

Abstract
The renal mineralocorticoid receptor (MR) is a steroid hormone receptor essential for maintaining electrolyte homeostasis. Its role in mediating effects of aldosterone was likely vital in enabling the evolution of terrestrial life. Dysregulated aldosterone-MR signaling has been identified as the cause of multiple clinical diseases, suggesting the physiological importance of the MR. While the physiology of this pathway has been studied for over 60 years, only more recently have genetic mouse models been available to dissect its function in vivo. This review will focus on recent advances in our knowledge of MR function with an emphasis on these models.
Keywords: mineralocorticoid, Na+ Cl− cotransporter, homeostasis, potassium, sodium
the mineralocorticoid receptor (MR) is essential for blood pressure regulation and electrolyte homeostasis. Physiologically, the MR mediates effects of the hormone aldosterone on electrolyte balance, promoting Na+ retention and K+ excretion. The evolution of the aldosterone-MR relationship likely occurred in response to dramatic changes in ionic stress experienced when organisms transitioned from aquatic to terrestrial life. In the ocean, aquatic organisms had the burden of fighting constant salt loading, whereas the prospect of a terrestrial existence presented the opposite problem, preventing their emergence from the sea. As the ability to synthesize aldosterone first appeared in tetrapods (3), the aldosterone-MR axis was likely part of the solution to maintain ion balance during this transition from water to land. The physiological importance of the MR and aldosterone is supported by the numerous clinical syndromes caused by dysregulated MR signaling [e.g., Addison's disease, primary aldosteronism, Liddle syndrome (17), and pseudohypoaldosteronism type I (PHA I) (4)].
The MR is expressed in multiple tissues and has physiological functions throughout the body (12), but it mediates aldosterone effects on ion homeostasis predominantly through activity in the kidney (16). Furthermore, while the MR is expressed along most of the nephron, aldosterone signaling occurs mainly along the aldosterone-sensitive distal nephron (ASDN) (16), which includes the connecting tubule (CNT), collecting duct (CD), and perhaps part of the late distal convoluted tubule (DCT). Specific localization of aldosterone effects along the nephron is related to ASDN-specific expression of the enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) (2, 9, 11, 18, 25), which converts cortisol to cortisone. Both cortisol and aldosterone can activate the MR and they have equal affinities for the receptor. Because cortisol circulates at 1,000 times the concentration of aldosterone, in most cells, cortisol outcompetes aldosterone for MR binding; however, in the ASDN, aldosterone effects are unmasked by 11β-HSD2 (6).
The essential transport elements of the ASDN are now established and include the basolateral Na-K-ATPase, which moves 3 Na+ out of the cell in exchange for 2 K+ in, keeping the intracellular [Na+] low. At the apical membrane, the epithelial Na+ channel (ENaC) mediates Na+ entry, down its electrochemical gradient. It lies in parallel with the renal outer medullary K+ channel (ROMK). When plasma [K+] rises, the cells in the ASDN respond by increasing K+ secretion, even if plasma aldosterone is kept constant. A rise in peritubular [K+] directly stimulates the Na-K-ATPase, as well as ENaC and ROMK, all of which favor K+ secretion (7). Aldosterone amplifies this response, as its secretion is also stimulated by rises in plasma [K+]. As a steroid hormone, aldosterone binds the MR in the cytosol, followed by translocation to the nucleus where the complex functions as a transcription factor. While there are many aldosterone-sensitive genes, most textbooks describe ENaC as the canonical final common pathway of aldosterone-MR signaling in the CNT/CD (5). Mechanistic details are complicated, but ultimate effects include increased ENaC transcription, apical abundance, and open probability (13). Increased ENaC activity increases Na+ reabsorption and the magnitude of the negative lumen potential, further enhancing K+ secretion through apical K+ channels (10).
More recently, aldosterone has been shown to regulate the thiazide-sensitive Na+ Cl− cotransporter (NCC). While aldosterone infusion increases (8, 23), and its absence reduces (21), NCC abundance and activity in animals, several observations raise questions as to whether the DCT MR mediates these effects. First, Liddle syndrome and PHA I, which are caused by disrupted ENaC function, phenocopy hyper- and hypoaldosterone states, suggesting the physiological effects of aldosterone are predominantly due to effects on ENaC and not NCC. Second, it has recently been observed that changes in plasma [K+] affect NCC abundance and function (20). As dysregulated aldosterone signaling almost always affects plasma [K+] via effects on the CNT/CD, observed NCC changes in hyper- and hypoaldosterone states may be secondary to plasma [K+] disturbances.
