

Abstract
Infiltrating macrophages are critically involved in pathogenic angiogenesis such as neovascular age-related macular degeneration (nAMD). Macrophages originate from circulating monocytes and three subtypes of monocyte exist in humans: classical (CD14+CD16-), non-classical (CD14-CD16+) and intermediate (CD14+CD16+) monocytes. The aim of this study was to investigate the role of circulating monocyte in neovascular age-related macular degeneration (nAMD). Flow cytometry analysis showed that the intermediate monocytes from nAMD patients expressed higher levels of CX3CR1 and HLA-DR compared to those from controls. Monocytes from nAMD patients expressed higher levels of phosphorylated Signal Transducer and Activator of Transcription 3 (pSTAT3), and produced higher amount of VEGF. In the mouse model of choroidal neovascularization (CNV), pSTAT3 expression was increased in the retina and RPE/choroid, and 49.24% of infiltrating macrophages express pSTAT3. Genetic deletion of the Suppressor of Cytokine Signalling 3 (SOCS3) in myeloid cells in the LysM-Cre+/-:SOCS3fl/fl mice resulted in spontaneous STAT3 activation and accelerated CNV formation. Inhibition of STAT3 activation using a small peptide LLL12 suppressed laser-induced CNV. Our results suggest that monocytes, in particular the intermediate subset of monocytes are activated in nAMD patients. STAT3 activation in circulating monocytes may contribute to the development of choroidal neovascularisation in AMD.
Keywords: Macrophage, monocyte, angiogenesis, retina, age-related macular degeneration, cytokine
Introduction
Macrophages are known to play an important role in pathogenic angiogenesis [1] in conditions such as malignant tumour [2], rheumatoid arthritis [3], diabetic retinopathy [4] and age-related macular degeneration (AMD) [5]. Macrophages are heterogeneous in phenotype and functions [6]. Naïve macrophages can be polarized into inflammatory (M1) macrophages upon IFN-γ and/or LPS stimulation, or the alternative activated wound-healing/immune regulatory (M2) subtype upon Th2 cytokines such as IL-4 or IL-10 stimulation [6]. M2 macrophages are known to play an important role in angiogenesis [6], although, M1 macrophages can also produce angiogenic growth factors (e.g., VEGF) under hypoxic conditions [7, 8]. The tissue microenvironment critically controls the phenotype and function of macrophages [9, 10].
Tissue macrophages originate from blood monocytes, which are also heterogeneous. Human monocytes can be classified into three subsets based on the surface CD16 and CD14 antigens: the “classical” (CD14+CD16-), “non-classical” (CD14-CD16+) [11], and the intermediate CD14+CD16+ [12, 13]. Different subsets of monocytes respond differently to inflammatory stimuli [14-16] and may contribute differently to human disease [17]. The classical monocytes, which account for 70-80% of circulating monocytes, express high levels of CD14, a co-receptor of LPS, and are potent producers of pro-inflammatory cytokines in response to bacterial infection [14, 15]. They play an important role in bacterial defence, inflammation and atherosclerosis [18, 19]. In contrast, non-classical monocytes, accounting for 10% of total monocytes, express high levels of CD16, an Fcγ receptor, and might respond preferentially to immunoglobulin related stimuli. These cells exhibit a “patrolling” (crawling) behaviour along vessel walls and react strongly against viruses [12, 20]. The intermediate monocytes have been linked to antigen presentation as well as angiogenesis [15]. In particular, the Tie-2 expressing monocytes (TEM), a subset of intermediate monocytes, are known to be the main progenitors that give rise to angiogenic macrophages [21] and promote angiogenesis in tumours [22, 23]. The intermediate monocytes and TEM may also be critically involved in myocardial remodelling [24] or liver regeneration [25].
Age-related macular degeneration (AMD) is progressive degeneration of the macula in people older than 60 years [5]. In the early stages, AMD is characterized by the presence of drusen –seen on colour fundus photography as yellowish deposits in the macula and pigmentary irregularities consisting of hypo- or hyper-pigmentation. Over time the lesions may progress into a form of macular atrophy known as geographic atrophy (GA) or neovascularization, a manifestation termed neovascular AMD (nAMD) [5]. GA is caused by the loss of retinal pigment epithelia (RPE), photoreceptors and the choriocapillaris, whereas nAMD is the growth of abnormal blood vessels into the macula resulting in haemorrhage or the leakage of blood fluids [5]. Both GA and nAMD lead to devastating visual loss and AMD poses a major social and economic burden to modern societies [26, 27].
