

Significance
The advances in whole-genome sequencing have greatly facilitated the identification of mismatch repair (MMR) gene mutations in suspected Lynch syndrome (LS) cases. The bottleneck in diagnosing LS patients now lies in determining whether MMR gene variants are pathogenic or polymorphic. In some cases the mutation is clearly deleterious for gene function. However, many suspected LS patients carry single missense mutations in one of the MMR genes, and the functional implications of these missense mutations are often elusive. To help clinicians diagnose and treat patients, we present a site-specific mutagenesis screen that takes advantage of oligonucleotide-directed gene-modification and is capable of detecting pathogenic mutator S homolog 2 (MSH2) variants in three simple steps.
Keywords: Lynch syndrome, DNA mismatch repair, MSH2, variants of uncertain significance, site-directed mutagenesis
Abstract
Single-stranded DNA oligonucleotides can achieve targeted base-pair substitution with modest efficiency but high precision. We show that “oligo targeting” can be used effectively to study missense mutations in DNA mismatch repair (MMR) genes. Inherited inactivating mutations in DNA MMR genes are causative for the cancer predisposition Lynch syndrome (LS). Although overtly deleterious mutations in MMR genes can clearly be ascribed as the cause of LS, the functional implications of missense mutations are often unclear. We developed a genetic screen to determine the pathogenicity of these variants of uncertain significance (VUS), focusing on mutator S homolog 2 (MSH2). VUS were introduced into the endogenous Msh2 gene of mouse embryonic stem cells by oligo targeting. Subsequent selection for MMR-deficient cells using the guanine analog 6-thioguanine allowed the detection of MMR-abrogating VUS. The screen was able to distinguish weak and strong pathogenic variants from polymorphisms and was used to investigate 59 Msh2 VUS. Nineteen of the 59 VUS were identified as pathogenic. Functional assays revealed that 14 of the 19 detected variants fully abrogated MMR activity and that five of the detected variants attenuated MMR activity. Implementation of the screen in clinical practice allows proper counseling of mutation carriers and treatment of their tumors.
The DNA mismatch repair (MMR) system is essential for genome fidelity: It corrects mismatches that may arise during erroneous DNA replication and induces apoptosis if adducts caused by certain DNA-damaging agents cannot be repaired. Newly replicated DNA is scanned for base–base mispairings and loops of unpaired bases by heterodimers composed of MMR proteins mutator S homolog 2 and 6 (MSH2/MSH6) or mutator S homolog 2 and 3 (MSH2/MSH3), respectively. Upon encountering a mismatch, the MSH heterodimers recruit another heterodimer composed of mutator L homolog 1 and postmeiotic segregation increased 2 (MLH1/PMS2) to coordinate downstream repair events (1, 2). Exposure to certain DNA-damaging agents, such as methylating agents and the nucleotide analog 6-thioguanine (6TG), creates lesions in the genome that give rise to mismatches when replicated (3, 4). The DNA MMR system recognizes these mismatches and induces cell death to remove them. Several models propose how DNA MMR activates cell death. One model suggests that DNA MMR recognizes the mismatch and repetitively removes the incorporated nucleotide rather than the lesion itself, creating a cycle of futile repair which ultimately leads to DNA breakage and cell death (3). Another possibility is that MSH2/MSH6 and MLH1/PMS2 bind at the site of the damaged base and act as molecular scaffolds that activate downstream DNA damage-response pathways that result in apoptosis (5, 6). In the absence of a functional DNA MMR system, cells have an increased rate of spontaneous mutagenesis and elevated resistance to DNA-methylating agents.
A dysfunctional DNA MMR system is the underlying cause of Lynch syndrome (LS). LS is an autosomal-dominant cancer predisposition that is characterized by the early onset of colorectal cancer and cancers at extracolonic sites such as the endometrium, ovaries, and stomach. It is caused by mutations in MSH2, MLH1, MSH6, or PMS2 DNA MMR genes that destroy gene function. Patients usually have a heterozygous germline mutation in one of the DNA MMR genes. Upon somatic loss of the wild-type allele, MMR-deficient cells arise, and a general mutator phenotype develops that increases the chance of mutations arising in oncogenes and tumor-suppressor genes and hence the development of malignancies (7).
Germline mutations in MSH2 account for about 40% of LS cases. In addition to nonsense and frameshift mutations that truncate the protein and clearly abrogate MMR function, a significant portion of LS-associated MSH2 sequence variants is composed of missense mutations that affect only a single amino acid (8). Missense mutations in MMR genes also are seen frequently in cancer cases that cannot be ascribed unambiguously to LS. Without reliable segregation data or functional information, whether such sequence variants contribute to cancer risk remains uncertain. To help clinicians diagnose LS and offer appropriate counseling and treatment, a method of examining the functional implications of these MSH2 variants of uncertain significance (VUS) is needed (9).
With this aim, a number of in silico algorithms as well as functional assays have been developed. The in silico algorithms take into account evolutionary conservation as well as physicochemical differences between amino acids to predict the consequences of specific mutations. Although such computer methods can identify pathogenic MMR gene variants, validation often is required to diagnose LS patients (10–13). The functional studies generally investigate the consequences of MMR VUS using assays based on ectopic expression of mutant MMR genes in MMR-deficient yeast, bacteria, or human cells or on in vitro-reconstituted MMR reactions (14–23). A possible caveat regarding such studies is that they often are performed in distantly related species, and the effect of the mutations may be masked by the unstable genetic background of MMR-deficient cells or over/under-representation of the ectopically expressed or in vitro-studied protein (9).
To study the phenotype of VUS expressed at physiological levels within a normal cellular context, we have developed a site-directed mutagenesis screen in mouse embryonic stem cells (mESCs) that assesses the phenotype of endogenous variant Msh2 alleles. The endogenous Msh2 gene is site-specifically mutated using the oligonucleotide-directed gene modification (oligo-targeting) technique we recently developed (24). This gene-modification technique is capable of substituting a single base pair at any desired location in the genome using 25- to 35-nt oligodeoxyribonucleotides (ssODN) that are complementary to an endogenous target sequence except for one or two centrally located nucleotides that comprise the desired modification. Upon transfection, the ssODN anneals to its chromosomal complement, creating a mismatch at the position of the mutating nucleotide(s). Recognition of this mismatch by DNA MMR leads to abortion of the gene-modification reaction (25). However, we found that mismatch recognition can be avoided when the mutating nucleotides in the ssODN are present as locked nucleic acids yielding base-pair substitution frequencies of 10−3–10−4 in MMR-proficient cells (24). The efficiency of oligo targeting is lower than the recently reported efficacies of nuclease-assisted (in particular CRISPR/Cas9-assisted) gene modification. Nonetheless, mutations that abrogate MMR activity can be identified effectively because of the resistance of MMR-deficient cells to methylating agents. Here we demonstrate that gene modification by short ssODNs can be used efficiently to fulfill a specific clinical need: the functional interrogation of variants of DNA MMR genes to establish whether they are causative for LS. We present the genetic screen, demonstrate that it is capable of distinguishing pathogenic MSH2 variants from polymorphisms, and analyze the phenotype of 59 MSH2 VUS found in suspected LS patients.