Over the last two decades, several mouse models have been developed to elucidate the role of aldosterone-MR signaling in vivo. These models have been instrumental in demonstrating the importance of the MR in vivo. The first of these models, developed by Schütz and colleagues (1), employed constitutive, total-body MR deletion. While not embryonic lethal, these animals died within the first 2 wk of life and displayed Na+ wasting and K+ retention. To determine whether the severe phenotype was due to loss of MR function in the CNT and CD, Berger and colleagues deleted the MR in principal cells, both constitutively (14), and in an inducible manner (15), using the aquaporin-2 promoter. Both of Berger's CNT/CD models displayed relatively mild phenotypes at baseline. Na+ wasting and increased urine volume only became apparent during Na+ restriction; on a normal diet, the mice appeared relatively normal. Because these mice did not phenocopy the effects of total body MR deletion, the authors suggested that MR effects in segments other than the CNT and CD likely explain differences between the models.
To determine whether the different phenotypes after total body deletion and CNT/CD deletion were due to MR effects along other nephron segments, we have recently deleted the MR along the entire nephron, in an inducible fashion, using the Pax8/LC1 CRE/Lox system (19). After MR deletion with doxycycline, the kidney-specific (KS) MR−/− animals survived but demonstrated weight loss, Na+ wasting, elevated plasma [K+], and reduced blood pressure, even on a normal Na+ and K+ diet. Additionally, they had reduced abundance of NCC, phosphorylated NCC (pNCC, the activate form), and total αENaC. While β and total γENaC levels remained unchanged, abundance of the cleaved form of γENaC was greatly reduced. Stress with either a low Na+ or high K+ diet greatly exacerbated the weight loss, Na+ wasting, and hyperkalemia experienced by the KS MR−/− mice. Furthermore, both of these dietary manipulations dramatically reduced pNCC abundance, had no effect on α or γENaC, and increased βENaC abundance compared with baseline in the KS MR−/− mice.
These data suggest that the MR is required for αENaC synthesis and γENaC cleavage, but not for synthesis of the β subunit. βENaC abundance is likely increased following treatment with low-Na+ and high-K+ diets because of MR-independent regulation. Regarding NCC, data are not only consistent with a direct role for MR in the DCT in regulating NCC, but also with effects on NCC being secondary to elevated plasma [K+]. To determine whether aldosterone affects NCC independent of altered plasma [K+], we next showed that aldosterone-infused wild-type animals had elevated NCC and pNCC abundance and reduced plasma [K+]. Consumption of a high-K+ diet prevented effects of aldosterone infusion on both plasma [K+] and NCC, suggesting that aldosterone infusion is not sufficient to affect NCC and changes are likely due to reduced plasma [K+].
K+ disturbances are becoming more widely accepted as dominant forces on NCC activity (20, 22, 24). Observations described herein provide evidence that aldosterone effects on NCC in disease models are predominantly secondary to changes in plasma [K+] and not direct effects on the MR in NCC-expressing cells (Fig. 1). If true, this would suggest that altered NCC activity in (pseudo-) hyper- and (pseudo-) hypoaldosterone states are homeostatic with respect to K+ balance at the expense of Na+ balance.
Fig. 1.
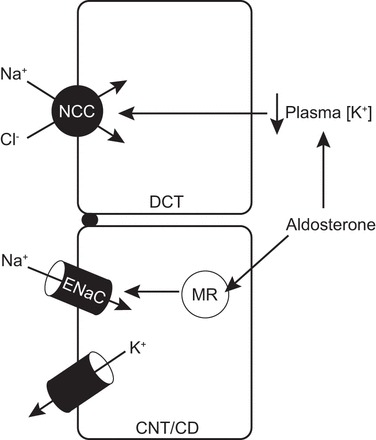
Model of how aldosterone affects Na+ Cl− cotransporter (NCC) in (pseudo-) hyperaldosterone states. Canonical aldosterone signaling increases epithelial Na+ channel (ENaC)-mediated Na+ reabsorption along the connecting tubule (CNT)-collecting duct (CD) (CNT/CD), driving electrogenic K+ secretion, which reduces plasma [K+]. Plasma [K+] secondarily signals NCC in the distal convoluted tubule (DCT) via altered membrane potential and intracellular [Cl−] (not shown). Opposite effects would occur to suppress NCC activity in (pseudo-) hypoaldosterone states.