The pathogenesis of AMD is not fully understood, although inflammation is known to play an important role. Genetic studies have identified a strong link between the risk of AMD and the polymorphisms of various immune related genes involving the complement pathways, Toll-like receptors (TLRs) and chemokine receptors (CX3CR1) [28-30]. Increased systemic immune activation evidenced by higher plasma levels of C-reactive protein [31, 32], complement fragments [33-35] and inflammatory cytokines (e.g., IL-1β, IL-6, IL-17) [36], and higher numbers of circulating neutrophils [37, 38] has been observed in AMD patients. Infiltrating macrophages have been detected at the lesion site of nAMD [39, 40], and in vivo studies in animal models have shown that depletion of monocytes/macrophages suppresses the development of choroidal neovascularization (CNV) [41]. These results suggest that macrophages may contribute critically to retinal angiogenesis in nAMD. However, which monocyte precursors give rise to macrophages in nAMD, how they infiltrate the macula and what controls their angiogenic phenotype remain unknown. In this study, we investigated the role of circulating monocytes in nAMD.
Materials and Methods
Study Participants
The study adhered to the tenets of the Declaration of Helsinki on research into human volunteers and the design was approved by the Northern Ireland HSC Trust Research Governance Permission through the Integrated Research Application System (IRAS). Written informed consent was obtained from all participants.
Participants were recruited from the macular disease clinics in Belfast Health and Social Care Trust, UK. Spouses or relatives who accompanied patients and who were confirmed to have no retinal disease by fundus photography and optical coherence tomography (OCT) were recruited as controls. Forty-nine participants, including 27 nAMD patients and 22 age-matched controls were recruited to the study. All participants were above 50 years of age and structured questionnaires were used to ascertain a history of medical conditions, current medication, family history of AMD, smoking habits and body mass index (BMI). Participants with systemic inflammatory or autoimmune disease and those on steroid therapy were excluded from the study.
The diagnosis of nAMD was by clinical examination and confirmed by multimodal imaging using a combination of colour, autofluorescence, OCT and fluorescein and indocyanine green angiography. Peripheral blood samples were drawn into tubes containing ethylenediaminetetraacetic acid and processed within 3h of collection.
Flow Cytometry
Blood samples (30 µl) were incubated with fluorochrome-labelled antibodies in a total volume of 100 µl FACS buffer (PBS/1% fetal calf serum (FCS)) for 45min. Red blood cells were removed and samples fixed with lysis/fix solution (BD Biosciences, Oxford, UK). All samples were examined by flow cytometry (FACS CANTO II; BD Biosciences), and data analysed using the FlowJo software (Tree Star, Ashland, OR, USA). The following antibodies were used: CD14-APC-Cy7, CD16-Pacific Blue, CD62L-APC, MHC-II (HLA-DR, DP, DQ)-FITC (BD Biosciences), CCR2-PerCP, CX3CR1-PE-Cy7 (BioLegend UK Ltd., London, UK) and HLA-DR-PE (eBioscience, Hatfield, UK).
Detecting STAT3 Activation in Circulating Leukocytes
Blood samples (100 µl) were incubated with recombinant human IL-6 (100 ng/ml; BD Biosciences) at 37°C for 15min. Red blood cells were removed and samples fixed with Lyse/Fix buffer (BD Biosciences). After thorough washing, cells were incubated with Human TruStain FcR Blocking Solution (BioLegend) for 10min followed by incubation with CD16-PE and HLA-DR-APC (BD Biosciences). Cells were permeabilised using pre-chilled Perm buffer III solution (BD Biosciences) and then incubated again with TruStain FcR Blocking Solution, followed by incubation with FITC-conjugated anti-pSTAT3 or mouse IgG2a-FITC isotype control (BD Biosciences). All samples were examined by flow cytometry, and data analysed using the FlowJo software (see above).
PBMC Isolation and VEGF Measurement in Cell Culture Supernatants
Peripheral blood mononuclear cells (PBMC) were isolated from whole blood by Ficoll-Paque (Histopaque; Sigma-Aldrich, UK) density gradient centrifugation. Cells were cultured in RPMI 1640 medium containing 10% FCS and 1% penicillin-streptomycin under normoxia (21% oxygen) or hypoxia conditions (1% oxygen) for 16h, and the supernatants were collected and stored at -80°C until analysis.
