day 20 post tumor cell inoculation. The percentage of CD8+ T cells in mesenteric lymph nodes and spleens are shown in (A) and (B). Whole splenocytes and absolute splenic CD8+ T cells are shown in (C) and (D).
To address whether vvDD-CXCL11 treatment could enhance the systemic activation of tumor-specific T cells in secondary lymphoid organ, we isolated splenic CD8+ T cells from the 20-d tumor-bearing mice and re-stimulated with mitomycin C-treated AB12-luc or control CA51 cancer cells in the presence of irradiated naïve CD8+-depleted splenocytes from BALB/c mice in vitro for 48 h. The concentration of IFNγ was significantly increased in the supernatant of the cultured CD8+ T cells from vvDD-CXCL11 treated mice compared with those treated with vvDD (Fig. 5A, p < 0.0001) or PBS (p < 0.0001). This IFNγ response is quite tumor-specific since syngeneic tumor cell control (CA51) stimulated splenic CD8+ T cells at a significantly lower level (Fig. 5A, p < 0.0001).
Figure 5.
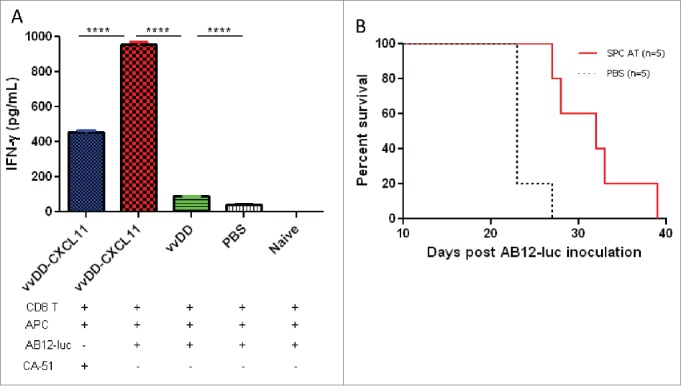
vvDD-CXCL11 elicited tumor-specific systemic immunity. Tumor-bearing BALB/c mice were treated as described in Fig. 2. Splenic CD8+ T cells (4 × 105 cells per assay) were isolated on day 20 post AB12-luc cell inoculation and restimulated with mitomycin C (MMC) treated AB12-luc (4 × 104) or control tumor CA51 cells (4 × 104) in the presence of 4,000-Rad-irradiated naive CD8− splenocytes (2 × 106) in 200 µL culture medium for 48 h. IFNγ in the culture supernatant was determined by ELISA (A). Splenocytes (3 × 107) from vvDD-CXCL11-cured mice 60 d post AB12-luc cell inoculation or PBS were adoptively transferred into naïve BALB/c mice and then challenged with AB12-luc (4 × 105) on the next day. Animal survival is presented using Kaplan-Meier survival curves (B). Symbol **** stands for p < 0.001.
To investigate whether vvDD-CXCL11 treatment resulted in the generation of a prolonged, protective antitumor effect, splenocytes from mice having been apparently cured after vvDD-CXCL11 treatment (survived > 60 d) were adoptively transferred into naïve BALB/c mice, and then challenged with AB12-luc cancer cells. The result showed that splenocyte-transfer extended the survival significantly compared to PBS control (Fig. 5B, p = 0.004). We believed that CD8+ T cells in the splenocytes made major contributions to the antitumor activity, and NK cell might make a minor contribution.
The antitumor immune response is dependent on CD8+ T cells and IFNγ
To investigate the roles of antitumor CD8+ T cells in vvDD-CXCL11 virotherapy, CD8+ T cells in vvDD-CXCL11 treated mice were depleted with an anti-CD8+ antibody. First, the antibody was injected daily on days 3, 4, 5 post tumor cell inoculation, and vvDD-CXCL11 was injected on day 5 post tumor cell inoculation. Mice in the CD8+ T cell depleted group receiving vvDD-CXCL11 treatment succumbed to their tumors earlier than vvDD-CXCL11 and PBS treated mice with intact CD8+ T cells (Fig. 6. p < 0.0001 and p < 0.0001, respectively). We also injected anti-CD8+ antibody to vvDD-CXCL11-treated mice daily on days 10, 11 and 12 post tumor cell inoculation (days 5, 6, and 7 post virus injection) and found that the antibody injection also completely abolished the therapeutic effect, resulting in earlier death than the control CD8+ intact mice treated with vvDD-CXCL11 or PBS (p < 0.0001 and p < 0.0001, respectively). In contrast, CD4+ T cell depletion beginning day 10 post tumor inoculation (day 5 post virus injection) did not significantly impact the therapeutic effect (p = 0.34). We also asked whether IFNγ was required for the therapeutic effect. The circulating IFNγ was neutralized by injection of anti-IFNγ Ab every 2 d beginning on day 10 post tumor cell inoculation for a total of 4 injections, and this resulted in the abolishment of the therapeutic effect, compared with the non-IFNγ depleted vvDD-CXCL11 treated mice (p < 0.01). In summary, these results demonstrate the essential roles that CD8+ T cells and IFNγ play in the efficacy of oncolytic virotherapy by CXCL11-armed oncolytic poxvirus.