Results
Genetic Screen to Identify Pathogenic MSH2 VUS.
To assess the phenotype of MSH2 VUS found in suspected LS patients, we developed a site-directed mutagenesis screen in mESCs. mESCs provide a good study model because the murine MSH2 protein shares 93% homology with its human counterpart and mouse models can be made from these cells if VUS need to be studied in vivo. The genetic screen is composed of three steps (Fig. 1): (A) site-directed mutagenesis to introduce the specific mutation into mESCs; (B) selection for cells that became MMR-deficient; and (C) sequencing to confirm that selected MMR-deficient cells contain the mutation of interest.
Fig. 1.

Genetic screen for the identification of pathogenic MSH2 variants. (A) Msh2+PUR/∆ mESCs were exposed to ssODNs that introduce the mutation of interest into the one endogenous Msh2 allele with an efficiency of 10−3–10−4. (B) The mESCs subsequently were exposed to 6TG. Cells that lost MMR activity form 6TG-resistant colonies. To remove cells that became MMR-deficient because of the loss-of-heterozygosity events, puromycin selection was performed simultaneously. (C) 6TG/puromycin-resistant colonies were selected and expanded. Sequence analysis was used to confirm the presence of the introduced mutation in the 6TG/puromycin-resistant cells.
For this screen we generated an Msh2+PUR/Δ mESC line in which one of the endogenous Msh2 alleles was completely deleted (Δ) and a puromycin-resistance gene was introduced adjacent to the remaining Msh2 allele that retained wild-type activity (+PUR) (Fig. S1). Hence, introduction of a mutation into the single endogenous Msh2 allele in Msh2+PUR/Δ mESCs ensured exclusive expression of the variant allele and immediate disclosure of its effect on MMR activity. Msh2 was site-specifically mutated using our oligo-targeting technique (24). Upon exposure of Msh2+PUR/Δ mESCs to a specific ssODN (which can have either sense or antisense polarity), the desired mutation was introduced into 1 in every 1,000–10,000 cells. To determine whether MMR was abrogated in this small subset, cells were exposed to 6TG that is toxic only when DNA MMR is active (4). The appearance of 6TG-resistant colonies indicates that MMR-deficient cells were generated in the ssODN-exposed mESC culture. Should the mutation not affect MMR, no colonies are expected to appear. However, 6TG-resistant colonies also may arise from cells that lost the Msh2 wild-type allele by loss of heterozygosity. To select against events resulting in loss of heterozygosity, puromycin selection was performed simultaneously. Furthermore, the presence of the planned mutation in the 6TG/puromycin-resistant colonies had to be confirmed by sequence analysis because we did encounter some Msh2+PUR/Δ mESCs that managed to survive the selection despite wild-type MSH2 activity.
Fig. S1.

Generation of Msh2+PUR/∆ mESCs. In Msh2+PUR/∆ mESCs one of the endogenous Msh2 alleles was deleted (∆), and a puromycin-resistance gene was introduced in front of the remaining Msh2 allele (+PUR). (A) Vectors containing a loxP site, a selection marker, and 6 kb homologous to the sequence either upstream or downstream of the Msh2 gene were created to introduce loxP sites flanking one of the Msh2 alleles. (B) The 3′ loxP vector was linearized by KpnI digestion and electroporated into wild-type 129/OLA mESCs. (C) Geneticin-resistant colonies were analyzed for integration of the 3′ loxP vector into its target site by Southern blot analysis. The genomic DNA was BglII-digested, and fragments were visualized using a probe (indicated by a yellow star). (D) Southern blot showing the wild-type 9.6-kb fragment in unmodified (Msh2+/+) cells. The presence of an additional 8.6-kb fragment indicates correct integration of the 3′ loxP vector (LoxP integrated). (E) Cells with successful 3′ loxP vector integration were electroporated with the BamHI-linearized 5′ loxP vector. (F) Puromycin-resistant colonies were analyzed for integration of the 5′ loxP vector into the target site by Southern blot analysis. The genomic DNA was NsiI-digested, and fragments were visualized using a probe (indicated by a yellow star). (G) Southern blot showing the wild-type 9.9-kb fragment in unmodified (Msh2+/+) cells. The presence of an additional 20.4-kb fragment indicates correct integration of the 5′ loxP vector (LoxP integrated). The 5′ loxP vector could integrate adjacent to either the wild-type or the 3′ loxP-modified Msh2 allele. (H) To delete the floxed Msh2 allele and create Msh2+/∆ mESCs, CMV-Cre recombinase was transiently transfected into six independent clones. With the 5′ and 3′ loxP vectors flanking the same Msh2 allele, recombination between loxP sites gives rise to deletion of the complete Msh2 gene plus the tk gene and hence confers Ganciclovir resistance. If the 5′ and 3′ loxP vectors were present on different Msh2 alleles, Cre-mediated interchromosomal recombination would retain the tk gene. Therefore, Ganciclovir resistance indicates the deletion of the floxed Msh2 allele. Of the six clones tested, three gave rise to Ganciclovir-resistant colonies; in each of these colonies the presence of a single loxP site was confirmed by sequencing PCR products obtained using primers adjacent to Msh2 (arrows). (I) Msh2+/∆ mESCs were electroporated again with the 5′ loxP vector to introduce a puromycin-resistance gene in front of the remaining Msh2 allele. (J) Puromycin-resistant colonies were analyzed by Southern blot analysis for integration of the 5′ loxP vector into its target site. The genomic DNA was NsiI-digested, and fragments were visualized using a probe (indicated by a yellow star). (K) Southern blot showing the wild-type 9.9-kb fragment in unmodified Msh2+/∆ cells; the presence of a 20.4-kb fragment indicates correct integration of the 5′ loxP-Pur vector (Msh2+PUR/∆). The 5′ loxP vector may integrate adjacent to either the Msh2+ or the Msh2∆ allele. However, only integration into the Msh2+ allele generates a 20.4-kb fragment, because the Msh2∆ allele has lost the DNA sequence that could bind the probe.
Distinguishing Pathogenic from Nonpathogenic Variants.
To demonstrate the efficacy of this approach to distinguish pathogenic from nonpathogenic MSH2 missense mutations, we tested 10 proven pathogenic MSH2 variants and 10 proven MSH2 polymorphisms. The variants were chosen based on clinical and in vitro data (Tables S1 and S2). Thirty-five–nucleotide ssODNs were used to introduce the pathogenic and nonpathogenic sequence alterations. In both cases, colonies appeared upon 6TG selection; however, the colonies of nonpathogenic ssODNs usually were smaller and fewer in number. Sequencing revealed all 10 pathogenic mutations were present in several of the 6TG-resistant colonies, whereas not one of the polymorphisms was detected (Fig. 2 A and B and Fig. S2). This result demonstrates that the readout of our screen, i.e., the combination of 6TG selection and sequencing of colonies, can distinguish pathogenic from nonpathogenic Msh2 variants.
Table S1.