For example, in PHA I, reduced ENaC activity promotes Na+ wasting and K+ retention. If Na+ were the dominant force on NCC, one would expect its activity to increase secondary to elevated circulating angiotensin II, and perhaps aldosterone, levels. However, with K+ dictating NCC function, its activity is likely reduced to promote distal Na+ flow in an attempt to drive ENaC-mediated K+ secretion. Unfortunately, ENaC function is impaired and this exacerbates Na+ wasting without ameliorating the hyperkalemia. This may explain the profound Na+ wasting seen in PHA I patients that appears disproportionate to the 2–3% of the filtered Na+ load normally reabsorbed by ENaC. If NCC is also turned off, patients may be wasting closer to 15% of the filtered Na+ load, causing the severe phenotype.
While this has yet to be shown in human disease, animal models support this hypothesis. Specifically, amiloride-treated mice had greatly reduced NCC abundance, an effect that was negated by maintaining plasma [K+] in the normal range with a low-K+ diet (20). A similar line of logic would apply to a hypoaldosterone state, such as Addison's disease, and the opposite scenario would occur in Liddle syndrome or primary hyperaldosteronism.
Details remain to be worked out, and it could be that both the DCT MR and plasma [K+] are able to affect NCC by their own independent mechanisms in vivo. A DCT-specific MR knockout mouse model could elegantly address this, but unfortunately, it has been technically difficult to generate such a model. Also remaining unanswered is why the total body MR knockout mouse dies so early in life. It seems likely that it is either because the renal MR is essential in the perinatal period, or because extrarenal MR is required for life, either perinatally or in adulthood. Just as the genetic mouse models described have enhanced our understanding of the MR, perhaps future ones can provide answers to these questions.
GRANTS
This work was funded by grants from the National Institutes of Health (2R01DK051496-15A1 and 5T32DK067864-10 to D. H. Ellison) and the Department of Veterans Affairs (1I0BX002228-01A1 to D. H. Ellison). A. S. Terker was the recipient of an American Heart Association predoctoral fellowship award (3PRE14090030). This work was performed by A. S. Terker in partial fulfillment for a PhD in Cell & Developmental Biology from Oregon Health and Science University.
REFERENCES
- 1.Berger S, Bleich M, Schmid W, Cole TJ, Peters J, Watanabe H, Kriz W, Warth R, Greger R, Schutz G. Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proc Natl Acad Sci USA 95: 9424–9429, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bostanjoglo M, Reeves WB, Reilly RF, Velazquez H, Robertson N, Litwack G, Morsing P, Dorup J, Bachmann S, Ellison DH. 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J Am Soc Nephrol 9: 1347–1358, 1998. [DOI] [PubMed] [Google Scholar]
- 3.Bridgham JT, Carroll SM, Thornton JW. Evolution of hormone-receptor complexity by molecular exploitation. Science 312: 97–101, 2006. [DOI] [PubMed] [Google Scholar]
- 4.Chang SS, Grunder S, Hanukoglu A, Rosler A, Mathew PM, Hanukoglu I, Schild L, Lu Y, Shimkets RA, Nelson-Williams C, Rossier BC, Lifton RP. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nature Genetics 12: 248–253, 1996. [DOI] [PubMed] [Google Scholar]
- 5.Eaton DC, Pooler JP. Vander's Renal Physiology (8th ed) Columbus, OH: McGraw-Hill, 2013. [Google Scholar]
- 6.Funder JW, Pearce PT, Smith R, Smith AI. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science 242: 583–585, 1988. [DOI] [PubMed] [Google Scholar]
- 7.Gennari FJ, Segal AS. Hyperkalemia: An adaptive response in chronic renal insufficiency. Kidney Int 62: 1–9, 2002. [DOI] [PubMed] [Google Scholar]