Figure 6.
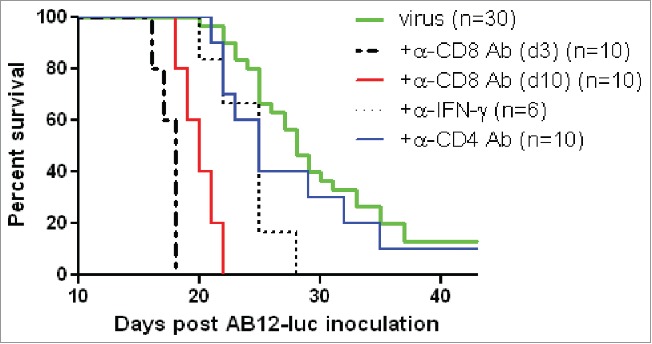
vvDD-CXCL11 elicited antitumor effects were dependent on CD8+ T cell and IFNγ. AB12-luc cancer cells (4 × 105) were inoculated i.p. into naive BALB/c mice on day 0 and tumor-bearing mice were injected i.p. with vvDD-CXCL11 (labeled as “virus”) at 1.0 × 108 pfu/mouse on day 5. For CD8+ depletion, anti-CD8+ Ab was injected i.p. on days 3, 4 and 5 or days 10, 11 and 12. For CD4+ depletion, anti-CD4+ Ab was injected i.p. on days 10, 15 and 19. For IFNγ neutralization, anti-IFNγ Ab was injected i.p. on days 10, 12, 14 and 16. Animal survival is presented using Kaplan–Meier survival analysis (n = 6 to 30 per group as shown). The p values between groups are presented in the main text in Results.
Discussion
Immunotherapy has shown great promise as a novel approach to cancer treatment.1-4 However, while it shows great promise, two major challenges remain: (1) how to reverse the immunosuppressive TME and break immune tolerance; and (2) how to efficiently educate T cells to traffic to the battlefield – the tumor site.30-32 The use of OVs expressing immune adjuvants can address both of these challenges. OVs function through multiple mechanisms to kill cancer cells, including direct cytotoxicity mediated by the virus, which is known to be immunogenic (immunogenic cell death [ICD].6,33-37 As for VV-induced lytic cancer cell death, it has been shown to elicit the release of immunogenic endoplasmic reticulum chaperone gp96,38 HMGB1 and ATP5,39,40 – danger signals for initiation of immune responses. Through ICD, in addition to the release of potent danger signals (signal 0), OVs may prompt the release and presentation of tumor-associated antigens to professional antigen presenting cells, thus activating DCs and eliciting a potent adaptive antitumor immune response, breaking tolerance.14,33-35,37,41,42, The OV-directed expression of a CXCR3 ligand in this study was designed to then attract activated, tumor-specific T-cells (induced by OV-mediated ICD) into the TME.18 Our data suggests that CXCL11 is not only important for the T-cell attraction (effector phase), but also for the development of adaptive immunity (induction phase). This finding was unexpected and could apply to other vectors and across many histologies, and represent a significant advance in the use of OVs for in situ vaccination and the development of an abscopal effect.
Our results demonstrate that T cells were increased in the TME after vvDD-CXCL11 treatment, consistent with the function of the CK to promote trafficking of active CD8+ T and Th1 cells into inflamed or tumor tissues.18,23-27 In addition, we have observed two novel findings after treatment with this CXCL11-expressing oncolytic virus. First, vvDD-CXCL11, but not the parental virus, modulated the TME into an immunologically favorable one for antitumor immunity, and we showed that only vvDD-CXCL11 led to sustained systemic immunity against the tumor. In our studies, modulation of the TME was achieved via CXCL11-mediated inhibition of suppressor factors, such as decreased TGF-β, COX2, and CCL22. We expect that the infection, replication and the ICD induced by the backbone oncolytic virus, provided not only danger signals and antigens for antitumor immunity, but also acute proinflammatory signals that may have a direct impact on the TME. However, the additional inhibition of suppressor factors by CXCL11 expression improved the antitumor response and systemic immunity. The mechanism of this inhibition requires further investigation. Perhaps the infiltration of activated T cells and expression of cytokines such as IFNγ and TNF-α, promoted by the CXCL11, are involved in the regulation of these important suppressor molecules in the TME.