Pathology, in silico data, and MMR activity according to literature for pathogenic MSH2 mutations used in the proof-of-principle study and our results
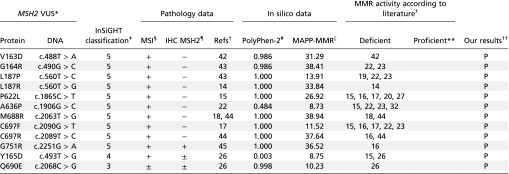 |
Msh2 variant amino acid and base change. Nucleotide numbering is based on cDNA where +1 is the A of the ATG translation initiation codon.
Literature reporting whether the studied MSH2 variants led to MMR deficiency or proficiency.
Classification according to InSiGHT: 3, unclassified; 4, likely pathogenic; 5, pathogenic.
MSI data: +, instable; −, stable; ±, discrepancies between tumor data.
Immunohistochemistry (IHC): +, stain for MSH2; −, MSH2 not detected; ±, MSH2 absence and presence seen in different tumors.
PolyPhen scores were calculated on genetics.bwh.harvard.edu/pph2/. Scores >0.2 are considered probably damaging. NA, not available.
MAPP-MMR scores were calculated on mappmmr.blueankh.com/. Scores >4.55 are predicted to affect MMR function. NA, not available.
Indicates no references were found that report MMR activity for these variants.
The conclusions from this study: P, pathogenic.
Table S2.
Pathology, in silico data, and MMR activity according to literature for nonpathogenic MSH2 variants used in the proof-of-principle study and our results
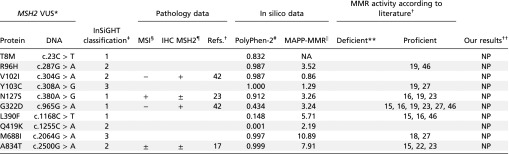 |
Msh2 variant amino acid and base change. Nucleotide numbering is based on cDNA where +1 is the A of the ATG translation initiation codon.
Literature reporting whether the studied MSH2 variants led to MMR deficiency or proficiency.
Classification according to InSiGHT: 1, not pathogenic; 2, likely not pathogenic; 3, unclassified.
MSI data: +, instable; −, stable; ±, discrepancies between tumor data.
Immunohistochemistry (IHC): +, stain for MSH2; −, MSH2 not detected; ±, MSH2 absence and presence seen in different tumors.
PolyPhen scores were calculated on genetics.bwh.harvard.edu/pph2/. Scores >0.2 are considered probably damaging. NA, not available.
MAPP-MMR scores were calculated on mappmmr.blueankh.com/. Scores >4.55 are predicted to affect MMR function. NA, not available.
Indicates no references were found that report defective MMR activity for these variants.
The conclusions from this study: NP, not pathogenic.
Fig. 2.

Identification of pathogenic Msh2 variants by sequencing 6TG-resistant colonies. (A) Proven pathogenic mutations in the proof-of-principle study. (B) Proven nonpathogenic variants in the proof-of-principle study. (C) Newly identified pathogenic mutations among 59 tested VUS. The “Variant” and “Nucleotide change” columns display the amino acid change as well as the location and the one- or two-base change introduced by either sense (upper bars) or antisense (lower bars) ssODNs. The bars in the “Sequenced colonies carrying mutation” column illustrate the number of 6TG/puromycin-resistant colonies sequenced per sense or antisense ssODN tested. We always aimed to sequence 12 colonies unless fewer survived the 6TG/puromycin selection. Each box in the bars represents one sequenced colony. Gray boxes represent colonies carrying the mutation of interest; white boxes represent colonies in which the wild-type Msh2 sequence was maintained. NA indicates the targeting was not performed.
Fig. S2.

Sequences of pathogenic mutations detected in the proof-of-principle study. One-letter amino acid codes are given below the nucleotide sequences.
In addition to the 10 overtly pathogenic variants, we also tested two partially pathogenic mutations: MSH2-Y165D and MSH2-Q690E. mESCs expressing these variants had been generated previously and had been shown to be only partially resistant to 6TG (26). Under the selection conditions applied thus far (termed “method 1”), our screen did not detect the two variants. Therefore we sought to create a second, more sensitive screening method to distinguish partially pathogenic from nonpathogenic Msh2 variants. The oligo-targeting efficiency was recently improved to 10−3 using 25-nt ssODNs and a different transfection reagent. Combining this protocol with a selection scheme in which the concentration of 6TG was lowered and the exposure time extended (termed “method 2”), allowed the recognition of the partially pathogenic MSH2-Y165D and MSH2-Q690E while true polymorphisms remained undetectable (Fig. 2A and Fig. S2). Thus, sequencing of the 6TG-resistant colonies using screening method 2 can identify both fully and partially pathogenic Msh2 VUS.
Screening VUS.
Having proven the genetic screen can identify pathogenic and partially pathogenic Msh2 variants, we went on to screen 59 MSH2 VUS. All 59 VUS have been found in suspected LS patients and were brought to our attention through contact with the clinic or by inspection of the International Society for Gastrointestinal Hereditary Tumors (InSiGHT) database. Of the 59 VUS, 19 were detected in 6TG-resistant colonies by sequence analysis and therefore were identified as pathogenic (Fig. 2C and Fig. S3). We initially used screening method 1, which identified 14 of these 19 mutations. After rescreening the remaining 45 VUS using screening method 2, which was optimized for the detection of partially deleterious variants, five additional variants were found: V63E, G162R, D603N, G674A, and G759E. These five variants may have an intermediate pathogenic phenotype. To determine the extent of the MMR-attenuating effect of the deleterious Msh2 missense mutations identified, we performed Western blot analyses and functional assays assessing response to DNA-damaging agents and microsatellite instability (MSI).
Fig. S3.

Sequences of the detected Msh2 variants. Nucleotide and one-letter amino acid codes are noted.
Phenotypic Assessment of Identified Pathogenic Msh2 Variants.
To confirm the pathogenic variants were identified because of their reduced sensitivity to methylating agents, we determined clonogenic survival of variant cell lines in response to the DNA methylating agents 6TG (Fig. 3) and N′-methyl-N′-nitro-N′-nitrosoguanidine (MNNG) (Fig. S4). Msh2+PUR/Δ mESCs clearly showed reduced colony formation upon exposure to increasing doses of 6TG and MNNG. Fourteen of the 19 variant cell lines behaved like the 6TG- and MNNG-resistant mESCs expressing MSH2-P622L that previously had been proven to be pathogenic (27). Variants V63E, G162R, D603N, G674A, and G759R conferred partial sensitivity to 6TG. This partial sensitivity explains why they could be detected only by using screening method 2.
Fig. 3.

6TG toxicity in Msh2 mutant mESCs. The colony-forming capacity of the detected pathogenic Msh2 variant cell lines, Msh2+PUR/∆ mESCs, and the Msh2P622L/Δ pathogenic control was determined in response to increasing doses of 6TG. The 6TG tolerance of mutant cell lines should be compared with the MMR-proficient Msh2+PUR/∆ and MMR-deficient Msh2P622L/Δ mESCs in the same experiment because slight differences in the 6TG concentrations result in small interexperiment variances.
Fig. S4.