- 8.Kim GH, Masilamani S, Turner R, Mitchell C, Wade JB, Knepper MA. The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proc Natl Acad Sci USA 95: 14552–14557, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kyossev Z, Walker PD, Reeves WB. Immunolocalization of NAD-dependent 11 beta-hydroxysteroid dehydrogenase in human kidney and colon. Kidney Int 49: 271–281, 1996. [DOI] [PubMed] [Google Scholar]
- 10.Magill SB. Pathophysiology, diagnosis, and treatment of mineralocorticoid disorders. Compr Physiol 4: 1083–1119, 2014. [DOI] [PubMed] [Google Scholar]
- 11.Naray-Fejes-Toth A, Fejes-Toth G. Expression cloning of the aldosterone target cell-specific 11 beta-hydroxysteroid dehydrogenase from rabbit collecting duct cells. Endocrinology 136: 2579–2586, 1995. [DOI] [PubMed] [Google Scholar]
- 12.Nguyen Dinh Cat A, Jaisser F. Extrarenal effects of aldosterone. Curr Opin Nephrol Hypertens 21: 147–156, 2012. [DOI] [PubMed] [Google Scholar]
- 13.Palmer LG, Patel A, Frindt G. Regulation and dysregulation of epithelial Na+ channels. Clin Exp Nephrol 16: 35–43, 2012. [DOI] [PubMed] [Google Scholar]
- 14.Ronzaud C, Loffing J, Bleich M, Gretz N, Grone HJ, Schutz G, Berger S. Impairment of sodium balance in mice deficient in renal principal cell mineralocorticoid receptor. J Am Soc Nephrol 18: 1679–1687, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Ronzaud C, Loffing J, Gretz N, Schutz G, Berger S. Inducible renal principal cell-specific mineralocorticoid receptor gene inactivation in mice. Am J Physiol Renal Physiol 300: F756–F760, 2011. [DOI] [PubMed] [Google Scholar]
- 16.Rossier BC, Staub O, Hummler E. Genetic dissection of sodium and potassium transport along the aldosterone-sensitive distal nephron: importance in the control of blood pressure and hypertension. FEBS Lett 587: 1929–1941, 2013. [DOI] [PubMed] [Google Scholar]
- 17.Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, Gill JR Jr, Ulick S, Milora RV, Findling JW, Canessa CM, Rossier BC, and Lifton RP. Liddle's syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell 79: 407–414, 1994. [DOI] [PubMed] [Google Scholar]
- 18.Smith RE, Li KX, Andrews RK, Krozowski Z. Immunohistochemical and molecular characterization of the rat 11 beta-hydroxysteroid dehydrogenase type II enzyme. Endocrinology 138: 540–547, 1997. [DOI] [PubMed] [Google Scholar]
- 19.Terker AS, Lazelle RA, Yang CL, Ellison DH. Induced pan-nephron mineralocorticoid receptor knockout causes Na+ wasting and K+ retention. FASEB J 29: 968–962., 2015. [Google Scholar]
- 20.Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL, Ellison DH. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21: 39–50, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Todkar A, Picard N, Loffing-Cueni D, Sorensen MV, Mihailova M, Nesterov V, Makhanova N, Korbmacher C, Wagner CA, Loffing J. Mechanisms of renal control of potassium homeostasis in complete aldosterone deficiency. J Am Soc Nephrol 26: 425–438, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Lubbe N, Moes AD, Rosenbaek LL, Schoep S, Meima ME, Danser AH, Fenton RA, Zietse R, Hoorn EJ. K+-induced natriuresis is preserved during Na+ depletion and accompanied by inhibition of the Na+-Cl- cotransporter. Am J Physiol Renal Physiol 305: F1177–F1188, 2013. [DOI] [PubMed] [Google Scholar]
- 23.Velazquez H, Bartiss A, Bernstein P, Ellison DH. Adrenal steroids stimulate thiazide-sensitive NaCl transport by rat renal distal tubules. Am J Physiol Renal Fluid Electrolyte Physiol 270: F211–F219, 1996. [DOI] [PubMed] [Google Scholar]
- 24.Vitzthum H, Seniuk A, Schulte LH, Muller ML, Hetz H, Ehmke H. Functional coupling of renal K+ and Na+ handling causes high blood pressure in Na+ replete mice. J Physiol 592: 1139–1157, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou MY, Gomez-Sanchez EP, Cox DL, Cosby D, andGomez-Sanchez CE. Cloning, expression, and tissue distribution of the rat nicotinamide adenine dinucleotide-dependent 11 beta-hydroxysteroid dehydrogenase. Endocrinology 136: 3729–3734, 1995. [DOI] [PubMed] [Google Scholar]