Many previous studies have shown that danger signal molecules, most of them TLR ligands, can reactivate tumor-infiltrated DCs and then rescue the function of CD8+ T cells in the TME.3,43 We believe that favorable modulation of the TME, as indicated by more attracted CTL cells and decreased CCL22, TGFβ, and COX2, leads to the enhanced cytotoxic activities of tumor-specific T cells, leading to more acute release of danger signals and TAAs, which in turn reactivates tumor-associated DCs and T cells, resulting in a robust systemic antitumor immunity. 44 Investigators have previously studied a vaccinia expressing CXCL11 in non-tumor bearing naive mice, and demonstrated enhanced safety and improved, sustained, systemic anti-viral immunity, resulting in lower viral titers in normal tissue.25 We have confirmed this finding as we have seen lower viral titers from a number of normal tissues after viral delivery in MC38-tumor-bearing mice (Francis et al., manuscript in preparation).
It is important to point out that murine AB12 mesothelioma, just like human mesothelioma, is relatively immunogenic and thus quite susceptible to immunotherapy.29 We did observe weaker therapeutic effect of vvDD-CXCL11 virus in MC38-luc colon cancer model, probably due to the fact that MC38 cells possess low immunogenicity and MC38 tumor is infiltrated with high levels of inhibitory receptors (PD-1, LAG-3)-expressing T lymphocytes which synergistically promote tumoral immune escape.45 In this case, vvDD-CXCL11 in combination with immune checkpoint blockade may be required in order to elicit a potent antitumor immunity in the MC38 tumor model.
OVs have demonstrated promising results in the treatment of cancer, especially by local or regional administration, in preclinical models and clinical trials with T-VEC and Pexa-Vec.46,47 We have recently published our results with a phase I clinical trial using an intratumoral injection of the tumor-selective oncolytic VV (vvDD), which is the backbone virus used in the current study. The virus replicated well in tumor tissues and appeared to be tumor specific, leading to the complete resolution of some of the injected lesions, without affecting surrounding normal tissues.48 This is the ideal situation for expression of immunologic adjuvants that can induce systemic immunity. The anti-vaccinia immune response cleared the virus within two weeks in the clinical trial, but we could not demonstrate systemic antitumor immunity based on circulating PBMC, ELISPOT response to CEA or survivin, two commonly expressed tumor antigens. We also did not see immunologic responses in non-injected lesions. Here we provide evidence that favorable immunological changes in the TME initiated by vvDD-CXCL11 can elicit and maintain potent systemic antitumor immunity. The absolute number of CD8+ T cells in spleens and the percentage of CD8+ T cells in mesenteric lymph nodes were higher in the vvDD-CXCL11 treated mice at day 20 post tumor cell injection. More importantly, splenic CD8+ T cells from the vvDD-CXCL11 treated mice at 20 d post tumor cell injection secreted more IFNγ in response to re-stimulation with tumor. We also demonstrated that CD8+ T cell and IFNγ were necessary for mounting the antitumor response. Finally, we showed that adoptive transfer of splenocytes from vvDD-CXCL11 treated mice could elicit an antitumor response. These results clearly demonstrated that the specific antitumor immunity elicited by the CXCL11-armed oncolytic virus contribute significantly to the overall therapeutic efficacy.
Many investigators have explored the expression of cytokine or CK from OVs. In fact, the two OVs with the most promising clinical results in clinical trials, T-VEC and Pexa-Vec, are both armed with human GM-CSF gene.46,47 We have also recently reported our results with vvDD armed with a CK gene encoding CCL5/RANTES.28 These viruses were shown to be effective in combination with active cancer vaccines or adoptive immune cell transfer by utilizing the well-characterized property of the CKs to attract activated T cells and CIK cells into cancer tissues.28 As we have stated earlier, CXCL11 have been shown previously to attract activated T (Th1) and NK cells, while CCL5 attracts both resting and activated immune cells (T, NK, monocytes, and immature DCs). Thus, we believe that CXCL11 is a superior CK. Another key contribution of this study is that we have studied CXCL11 in the initiation phase of the antitumor immune response. Previously, few, if any, studies have explored the mechanisms of CK-armed OVs to elicit adaptive antitumor immunity. This property of vvDD-CXCL11 to elicit initiation phase of the immunity is unique in our experience.