MNNG clonogenic survival assay. The colony-forming capacity of the detected Msh2 mutant mESCs and the Msh2+PUR/∆ and Msh2P622L/Δ control cell lines was determined in response to increasing doses of the methylating agent MNNG. Msh2+PUR/∆ cells appeared to die at slightly different MNNG doses in various experiments. This variance was caused by small differences in MNNG concentrations in the experiments. Hence the MNNG tolerance of the variant cells lines should be compared with the MMR-proficient Msh2+PUR/∆ and MMR-deficient Msh2P622L/Δ mESCs in the same experiment.
The abundance of variant MSH2 protein was quantified with respect to Msh2+/+ mESCs. Msh2+PUR/Δ mESCs contained 60% of the MSH2 seen in Msh2+/+ cells (Fig. S5). MSH2 levels dropped to 1–4% in 15 of the variant cell lines, comparable to the amount of protein in Msh2P622L/Δ cells. MSH6 levels mirrored the decrease in MSH2 in all variant cell lines because MSH6 is less stable without its heterodimer partner. Cell lines Msh2V63E/Δ, Msh2D603N/Δ, Msh2G674A/Δ, and Msh2S723F/Δ had relatively high MSH2 levels of 33%, 25%, 33%, and 21%, respectively (Fig. 4).
Fig. S5.

Western blot analysis of the Msh2+PUR/∆ cell line. MSH2 levels in Msh2+PUR/∆ mESCs were quantified with respect to Msh2+/+ cells and compared with levels in Msh2+/∆ and Msh2−/− mESCs. MSH6 and CDK4 levels were analyzed also. CDK4 functioned as the loading control.
Fig. 4.

Western blot analysis of detected Msh2 VUS. MSH2, MSH6, and CDK4 levels were analyzed in whole-cell lysates. CDK4 functioned as the loading control. Relative MSH2 levels compared with Msh2+/+ mESCs are shown as percentages. Msh2−/− and Msh2P622L/Δ cells were used as pathogenic controls.
MSI is a hallmark of MMR deficiency. In the absence of MMR, DNA polymerase slippage errors at highly repetitive sequences are not corrected, causing microsatellites to vary drastically in size (7). To obtain a quantitative readout for MSI in the 6TG-resistant Msh2 variant mESCs, we introduced a single copy of a slippage reporter into the Rosa26 locus of MMR-proficient and mutant mESCs. The slippage reporter was composed of a neomycin-resistance gene (neo) that was rendered out of frame by the insertion of a (G)10 repeat. For the neo gene to become in frame, DNA polymerase slippage errors, such as the deletion of one G or the insertion of two Gs, need to go unnoticed by the MMR system. Hence, the number of cells that survived Geneticin selection because of neo-restoring slippage events indicates the MMR capacity of the cells (28). The slippage rate (i.e., the chance of a slippage event occurring during one cell division) in the Msh2 variant mESCs ranged from 4.7 × 10−5 to 1.6 × 10−3. This rate is 65–2,200 times higher than the slippage rate of 7.3 × 10−7 observed in Msh2+PUR/Δ mESCs (Fig. 5). The majority of the variant mESCs showed slippage rates similar to that of the MSH2-P622L pathogenic control (9.3 × 10−4). Variants V63E, G162R, and D603N conferred lower slippage rates of 4.7 × 10−5, 1.5 × 10−4, and 1.6 × 10−4, respectively, more comparable to that of the partially pathogenic control MSH2-Y165D. The other two variants that could be detected only with screening method 2, G674A and G759E, conferred slippage rates similar to those conferred by several of the pathogenic variants revealed by screening method 1. Although the slippage rates of the studied Msh2 VUS varied, the data clearly show that they all increased mutagenesis. As summarized in Table 1, all variants detected in 6TG-resistant colonies by sequence analysis showed abrogated or attenuated MMR capacity and therefore can be classified unambiguously as pathogenic.
Fig. 5.

MSI analysis of detected Msh2 mutant mESCs. A slippage reporter composed of a neo gene that was rendered out of frame by a (G)10 repeat, was introduced into the Msh2 mutant mESCs. The relative slippage rates could be calculated by the number of cells that became Geneticin-resistant because of a slippage event bringing the neo gene in-frame. Slippage rates were compared with the MMR-proficient Msh2+PUR/∆ cell line, the MMR-deficient control P622L, and the partially pathogenic control Y165D. Statistical differences were calculated using an unpaired t test with Welch’s correction. Asterisks indicate values significantly higher than those in the MMR-proficient Msh2+PUR/∆ control: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Table 1.
Overview of results from functional assays performed with detected pathogenic variants
| MSH2 VUS* | Sensitivity to 6TG† | MSH2 protein levels, %‡ | MSI§ |
| Msh2+PUR/∆ | S | 60 | L |
| P622L | R | 1.5 | H |
| V63E | I | 33 | M |
| V161D | R | 1 | H |
| G162R | I | 1 | M |
| L173P | R | 1 | H |
| L173R | R | 4 | H |
| C333Y | R | 1 | H |
| L341P | R | 1 | H |
| V342I | R | 3 | H |
| P349L | R | 3 | H |
| P349R | R | 1 | H |
| D603N | I | 25 | M |
| G674A | I | 33 | H |
| G674R | R | 4 | H |
| G692R | R | 1 | H |
| P696L | R | 3 | H |
| C697Y | R | 4 | H |
| S723F | R | 21 | H |
| G759E | I | 1 | H |
| E878D | R | 2 | H |
The variants are annotated according to their amino acid number and change.
The degree of 6TG tolerance of the mutant cell lines was classified as sensitive (S), similar to Msh2+PUR/∆ cells; resistant (R), similar to Msh2P622L/Δ mESCs; and intermediate (I) i.e., the mutant cell lines are less resistant than Msh2P622L/Δ cells but not as sensitive as Msh2+PUR/∆ mESCs.
The abundance of MSH2 in the variant cell lines was quantified (%) with respect to the MSH2 levels in Msh2+/+ mESCs.
The results from the MSI assay were divided into three groups: H, high slippage rate (≥ 3 × 10−4); M, medium slippage rate (<3 × 10−4 and >8 × 10−7); and L, low slippage rate (<8 × 10−7).
Discussion
We present a genetic screen that assesses the MMR-abrogating effect of endogenously expressed Msh2 VUS in mESCs. Missense mutations were site-specifically introduced into cultures of mESCs that contained only one endogenous Msh2 allele to obtain cells that expressed only the mutant protein. The mutagenized cell cultures subsequently were exposed to 6TG to investigate whether mutant cells had lost MMR capacity, in which case 6TG-resistant colonies appear. The presence of the planned mutation in the 6TG-resistant cells was confirmed by sequence analysis. Resistance to 6TG has been used previously to select for MMR deficiency in cell cultures that were randomly mutagenized with ethylnitrosourea yielding a catalog of deleterious codon substitutions (29). By using oligo targeting, we can interrogate variants of interest directly. To demonstrate that our approach was capable of distinguishing pathogenic MSH2 missense mutations from polymorphisms, we tested 12 proven pathogenic and 10 nonpathogenic MSH2 variants. All 12 pathogenic variants, but none of the polymorphisms, were detected in 6TG-resistant colonies. The proof-of-principle study manifested that the oligonucleotide-directed mutagenesis screen was capable of identifying both fully and partially pathogenic Msh2 VUS. However, despite this good performance and the high level of amino acid conservation, possible functional differences in the human and mouse MSH2 proteins may be a limitation of the use of mESCs.