Cytometric Bead Array (CBA; BD Biosciences) was used to measure VEGF in cell culture supernatants according to the manufacturer’s instructions. Briefly, beads coated with anti-VEGF antibodies were mixed and incubated with cell culture supernatants or standards for 1h, followed by incubation for 2h with PE-conjugated anti-VEGF detection. Samples were analysed by flow cytometry (FACS CANTO II; BD Biosciences). VEGF concentrations were calculated using the FCAP Array software version 3.0 (BD Biosciences). The total protein concentration was measured using a Pierce BCA protein assay kit (Thermo Fisher Scientific, Renfrew, UK). The concentration of VEGF was normalised to the total protein concentration (pg/mg total protein).
Real Time RT-PCR
Total RNAs were extracted from PBMC using Qiagen’s RNeasy Mini kit (Qiagen, Manchester, UK) and then reverse transcripted into cDNA using a Superscript II Reverse Transcriptase kit (Invitrogen, Renfrew, UK) as per manufacturer’s instructions. The cDNA samples were stored at -20°C until further use.
Real time PCR was carried out in a total reaction volume of 10 µl containing 2 µl of cDNA, 5 µl of 2X SYBR green (Fermentas, Ontario, Canada), 0.5 µl of 10 mM forward (FP) and reverse primer (RP) and 2.5 µl RNase/DNase free water. Reactions were performed in triplicates. Thermal cycling was completed using the LightCycler 480 Real-Time PCR System (Roche Diagnostics Ltd, West Sussex, UK). Expression of the target genes relative to the reference gene was calculated using the advanced relative quantification analysis module of the LightCycler 480 software 1.5 (Roche Diagnostics Ltd) based on the delta delta CT method. 60S acidic ribosomal protein P0 (RPLP0 or HuP0) was used as a reference gene as it was shown to be a suitable reference gene for gene expression studies in PBMC cultures [42]. The following primers were used: IL-6R FP: 5′-ATCGGGCTGAACGGTCAAA G-3′, IL-6R RP: 5′-GGCGTCGTGGATGACACAG-3′, gp130 FP: 5′-GCAACACACAAGTTTGCTGATT-3′, gp130 RP: 5′-CCTTCCCAAGGGCATTCTCTG-3′, SOCS3 FP: 5′-CCTGCGCCTCAAGACCTTC-3′, SOCS3 RP: 5′-GTCACTGCGCTCCAGTAGAA-3′, RPLP0 FP: 5′-CCATTCTATCATCAACGGGTACAA-3′, RPLP0 RP: 5′-AGCAAGTGGGAAGGTGTAATCC-3′.
Animals
Eight to twelve weeks old C57BL/6J, CX3CR1gfp/+, and LysM-Cre+/-:SOCS3fl/fl mice were provided by the Biological Research Unit of the Queen’s University Belfast. All animals were housed in a temperature and light-controlled environment with 12h-light-dark cycle. All procedures were complied with the UK Animals (Scientific Procedures) Act 1986, and the protocols were approved by the Animal Welfare & Ethical Review Board of the Queen’s University Belfast.
Laser-Induced Choroidal Neovascularization
CNV was induced in mice as previously described [43]. Briefly, mice were anesthetized with an intraperitoneal injection of 75 mg/kg ketamine and 7.5 mg/kg xylazine. The pupils were dilated with 1% tropicamide (Chauvin Pharmaceuticals Ltd., Essex, UK). Three 532nm diode laser spots (150mW, 100msec, 100µm, HGM Elite 532 Green Laser, Litechnica) were applied to each fundus. The rupture of Bruch’s membrane was confirmed by the appearance of a vaporization bubble at the time of laser treatment. At different times after CNV induction, eyes were collected for Western blotting (on day 1 and 3) or immunohistochemistry investigation (on day 5, 7, 10 and 14).
In Vivo Treatment
Immediately after CNV induction, mice were treated with an intraperitoneal injection of a STAT3 inhibitor LLL12 (once daily, 5 mg/kg, 57313, Merck Chemicals Ltd., Nottingham, UK) or vehicle (Dimethyl Sulfoxide, DMSO). LLL12 specifically inhibits STAT3 phosphorylation (Tyr705) and activation [44]. On day 7, fluorescein angiography was performed as previously described [43, 45]. The animals were then sacrificed and eyes collected and fixed in 2%PFA for further investigation.