In summary, our current study has revealed two novel properties of a CXCL11-armed oncolytic virus: modulation of the immunosuppressive TME to an immunosupportive one, and elicitation of potent and sustained systemic antitumor immunity. The antitumor immunity, together with potent oncolysis, contributes to the superior overall efficacy of vvDD-CXCL11-mediated therapy. The current findings suggest that the ability of different viral vectors to induce CXCL11 (and other CXCR3 ligands) may be used as a predictive factor of their effectiveness as cancer vaccines and indicate the possibility of using intratumoral expression of CXCL11 to enhance the effects of virus-based vaccines against cancer. The results from our study and other previous studies in the literature, suggest that viruses expressing CXCR3 ligands could serve as potent vaccines for cancer and infectious diseases as well.
Materials and methods
Mice and cell lines
Female BALB/c and C57BL/6 mice were purchased from Taconic Biosciences, Inc. (Germantown, NY) and housed in specific pathogen-free conditions in the University of Pittsburgh animal facility. All animal studies were approved by the Institutional Animal Care and Use Committee. Murine malignant mesothelioma cell line AB12 was derived from an asbestos-induced BALB/c mouse,49 and it was a gift kindly provided by Dr. Steven M Albelda (University of Pennsylvania). AB12 cells were infected with firefly luciferase-carrying lentivirus and selected by blasticidin to generate a new subline AB12-luc. Murine colon adenocarcinoma cell line CA51 was derived from a tissue collection of the National Cancer Institute. All cell lines were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 1X penicillin/streptomycin solution (Invitrogen, Carlsbad, CA) in 37°C, 5% CO2 incubator.
Recombinant vvDD-CXCL11
vSC20, a vgf gene-deleted Western Reserve (WR) strain VV was used as the parental virus for homologous recombination. The shuttle plasmid carrying murine cxcl11 cDNA was created for homologous recombination of cxcl11 into the tk locus of vaccinia viral genome. To make the new virus vvDD-CXCL11, CV-1 cells were infected with vSC20 at multiplicity of infection (MOI) of 0.1 and then transfected with the shuttle plasmid. Selection of the new recombinant virus was based on expression of Discosoma red fluorescent protein (DsRed) 48 h post transfection. vvDD-DsRed, or vvDD in short, a double viral gene-deleted (tk- and vgf-) VV carrying (DsRed) gene at the tk locus, is the control virus for this work ( Fig. S1A).
Viral replication and CK expression in vitro
Viral replication assays were performed as previously described.50 Briefly, 1.0 × 105 AB12-luc or MC38-luc cells, were seeded in six-well plates overnight and infected with vvDD or vvDD-CXCL11 at MOIs of 0.1, 1.0, or 10 in 1 mL of 2% FBS-containing–DMEM for 2 h. Following infection, cells were cultured until harvesting at 48 h post viral infection. The cell pellets were homogenized using a FastPrep Cell Disrupter (Model FP120) (Qbiogene, Carlsbad, CA) to release virions, and viral load was determined by plaque assay. For analysis of CK expression, 3.0 × 105 AB12-luc cells were seeded in 24-well plates overnight and then infected with vvDD or vvDD-CXCL11 at MOI of 1 and then cultured for 24 h. Cell culture supernatant was harvested and the concentration of CXCL11 was determined using ELISA kit according to the manufacturer's instructions (R & D). Cell pellets were harvested and the total RNA was extracted using the RNeasy Kit (Qiagen). One microgram of RNA was used for cDNA synthesis, and 25 to 50 ng of subsequent cDNA was used to conduct mRNA expression analysis by TaqMan analysis on the StepOnePlus system (Life Technologies, Grand Island, NY). All the primers used for the analysis were purchased from Applied Biosystems by Life Technologies. The gene expression was normalized to HPRT1 and expressed as fold increase (2−or), where ΔCT = CT (Target gene)- CT (HPRT1).