We used the genetic screen to assess the pathogenicity of 59 MSH2 VUS found in suspected LS patients. All mutated amino acids were conserved between human and mouse MSH2 (Fig. S6). Sequence analysis detected 19 mutations in 6TG-resistant colonies. Their pathogenicity was confirmed by Western blot analyses and functional assays demonstrating fully abolished or attenuated MMR capacity.
Fig. S6.

Alignment of human (Upper Rows) and mouse (Lower Rows) MSH2 amino acid sequences demonstrating conservation of studied mutations. Asterisks mark differences between the two sequences. (A) The 12 pathogenic mutations (red) and 10 polymorphisms (green) used in the proof-of-principle study are highlighted in both the human and mouse sequences to demonstrate their conservation. At the yellow-colored residues, both a pathogenic and nonpathogenic mutation was detected. (B) The 59 studied VUS are highlighted, illustrating their conservation between humans and mice. Detected pathogenic variants are colored red, undetected variants are green, and residues in which both detected and undetected variants were identified are yellow.
The specificity of our screen can be defined as the chance that a variant that is picked up as deleterious does indeed affect MMR. We found an absolute concordance between the variants identified in our screen and defective MMR: Not one of the 10 proven polymorphisms was detected, but all 12 confirmed deleterious variants were identified, and all 19 VUS that were detected showed a defect in MMR. Hence, the false-positive frequency was <1/41, giving a specificity of >97.6%. We argue that the specificity of our screen may approach 100% because resistance to 6TG is a hallmark of MMR deficiency and hence, by definition, all Msh2 mutations detected in 6TG-resistant colonies by sequence analysis affect MMR activity.
We also can estimate the sensitivity of our screen. Sensitivity is a measure of the chance that a deleterious variant was not picked up; it can be defined as the ratio of true positives (deleterious mutations picked up by our test) to all deleterious mutations (true positives plus false negatives that were not picked up). All 12 variants that were a priori selected for pathogenicity were picked up in our screen. Hence, with the number of true positives being 12 and the number of false negatives <1, the screen had a sensitivity of >92.3%. However, we notice that one partially deleterious variant, Y165D, yielded only one clone carrying the planned mutation. Therefore the sensitivity may be lower for weak variants.
We argue that VUS picked up by our screen can be assigned unambiguously as deleterious for MMR and hence be placed in class 5 (proven pathogenic) of the five-tiered VUS classification scheme adopted by InSiGHT (30). Inclusion of more variants proven to be weakly pathogenic by independent criteria and assays may increase the sensitivity of our screening protocol, allowing assignment of nondetected variants to class 2 (likely nonpathogenic) or even class 1 (proven nonpathogenic).
The 59 studied VUS and the 22 variants used in the proof-of-principle study were dispersed across almost all domains of MSH2. The identified pathogenic mutations, however, clustered predominantly in three areas: the connector domain from amino acids 160–188, the lever domain between residues 332 and 350, and the ATPase domain from amino acids 621–760 (Fig. S7). Of the 16 variants located in the mismatch-binding domain, only V63E was found to be pathogenic. The other 15 variants do not appear to influence MMR activity. However, studies have shown that deletions and several missense mutations in this domain affect MSH2/MSH3 activity (19, 31). We cannot exclude the possibility that some of these 15 variants affect MSH2/MSH3 activity; such mutations cannot be detected in our screen because the MSH2/MSH3 complex is not involved in the toxicity of 6TG.
Fig. S7.

Location of the studied mutations in the MSH2 protein. The MSH2 domains are displayed in different colors (39–41) and are annotated according to their amino acid number and change. (A) The location of the variants used in the proof-of-principle study. The 12 pathogenic mutations are indicated by red lines above the MSH2 domains, and the 10 polymorphisms are indicated by green lines below the MSH2 domains. (B) The location of the 59 studied VUS is depicted: Pathogenic mutations are indicated by red lines above the MSH2 domains, and the undetected variants are indicated by green lines below the MSH2 domains.
Fourteen of the 19 pathogenic VUS were identified using screening method 1. Western blot analysis and functional assays revealed that 13 behaved similarly to the MSH2-P622L pathogenic control and likewise can be considered to abrogate MMR function fully. Only mutation S723F behaved differently, because the level of protein remained relatively high, suggesting that substitution S723F did not debilitate protein folding to the same extent as the other mutations. Functional assays showed Msh2S723F/Δ mESCs had the highest slippage rate in the MSI assay and were as resistant to DNA-methylating agents as Msh2P622L/Δ mESCs. The MMR-debilitating effect of this mutation most likely can be ascribed to its interference with ADP/ATP binding at the ATPase domain of MSH6 (Fig. S8).
Fig. S8.

Location of variants V63E, G162R, D603N, G674A, S723F, and G759E in MSH2. The MSH2–MSH6 heterodimer bound to G:T mispaired DNA, taken from Warren et al. (39), is presented in a ribbon diagram. MSH2, lilac; MSH6, gray; DNA, orange/blue/green; ADP, yellow/orange/red/blue. The location of residues Val63, Gly162, Asp603, Gly674, Ser723, and Gly759 are enlarged and colored green to indicate why substitutions may lead to MMR attenuation.
MSH2 VUS V63E, G162R, D603N, G674A, and G759E (see Fig. S8 for their position in the 3D structure) could be detected only using screening method 2 that was optimized for the identification of partially pathogenic variants. Hence one may expect these five VUS to behave like the partially pathogenic variant MSH2-Y165D we studied previously (26). Variants V63E, G162R, and D603N did seem to meet this expectation. Msh2V63E/Δ, Msh2G162R/Δ, and Msh2D603N/Δ mESCs experienced slippage events at rates similar to that in the partially pathogenic control and were less resistant to 6TG in the DNA damage-response assay than the fully pathogenic Msh2P622L/Δ control. Msh2V63E/Δ and Msh2D603N/Δ mESCs also had higher MSH2 levels than the majority of fully pathogenic variants described in this study. The MMR-attenuating effect of mutation V63E, located at the interface with MSH6, may be ascribed to interference with MSH6 function. Variant D603N resides in the area of MSH2 that interacts with EXO1, MSH3, and MSH6; hence it may hinder these interactions. The G162R mutation decreased MSH2 levels to 1% of that seen in Msh2+/+ cells. Although this low MSH2-G162R protein level was in line with tumor pathology data showing no MSH2 staining (32), it is striking, given the partially pathogenic phenotype seen for this variant in the functional assays. Perhaps the G162R mutation has a destabilizing effect that reduces the MSH2 protein level but not intrinsic MSH2 activity.
Although both the G674A and G759E variants were less resistant to 6TG than the fully pathogenic Msh2P622L/Δ cell line, their pathogenic phenotypes were stronger than the partially pathogenic Msh2Y165D/Δ control. Msh2G759E/Δ mESCs experienced slippage events at rates similar to those of several of the fully pathogenic variant cell lines detected in our study using screening method 1. This mutation also decreased MSH2 levels to 1% of those seen in Msh2+/+ cells such as the G162R variant that showed reduced slippage rates. Hence, the pathogenic phenotype of G759E appears to arise from an effect on both protein stability and protein function. Consistent with previous data describing MSH2-G674A as a separation-of-function mutation that was incapable of initiating MMR but nevertheless was sensitive to the genotoxic effect of 6TG (33), we found a high slippage rate in Msh2G674A/Δ mESCs. At variance however, we demonstrate mutation G674A did reduce 6TG toxicity. Our results indicate that for this mutant, also, the loss of MMR was associated with reduced sensitivity to 6TG. MSH2-G674A protein levels were relatively high, indicating that the pathogenicity of the mutation was not caused by protein destabilization but rather by its effects on protein function. The G674A mutation most likely interferes with ADP/ATP binding to the MSH2 ATPase domain.
It should be noted that two variants that in a yeast assay were found to cause a very weak MMR defect, T33P and D167H, were not detected in our assay (19). The weak mutator phenotype of the yeast T33P variant was attributed to disruption of MSH2/MSH3, explaining why it remained undetected in our screen.
The oligonucleotide-directed mutagenesis screen presented here is capable of distinguishing partially pathogenic from nonpathogenic MSH2 variants. The proof-of-principle study demonstrates that combining oligo targeting with 6TG selection and sequence analysis allows parallel detection of many pathogenic MSH2 variants. Furthermore, we show that all detected Msh2 VUS indeed lead to MMR abrogation in the functional assays. Importantly, the absence of false positives and the finding that the deleterious variants we identified contained only the planned mutation demonstrate that oligo targeting was highly accurate and did not lead to inadvertent mutations that may have generated 6TG-resistant cells.
Screening method 2 allows the detection of both fully and partially deleterious MSH2 variants and now is used routinely. Its relative simplicity allows the screen to be implemented in clinical genetics laboratories that are confronted with suspected LS-associated mutations. In the future, the applicability of the screen may be extended to characterize suspected LS-associated variants in the other DNA MMR genes, and it may be developed in human cells to investigate intronic variants or mutations at residues that are not conserved between mice and men.
Materials and Methods
Genetic Screen for the Identification of Pathogenic MSH2 Mutations.
Two methods were used to identify pathogenic MSH2 mutations. In method 1 mutations were introduced into Msh2+PUR/Δ mESCs (described in SI Materials and Methods) by oligo targeting using 35-nt ssODNs (Eurogentec) (24). For each mutation, 7 × 105 Msh2+PUR/Δ mESCs were seeded on six wells and on the following day were transfected with 3 μg ssODNs plus 27 μL TransFast transfection agent (Promega) in 1.4 mL serum-free medium. After 1 h, 4 mL of Buffalo rat liver (BRL)-conditioned medium (28) was added, and the cells were incubated for 3 d. Subsequently, 1.5 × 106 ssODN-exposed cells were seeded on 10-cm plates. 6TG (1.5 μM) (Sigma-Aldrich) and puromycin (1.8 μg/mL) selection started 24 h later. 6TG exposure lasted for 3 d; puromycin remained on the plates for 10 d; then the 12 largest 6TG/puromycin-resistant colonies were selected and expanded. The presence of the planned mutation was verified by sequence analysis.
Screening method 2 was optimized for the identification of partially pathogenic variants by using a 10-fold more efficient oligo-targeting technique (24), a reduced 6TG concentration, and an extended exposure time. Msh2+PUR/Δ mESCs (7 × 105) were seeded per six-well plate and were transfected on the following day with 3 μg 25-nt ssODNs and 7.5 μL TransIT-siQuest transfection agent (Mirus) in 250 μL of serum-free medium. After 3 d, 1.5 × 106 ssODN-exposed cells were seeded on 10-cm plates and subjected to 6TG (250 nM) and puromycin (1.8 μg/mL) selection for 10 d. The 12 largest 6TG/puromycin-resistant colonies were processed for sequencing.
DNA Damage-Response Assay.
6TG and MNNG DNA damage-response assays were performed as described in Wielders et al. (27).
Western Blot Analysis.
Western blot analyses were executed as described in Wielders et al. (26) using goat polyclonal antibody against CDK4 (1:2,000) (SC-260-G; Santa Cruz Biotechnology) and rabbit polyclonal antibodies against MSH2 (1:500) (34) and MSH6 (1:500) (35). IRDye 800CW donkey anti-goat IgG and IRDye 800CW goat anti-rabbit IgG antibodies (LI-COR) were used as secondary antibodies, and signals were visualized on an Odyssey imaging system (LI-COR), allowing quantification of the protein bands.
Microsatellite Instability Assay.
mESCs were electroporated (36) with the (G)10-neo Rosa26 targeting vector that encodes the pMC1-(G)10-neo gene downstream of a promoterless histidinol-resistance gene and integrates into the Rosa26 locus (28). Histidinol (3 mM; Sigma-Aldrich)-resistant clones were selected, expanded to 107 cells, and subsequently seeded on 10-cm plates at a density of 105 cells per plate for Geneticin selection (600 μg/mL). Successful integration of the (G)10-neo Rosa26 targeting vector was confirmed by Southern blot analysis. After 10 d of Geneticin selection, resistant colonies were counted and mutation rates were calculated using the formula: 0.6 × Geneticintotal = N × p × log (N × p), where Geneticintotal is the number of Geneticin-resistant colonies in a culture expanded to N cells, and p is the number of mutations per cell division (37). Experiments were performed in quadruplicate, and statistical differences were calculated using an unpaired t test with Welch’s correction.
SI Materials and Methods
Two targeting vectors were created to introduce loxP sites 5′ and 3′ of one of the endogenous Msh2 alleles in 129/OLA mESCs (Fig. S1). To create the 5′ loxP vector, a loxP site and the PUR gene were introduced into plasmid pSP73 (Promega) using standard cloning methods. Two 3-kb fragments upstream of the Msh2 gene were amplified from 129/OLA-derived genomic DNA by primer pairs 5′-CGTAACCGTTAACCCTCCAGGACAGGTAAGATTGAA-3′, 5′-CTGGAAGCAAAGACGGACAA-3′ and 5′-CCCCAAGTGTACCAGGACAG-3′, 5′-GAAGATCTGGTTGTCATACTCCAATTAACAAGAAC-3′ and the Phusion Hot Start II High-Fidelity DNA Polymerase (ThermoFisher Scientific). The 3-kb fragments subsequently were ligated into the unique HpaI and BamHI sites in the pSP73-loxP-PUR vector using the unique restriction sites present (BamHI) or engineered (underlined) (HpaI, BglII) at the ends of the fragments. The BamHI site at the center of the ligated 6-kb fragment remained unique. To create the 3′ loxP vector, a loxP site, neo gene, and thymidine kinase (tk) gene were introduced into a pSP73 backbone. Two 3-kb fragments downstream of the Msh2 gene were amplified from 129/OLA-derived genomic DNA using primer pairs 5′-GAAGATCTAGGAAGCAAGTGAGAGCTACAGG-3′, 5′-GGGGTACCCCTGACTATTCTGCACACCATTTT-3′ and 5′-GGGGTACCCCCTGCTCCGAAATAAACA-3′, 5′-GGAATTCCATATGCCACACAGCTAAGCATCCTACT-3′ and were ligated into the pSP73-loxP-neo-tk vector. Each primer contained a unique restriction site (underlined) that allowed the ends of the respective 3-kb fragments to be ligated into the unique BamHI and KpnI sites or NdeI and KpnI sites in the pSP73-loxP-neo-tk vector. The KpnI site at the center of the 6-kb fragment remained unique in the vector.