Tumor challenge
BALB/c or C57BL/6 mice were intraperitoneally inoculated with 4 × 105 Ab12-luc or 5 × 105 MC38-luc cells, respectively, and divided into three groups according to tumor growth condition based on live animal IVIS imaging 5 d post tumor cell injection, performed using a Xenogen IVIS 200 Optical In Vivo Imaging System (Caliper Life Sciences, Hopkinton, MA). Randomly grouped mice were intraperitoneally injected with vvDD, vvDD-CXCL11 at 1.0 × 108 pfu/100 μL or 100 μL PBS per mouse, respectively. In some experiments, anti-mouse CD8+ Ab at 250 µg/injection (clone 53–6.7; BioXCell, West Lebanon, NH), anti-mouse CD4+ Ab (clone GK1.5, Bio X Cell; 150µg/injection), or anti-mouse IFNγ Ab (clone XMG1.2, Bio X Cell; 200 µg/injection) were injected i.p. into mice to deplete CD8+ T cells, CD4+ T cells or neutralize circulating IFNγ.
Analyses of cells from tumor tissue
Tumor tissues were removed from mock-treated or virus-treated mice, weighed, and incubated in RPMI 1640 containing 2% FBS, 1 mg/mL collagenase, 0.1 mg hyaluronidase, and 200 U DNase I (All enzymes were from Sigma, St. Louis, MO) to make single cells 4 d post virus treatment. Single-cell suspensions were counted and stained with PerCP-cy5.5-α-CD45 (clone 30-F11, BioLegend, San Diego, CA), APC-α-CD8+ (clone 53–6.7, BioLegend) and APC-α-CD49 (BioLegend) post Fc receptor blockade by α-CD16/32 Ab to determine the populations of CD8+ T cells and NK cells by flow cytometry. 3.0 × 106 single cells were used for total RNA extraction and qRT-PCR assay as described above.
Systemic antitumor immune response
Splenocytes or mesentery lymph node cells were isolated 9 or 20 d post tumor cell injection from different groups of the treated mice and stained with APC-anti-mouse CD8+ Ab to determine CD8+ T cell population (post Fc receptor blockade by α-CD16/32 Ab) by flow cytometry. CD8+ T cells (4 × 105) were purified from splenocytes of different groups of treated or naive BALB/c mice using anti-mouse CD8+ microbeads following vendor's protocols (Miltenyi Biotec, San Diego, CA) and restimulated with mitomycin C-treated AB12-luc cells (4 × 104) or CA51 colon cells (tumor-specific control; 4 × 104) in the presence of 4,000-rad-irradiated naive CD8+-depleted BALB/c splenocytes (2 × 106) in 200 μL RPMI 1640 medium supplemented with 10% FBS at 37°C, 5% CO2 for 3 d The concentration of IFNγ in the culture supernatants was tested using ELISA kit according to the manufacturer's instructions (BioLegend).
As for adoptive cell transfer, single splenocytes were also isolated from tumor-bearing BALB/c mice treated with vvDD-CXCL11, which survived more than 60 d, and adoptively transferred into naive BALB/c mice by tail vein injection at 3.0 × 107 cells per mouse. The age-matched naive BALB/c mice received PBS injection as a control. The next day, these mice were challenged with 4 × 105 AB12-luc cancer cells intraperitoneally.
Long-term survival of mice
The health and survival of treated mice was closely monitored. All mice bearing peritoneal tumors were monitored via caliper measurements for changes in abdominal girth. Mice were dead naturally due to the disease or sacrificed when their abdominal girth exceeded 1.5 × the original measurement.
Statistics
Statistical analyses were performed using Student's t test (GraphPad Prism version 5). Animal survival is presented using Kaplan–Meier survival curves and was statistically analyzed using log rank test (GraphPad Prism version 5). Value of p < 0.05 is considered to be statistically significant, and all p values were two sided. In the figures, we have adopted the standard symbols in the statistics: * stands for p < 0.05; ** p < 0.01; *** p < 0.001; and NS: not significant.
Supplementary Material
Disclosure of potential conflict of interest
DLB is a shareholder of Sillajen Biotherapeutics, a company developing OVs. The other authors declared no conflicts of interest.
Funding
This study was supported by NIH grants P01CA132714 and R01CA155925. This project used the University of Pittsburgh Cancer Institute (UPCI) shared facilities that are supported in part by the NIH grant award P30CA047904. This work was also partially funded by generous support for mesothelioma research from the New Era Cap Company and from Valarie Koch.
References
- 1.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol (2012); 12:269-81; PMID:22437939; http://dx.doi.org/ 10.1038/nri3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity (2013); 39:61-73; PMID:23890064; http://dx.doi.org/ 10.1016/j.immuni.2013.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalinski P, Okada H. Polarized dendritic cells as cancer vaccines: directing effector-type T cells to tumors. Semin Immunol (2010); 22:173-82; PMID:20409732; http://dx.doi.org/ 10.1016/j.smim.2010.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012); 12:252-64; PMID:22437870; http://dx.doi.org/ 10.1038/nrc3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.John LB, Howland LJ, Flynn JK, West AC, Devaud C, Duong CP, Stewart TJ, Westwood JA, Guo ZS, Bartlett DL et al.. Oncolytic virus and anti-4-1BB combination therapy elicits strong antitumor immunity against established cancer. Cancer Res (2012); 72:1651-60; PMID:22315352; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2788 [DOI] [PubMed] [Google Scholar]
- 6.Diaconu I, Cerullo V, Hirvinen ML, Escutenaire S, Ugolini M, Pesonen SK, Bramante S, Parviainen S, Kanerva A, Loskog AS et al.. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res (2012); 72:2327-38; PMID:22396493; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2975 [DOI] [PubMed] [Google Scholar]
- 7.Willmon CL, Saloura V, Fridlender ZG, Wongthida P, Diaz RM, Thompson J, Kottke T, Federspiel M, Barber G, Albelda SM et al.. Expression of IFN-beta enhances both efficacy and safety of oncolytic vesicular stomatitis virus for therapy of mesothelioma. Cancer Res (2009); 69:7713-20; PMID:19773437; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, Ohtani H. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res (1998); 58:3491-4; PMID:9721846 [PubMed] [Google Scholar]
- 9.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN et al.. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med (2003); 348:203-13; PMID:12529460; http://dx.doi.org/ 10.1056/NEJMoa020177 [DOI] [PubMed] [Google Scholar]
- 10.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P et al.. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006); 313:1960-4; PMID:17008531; http://dx.doi.org/ 10.1126/science.1129139 [DOI] [PubMed] [Google Scholar]
- 11.Pages F, Galon J, Dieu-Nosjean MC, Tartour E, Sautes-Fridman C, Fridman WH. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene (2010); 29:1093-102; PMID:19946335; http://dx.doi.org/ 10.1038/onc.2009.416 [DOI] [PubMed] [Google Scholar]
- 12.Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene (2008); 27:5904-12; PMID:18836471; http://dx.doi.org/ 10.1038/onc.2008.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013); 14:1014-22; PMID:24048123; http://dx.doi.org/ 10.1038/ni.2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, He Y, Guo ZS. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer (2013); 12:103; PMID:24020520; http://dx.doi.org/ 10.1186/1476-4598-12-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res (2014); 2:295-300; PMID:24764576; http://dx.doi.org/ 10.1158/2326-6066.CIR-14-0015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer (2014); 14:559-67; PMID:24990523; http://dx.doi.org/ 10.1038/nrc3770 [DOI] [PubMed] [Google Scholar]
- 17.Pol J, Bloy N, Obrist F, Eggermont A, Galon J, Cremer I, Erbs P, Limacher JM, Preville X, Zitvogel L et al.. Trial Watch: Oncolytic viruses for cancer therapy. Oncoimmunology (2014); 3:e28694; PMID:25097804; http://dx.doi.org/ 10.4161/onci.28694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bromley SK, Mempel TR, Luster AD. Orchestrating the orchestrators: chemokines in control of T cell traffic. Nat Immunol (2008); 9:970-80; PMID:18711434; http://dx.doi.org/ 10.1038/ni.f.213 [DOI] [PubMed] [Google Scholar]
- 19.Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell (1999); 99:23-33; PMID:10520991; http://dx.doi.org/ 10.1016/S0092-8674(00)80059-8 [DOI] [PubMed] [Google Scholar]
- 20.Nesbeth Y, Scarlett U, Cubillos-Ruiz J, Martinez D, Engle X, Turk MJ, Conejo-Garcia JR. CCL5-mediated endogenous antitumor immunity elicited by adoptively transferred lymphocytes and dendritic cell depletion. Cancer Res (2009); 69:6331-8; PMID:19602595; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-4329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tannenbaum CS, Tubbs R, Armstrong D, Finke JH, Bukowski RM, Hamilton TA. The CXC chemokines IP-10 and Mig are necessary for IL-12-mediated regression of the mouse RENCA tumor. J Immunol (1998); 161:927-32; PMID:9670971 [PubMed] [Google Scholar]