The 3′ loxP vector was KpnI-linearized and electroporated into 129/OLA mESCs (36). Electroporated cells were seeded on gelatin-coated 10-cm plates at a density of 106 cells per plate and were subjected to Geneticin selection (400 μg/mL) (Life Technologies). After 10 d of Geneticin selection, individual resistant colonies were selected and expanded. DNA from the Geneticin-resistant colonies was BglII-digested and Southern blot analyzed with a probe that was amplified from 129/OLA-derived genomic DNA with primer pair 5′-CCCTGCATGTTCTTCCACA-3′ and 5′-ATGCAGAGCTAGGGAATGAGC-3′. Successfully targeted cells then were electroporated with BamHI-linearized 5′ loxP vector and selected for puromycin (1.8 μg/mL) (Sigma-Aldrich) resistance. DNA from puromycin-resistant clones was NsiI-digested for Southern blot analysis with a probe that was amplified from 129/OLA-derived genomic DNA with primer pair 5′-GGATACCACACAATAGGTCCAG-3′ and 5′-TGCTAGTGAATGGATGCAGA-3′.
The floxed-Msh2 mESCs were transiently transfected with CMV-Cre recombinase and hygromycin-resistance (HYG) vectors (38). More specifically, 7 × 105 mESCs were seeded on a gelatin-coated six-well plate in BRL-conditioned medium and on the following day were exposed to 1.4 mL of serum-free medium containing 3 μg CMV-Cre recombinase vector plus 0.12 μg HYG vector and 27 μL TransFast transfection agent (Promega). After 1 h, 4 mL BRL-conditioned medium was added, and the cells were incubated overnight. Subsequently, 104 cells were reseeded on a 10-cm plate in BRL-conditioned medium with 150 μg/mL hygromycin (Calbiochem). Hygromycin selection continued for 2 d. Five days post transfection 5 μM ganciclovir (Roche) was added to the medium. After 8 d of ganciclovir selection, resistant colonies were selected and expanded, and their DNA was sequenced with primers flanking the loxP sites to confirm the deletion of one of the Msh2 alleles and the creation of Msh2+/Δ mESCs. To introduce a PUR gene upstream of the remaining endogenous Msh2 allele, the 5′ loxP vector was electroporated into Msh2+/Δ mESCs as described above.
Acknowledgments
We thank Rob Dekker, Sandra de Vries, Tim Harmsen, Frans Hogervorst, Jarnick Lusseveld, Titia Sixma, and Thomas van Ravesteyn for technical assistance, valuable discussions, and critical reading of the manuscript. This work was supported by Dutch Cancer Society Grant NKI 2009-4477.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 3918.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1520813113/-/DCSupplemental.
References
- 1.Palombo F, et al. GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science. 1995;268(5219):1912–1914. doi: 10.1126/science.7604265. [DOI] [PubMed] [Google Scholar]
- 2.Palombo F, et al. hMutSbeta, a heterodimer of hMSH2 and hMSH3, binds to insertion/deletion loops in DNA. Curr Biol. 1996;6(9):1181–1184. doi: 10.1016/s0960-9822(02)70685-4. [DOI] [PubMed] [Google Scholar]
- 3.Mojas N, Lopes M, Jiricny J. Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA. Genes Dev. 2007;21(24):3342–3355. doi: 10.1101/gad.455407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swann PF, et al. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science. 1996;273(5278):1109–1111. doi: 10.1126/science.273.5278.1109. [DOI] [PubMed] [Google Scholar]
- 5.Brown KD, et al. The mismatch repair system is required for S-phase checkpoint activation. Nat Genet. 2003;33(1):80–84. doi: 10.1038/ng1052. [DOI] [PubMed] [Google Scholar]
- 6.Li GM. The role of mismatch repair in DNA damage-induced apoptosis. Oncol Res. 1999;11(9):393–400. [PubMed] [Google Scholar]
- 7.de la Chapelle A. Genetic predisposition to colorectal cancer. Nat Rev Cancer. 2004;4(10):769–780. doi: 10.1038/nrc1453. [DOI] [PubMed] [Google Scholar]
- 8.Peltomäki P, Vasen H. Mutations associated with HNPCC predisposition -- Update of ICG-HNPCC/INSiGHT mutation database. Dis Markers. 2004;20(4-5):269–276. doi: 10.1155/2004/305058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heinen CD, Juel Rasmussen L. Determining the functional significance of mismatch repair gene missense variants using biochemical and cellular assays. Hered Cancer Clin Pract. 2012;10(1):9. doi: 10.1186/1897-4287-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ali H, Olatubosun A, Vihinen M. Classification of mismatch repair gene missense variants with PON-MMR. Hum Mutat. 2012;33(4):642–650. doi: 10.1002/humu.22038. [DOI] [PubMed] [Google Scholar]
- 12.Chao EC, et al. Accurate classification of MLH1/MSH2 missense variants with multivariate analysis of protein polymorphisms-mismatch repair (MAPP-MMR) Hum Mutat. 2008;29(6):852–860. doi: 10.1002/humu.20735. [DOI] [PubMed] [Google Scholar]
- 13.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 14.Christensen LL, et al. Functional characterization of rare missense mutations in MLH1 and MSH2 identified in Danish colorectal cancer patients. Fam Cancer. 2009;8(4):489–500. doi: 10.1007/s10689-009-9274-4. [DOI] [PubMed] [Google Scholar]
- 15.Drost M, et al. A rapid and cell-free assay to test the activity of lynch syndrome-associated MSH2 and MSH6 missense variants. Hum Mutat. 2012;33(3):488–494. doi: 10.1002/humu.22000. [DOI] [PubMed] [Google Scholar]
- 16.Gammie AE, et al. Functional characterization of pathogenic human MSH2 missense mutations in Saccharomyces cerevisiae. Genetics. 2007;177(2):707–721. doi: 10.1534/genetics.107.071084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lützen A, de Wind N, Georgijevic D, Nielsen FC, Rasmussen LJ. Functional analysis of HNPCC-related missense mutations in MSH2. Mutat Res. 2008;645(1-2):44–55. doi: 10.1016/j.mrfmmm.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 18.Martín-López JV, et al. The hMSH2(M688R) Lynch syndrome mutation may function as a dominant negative. Carcinogenesis. 2012;33(9):1647–1654. doi: 10.1093/carcin/bgs199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez SL, Kolodner RD. Functional analysis of human mismatch repair gene mutations identifies weak alleles and polymorphisms capable of polygenic interactions. Proc Natl Acad Sci USA. 2010;107(11):5070–5075. doi: 10.1073/pnas.1000798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mastrocola AS, Heinen CD. Lynch syndrome-associated mutations in MSH2 alter DNA repair and checkpoint response functions in vivo. Hum Mutat. 2010;31(10):E1699–E1708. doi: 10.1002/humu.21333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ou J, et al. Functional analysis helps to clarify the clinical importance of unclassified variants in DNA mismatch repair genes. Hum Mutat. 2007;28(11):1047–1054. doi: 10.1002/humu.20580. [DOI] [PubMed] [Google Scholar]