- 22.Kanegane C, Sgadari C, Kanegane H, Teruya-Feldstein J, Yao L, Gupta G, Farber JM, Liao F, Liu L, Tosato G. Contribution of the CXC chemokines IP-10 and Mig to the antitumor effects of IL-12. J Leukoc Biol (1998); 64:384-92; PMID:9738666 [DOI] [PubMed] [Google Scholar]
- 23.Cole KE, Strick CA, Paradis TJ, Ogborne KT, Loetscher M, Gladue RP, Lin W, Boyd JG, Moser B, Wood DE et al.. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med (1998); 187:2009-21; PMID:9625760; http://dx.doi.org/ 10.1084/jem.187.12.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hensbergen PJ, Wijnands PG, Schreurs MW, Scheper RJ, Willemze R, Tensen CP. The CXCR3 targeting chemokine CXCL11 has potent antitumor activity in vivo involving attraction of CD8+ T lymphocytes but not inhibition of angiogenesis. J Immunother (2005); 28:343-51; PMID:16000952; http://dx.doi.org/ 10.1097/01.cji.0000165355.26795.27 [DOI] [PubMed] [Google Scholar]
- 25.Hamilton NH, Mahalingam S, Banyer JL, Ramshaw IA, Thomson SA. A recombinant vaccinia virus encoding the interferon-inducible T-cell alpha chemoattractant is attenuated in vivo. Scand J Immunol (2004); 59:246-54; PMID:15030574; http://dx.doi.org/ 10.1111/j.0300-9475.2004.01391.x [DOI] [PubMed] [Google Scholar]
- 26.Berencsi K, Meropol NJ, Hoffman JP, Sigurdson E, Giles L, Rani P, Somasundaram R, Zhang T, Kalabis J, Caputo L et al.. Colon carcinoma cells induce CXCL11-dependent migration of CXCR3-expressing cytotoxic T lymphocytes in organotypic culture. Cancer Immunol Immunother (2007); 56:359-70; PMID:16783574; http://dx.doi.org/ 10.1007/s00262-006-0190-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wendel M, Galani IE, Suri-Payer E, Cerwenka A. Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer Res (2008); 68:8437-45; PMID:18922917; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-1440 [DOI] [PubMed] [Google Scholar]
- 28.Li J, O'Malley M, Urban J, Sampath P, Guo ZS, Kalinski P, Thorne SH, Bartlett DL. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol Ther (2011); 19:650-7; PMID:21266959; http://dx.doi.org/ 10.1038/mt.2010.312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gregoire M. What's the place of immunotherapy in malignant mesothelioma treatments? Cell Adh Migr (2010); 4:153-61; PMID:20179421; http://dx.doi.org/ 10.4161/cam.4.1.11361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swartz MA, Iida N, Roberts EW, Sangaletti S, Wong MH, Yull FE, Coussens LM, DeClerck YA. Tumor microenvironment complexity: emerging roles in cancer therapy. Cancer Res (2012); 72:2473-80; PMID:22414581; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-0122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gajewski TF. Failure at the effector phase: immune barriers at the level of the melanoma tumor microenvironment. Clin Cancer Res (2007); 13:5256-61; PMID:17875753; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-0892 [DOI] [PubMed] [Google Scholar]
- 32.Abastado JP. The next challenge in cancer immunotherapy: controlling T-cell traffic to the tumor. Cancer Res (2012); 72:2159-61; PMID:22549945; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3538 [DOI] [PubMed] [Google Scholar]
- 33.Boozari B, Mundt B, Woller N, Struver N, Gurlevik E, Schache P, Kloos A, Knocke S, Manns MP, Wirth TC et al.. Antitumoural immunity by virus-mediated immunogenic apoptosis inhibits metastatic growth of hepatocellular carcinoma. Gut (2010); 59:1416-26; PMID:20675696; http://dx.doi.org/ 10.1136/gut.2009.196519 [DOI] [PubMed] [Google Scholar]
- 34.Donnelly OG, Errington-Mais F, Steele L, Hadac E, Jennings V, Scott K, Peach H, Phillips RM, Bond J, Pandha H et al.. Measles virus causes immunogenic cell death in human melanoma. Gene Ther (2013); 20:7-15; PMID:22170342; http://dx.doi.org/ 10.1038/gt.2011.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyamoto S, Inoue H, Nakamura T, Yamada M, Sakamoto C, Urata Y, Okazaki T, Marumoto T, Takahashi A, Takayama K et al.. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res (2012); 72:2609-21; PMID:22461509; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3185 [DOI] [PubMed] [Google Scholar]