- 22.Ollila S, et al. Pathogenicity of MSH2 missense mutations is typically associated with impaired repair capability of the mutated protein. Gastroenterology. 2006;131(5):1408–1417. doi: 10.1053/j.gastro.2006.08.044. [DOI] [PubMed] [Google Scholar]
- 23.Ollila S, Dermadi Bebek D, Jiricny J, Nyström M. Mechanisms of pathogenicity in human MSH2 missense mutants. Hum Mutat. 2008;29(11):1355–1363. doi: 10.1002/humu.20893. [DOI] [PubMed] [Google Scholar]
- 24.van Ravesteyn TW, et al. LNA modification of single-stranded DNA oligonucleotides allows subtle gene modification in mismatch-repair-proficient cells. Proc Natl Acad Sci USA. 2016;113:4122–4127. doi: 10.1073/pnas.1513315113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dekker M, Brouwers C, te Riele H. Targeted gene modification in mismatch-repair-deficient embryonic stem cells by single-stranded DNA oligonucleotides. Nucleic Acids Res. 2003;31(6):e27. doi: 10.1093/nar/gng027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wielders EAL, et al. Functional analysis of MSH2 unclassified variants found in suspected Lynch syndrome patients reveals pathogenicity due to attenuated mismatch repair. J Med Genet. 2014;51(4):245–253. doi: 10.1136/jmedgenet-2013-101987. [DOI] [PubMed] [Google Scholar]
- 27.Wielders EAL, Dekker RJ, Holt I, Morris GE, te Riele H. Characterization of MSH2 variants by endogenous gene modification in mouse embryonic stem cells. Hum Mutat. 2011;32(4):389–396. doi: 10.1002/humu.21448. [DOI] [PubMed] [Google Scholar]
- 28.Aarts M, Dekker M, de Vries S, van der Wal A, te Riele H. Generation of a mouse mutant by oligonucleotide-mediated gene modification in ES cells. Nucleic Acids Res. 2006;34(21):e147. doi: 10.1093/nar/gkl896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drost M, et al. Genetic screens to identify pathogenic gene variants in the common cancer predisposition Lynch syndrome. Proc Natl Acad Sci USA. 2013;110(23):9403–9408. doi: 10.1073/pnas.1220537110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Plon SE, et al. IARC Unclassified Genetic Variants Working Group Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29(11):1282–1291. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee SD, Surtees JA, Alani E. Saccharomyces cerevisiae MSH2-MSH3 and MSH2-MSH6 complexes display distinct requirements for DNA binding domain I in mismatch recognition. J Mol Biol. 2007;366(1):53–66. doi: 10.1016/j.jmb.2006.10.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loader S, Shields C, Levenkron JC, Fishel R, Rowley PT. Patient vs. physician as the target of educational outreach about screening for an inherited susceptibility to colorectal cancer. Genet Test. 2002;6(4):281–290. doi: 10.1089/10906570260471813. [DOI] [PubMed] [Google Scholar]
- 33.Lin DP, et al. An Msh2 point mutation uncouples DNA mismatch repair and apoptosis. Cancer Res. 2004;64(2):517–522. doi: 10.1158/0008-5472.can-03-2957. [DOI] [PubMed] [Google Scholar]
- 34.de Wind N, Dekker M, van Rossum A, van der Valk M, te Riele H. Mouse models for hereditary nonpolyposis colorectal cancer. Cancer Res. 1998;58(2):248–255. [PubMed] [Google Scholar]
- 35.de Wind N, et al. HNPCC-like cancer predisposition in mice through simultaneous loss of Msh3 and Msh6 mismatch-repair protein functions. Nat Genet. 1999;23(3):359–362. doi: 10.1038/15544. [DOI] [PubMed] [Google Scholar]
- 36.te Riele H, Maandag ER, Berns A. Highly efficient gene targeting in embryonic stem cells through homologous recombination with isogenic DNA constructs. Proc Natl Acad Sci USA. 1992;89(11):5128–5132. doi: 10.1073/pnas.89.11.5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dekker M, et al. Transient suppression of MLH1 allows effective single-nucleotide substitution by single-stranded DNA oligonucleotides. Mutat Res. 2011;715(1-2):52–60. doi: 10.1016/j.mrfmmm.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 38.te Riele H, Maandag ER, Clarke A, Hooper M, Berns A. Consecutive inactivation of both alleles of the pim-1 proto-oncogene by homologous recombination in embryonic stem cells. Nature. 1990;348(6302):649–651. doi: 10.1038/348649a0. [DOI] [PubMed] [Google Scholar]
- 39.Warren JJ, et al. Structure of the human MutSalpha DNA lesion recognition complex. Mol Cell. 2007;26(4):579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 40.Guerrette S, Wilson T, Gradia S, Fishel R. Interactions of human hMSH2 with hMSH3 and hMSH2 with hMSH6: Examination of mutations found in hereditary nonpolyposis colorectal cancer. Mol Cell Biol. 1998;18(11):6616–6623. doi: 10.1128/mcb.18.11.6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmutte C, Sadoff MM, Shim KS, Acharya S, Fishel R. The interaction of DNA mismatch repair proteins with human exonuclease I. J Biol Chem. 2001;276(35):33011–33018. doi: 10.1074/jbc.M102670200. [DOI] [PubMed] [Google Scholar]
- 42.Ward R, et al. Impact of microsatellite testing and mismatch repair protein expression on the clinical interpretation of genetic testing in hereditary non-polyposis colorectal cancer. J Cancer Res Clin Oncol. 2002;128(8):403–411. doi: 10.1007/s00432-002-0361-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mangold E, et al. Tumours from MSH2 mutation carriers show loss of MSH2 expression but many tumours from MLH1 mutation carriers exhibit weak positive MLH1 staining. J Pathol. 2005;207(4):385–395. doi: 10.1002/path.1858. [DOI] [PubMed] [Google Scholar]
- 44.Pastrello C, et al. Integrated analysis of unclassified variants in mismatch repair genes. Genet Med. 2011;13(2):115–124. doi: 10.1097/GIM.0b013e3182011489. [DOI] [PubMed] [Google Scholar]
- 45.Dieumegard B, et al. Extensive molecular screening for hereditary non-polyposis colorectal cancer. Br J Cancer. 2000;82(4):871–880. doi: 10.1054/bjoc.1999.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Papadopoulos N, Lindblom A. Molecular basis of HNPCC: Mutations of MMR genes. Hum Mutat. 1997;10(2):89–99. doi: 10.1002/(SICI)1098-1004(1997)10:2<89::AID-HUMU1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]