- 36.Guillerme JB, Boisgerault N, Roulois D, Menager J, Combredet C, Tangy F, Fonteneau JF, Gregoire M. Measles virus vaccine-infected tumor cells induce tumor antigen cross-presentation by human plasmacytoid dendritic cells. Clin Cancer Res (2013); 19:1147-58; PMID:23339127; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-2733 [DOI] [PubMed] [Google Scholar]
- 37.Fonteneau JF, Guillerme JB, Tangy F, Gregoire M. Attenuated measles virus used as an oncolytic virus activates myeloid and plasmacytoid dendritic cells. Oncoimmunology (2013); 2:e24212; PMID:23762802; http://dx.doi.org/ 10.4161/onci.24212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berwin B, Reed RC, Nicchitta CV. Virally induced lytic cell death elicits the release of immunogenic GRP94/gp96. J Biol Chem (2001); 276:21083-8; PMID:11279246; http://dx.doi.org/ 10.1074/jbc.M101836200 [DOI] [PubMed] [Google Scholar]
- 39.Guo ZS, Naik A, O'Malley ME, Popovic P, Demarco R, Hu Y, Yin X, Yang S, Zeh HJ, Moss B et al.. The enhanced tumor selectivity of an oncolytic vaccinia lacking the host range and antiapoptosis genes SPI-1 and SPI-2. Cancer Res (2005); 65:9991-8; PMID:16267024; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-1630 [DOI] [PubMed] [Google Scholar]
- 40.Whilding LM, Archibald KM, Kulbe H, Balkwill FR, Oberg D, McNeish IA. Vaccinia virus induces programmed necrosis in ovarian cancer cells. Mol Ther (2013); 21:2074-86; PMID:23985697; http://dx.doi.org/ 10.1038/mt.2013.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo ZS, Liu Z, Bartlett DL. Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity. Front Oncol (2014); 4:74; PMID:24782985; http://dx.doi.org/ 10.3389/fonc.2014.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Workenhe ST, Mossman KL. Oncolytic virotherapy and immunogenic cancer cell death: sharpening the sword for improved cancer treatment strategies. Mol Ther (2014); 22:251-6; PMID:24048442; http://dx.doi.org/ 10.1038/mt.2013.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao H, Peng Y, Hong Y, Huang L, Guo ZS, Bartlett DL, Fu N, Munn DH, Mellor A, He Y. Local administration of TLR ligands rescues the function of tumor-infiltrating CD8 T cells and enhances the antitumor effect of lentivector immunization. J Immunol (2013); 190:5866-73; PMID:23610140; http://dx.doi.org/ 10.4049/jimmunol.1203470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mantovani A, Romero P, Palucka AK, Marincola FM. Tumour immunity: effector response to tumour and role of the microenvironment. Lancet (2008); 371:771-83; PMID:18275997; http://dx.doi.org/ 10.1016/S0140-6736(08)60241-X [DOI] [PubMed] [Google Scholar]
- 45.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL et al.. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res (2012); 72:917-27; PMID:22186141; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, Cho M, Lim HY, Chung HC, Kim CW et al.. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med (2013); 19:329-36; PMID:23396206; http://dx.doi.org/ 10.1038/nm.3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS et al.. Talimogene Laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol (2015); 33:2780-8; PMID:26014293; http://dx.doi.org/ 10.1200/JCO.2014.58.3377 [DOI] [PubMed] [Google Scholar]
- 48.Zeh HJ, Downs-Canner S, McCart JA, Guo ZS, Rao UN, Ramalingam L, Thorne SH, Jones HL, Kalinski P, Wieckowski E et al.. First-in-man study of western reserve strain oncolytic vaccinia virus: safety, systemic spread, and antitumor activity. Mol Ther (2015); 23:202-14; PMID:25292189; http://dx.doi.org/ 10.1038/mt.2014.194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davis MR, Manning LS, Whitaker D, Garlepp MJ, Robinson BW. Establishment of a murine model of malignant mesothelioma. Int J Cancer (1992); 52:881-6; PMID:1459729; http://dx.doi.org/ 10.1002/ijc.2910520609 [DOI] [PubMed] [Google Scholar]
- 50.Thirunavukarasu P, Sathaiah M, Gorry MC, O'Malley ME, Ravindranathan R, Austin F, Thorne SH, Guo ZS, Bartlett DL. A rationally designed A34R mutant oncolytic poxvirus: improved efficacy in peritoneal carcinomatosis. Mol Ther (2013); 21:1024-33; PMID:23439499; http://dx.doi.org/ 10.1038/mt.2013.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.