Immunostaining of RPE/Choroidal Whole Mounts
RPE/choroidal whole mounts were prepared as described previously [43]. Briefly, tissues were permeabilised with 0.3% triton X-100 for 1h. The samples were then blocked with 6% BSA and incubated with biotinylated Griffonia Simplicifolia Lectin I-Isolectin B4 (1:100, Vector Laboratories Ltd., Peterborough, UK) and rabbit anti-mouse collagen IV (1:100, ABD Serotec Ltd, Oxford, UK), followed by FITC-conjugated Streptavidin (1:200, Dako, Denmark) and goat anti-rabbit AlexFluor 594 (1:200, Invitrogen, Paisley, UK) for 2h. Samples were observed by confocal microscopy (Eclipse TE200-U, Nikon UK Ltd., Surry, UK).
Immunoblotting
Retina and RPE-choroid were lysed in RIPA buffer with protease inhibitor cocktails and phosphatase inhibitors (Sigma-Aldrich). The protein concentration was determined by BCA kit (Thermo Scientific). Thirty microgram protein was loaded on a 10% SDS-PAGE gel and transferred to an Immobilon-FL PVDF membrane (Millipore, Watford, UK). The membranes were incubated sequentially with rabbit anti-STAT3 (79D7, 1:1000) or rabbit anti-pSTAT3 (Tyr705, D3A7, 1:1000) (both from Cell Signalling, Danvers, MA, USA), followed by goat anti-rabbit IgG (1:10,000, Li-COR Biosciences, Cambridge, UK). The membranes were imaged with Odyssey infrared imaging system (Li-COR Biosciences), and analysed by ImageStudioLite Software (Li-COR Biosciences). The mouse anti-β actin (C4, 1:10,000, Santa Cruz, Dallas, Texas, USA) was used as a housekeeping control protein.
Data Analysis
Data were analysed using the Statistical Package for the Social Sciences (version 21, SPSS Inc., Armonk, NY). The distribution of continuous variables was assessed for normality using the Kolmogorov-Smirnov test and logarithmic transformation was performed if necessary to achieve normal distribution. The data were then analysed using the Independent samples t-test or one-way ANOVA.
For the associations that were significant in the univariate analysis, multinomial logistic regression was performed to adjust for age and gender. All variables were also tested for association with family history of AMD, history of cardiovascular disease, history of hypertension, history of diabetes, smoking habits, BMI, taking of cardiovascular medication, vitamins and low dose aspirin using the Independent samples t-test, one-way ANOVA or Pearson’s correlation. If significant associations were identified, adjustments were made in the multinomial logistic regression analysis.
Data from animal studies were analysed using either unpaired t test or one-way ANOVA (for multiple group test). P values <0.05 were considered statistically significant.
Results
Monocyte Subsets in nAMD Patients and Controls
Leukocyte antigen CD14, CD16 and HLA-DR were used to identify blood monocytes (Fig. 1A). A gating strategy previously suggested by Heimbeck et al. [46, 47] was used in this study. Firstly, CD16+ and CD14+ cells were gated (R1, Fig. 1A). These cells were then plotted using CD16 and HLA-DR to exclude CD16+HLA-DR- neutrophils/NK cells (Fig. 1B). The remaining cells (R2) were then plotted using CD14 and CD16 to identify three subsets of monocytes: CD14+CD16- (classical), CD14+CD16+ (intermediate) and CD14CD16+ (non-classical) (Fig. 1C). The classical monocytes constituted 70-80%, whereas the non-classical and intermediate each constituted 10-15% of total monocyte. The percentage of different subsets of monocyte did not differ between nAMD and controls (Fig. 1D).
Fig. (1).
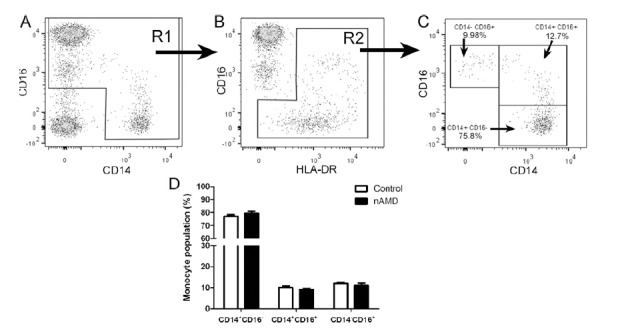
Monocyte subsets in nAMD patients. A-C gating strategy to identify true monocyte. CD14+ and CD16+ cells were gated in R1 (A). Cells in R1 were then plotted using CD16 and HLA-DR, and CD16+HLA-DR- cells were excluded (B). The remaining cells (R2) were true monocytes and were further plotted using CD16 and CD14 to identify the classical monocytes (CD14+CD16-), non-classical monocytes (CD14-CD16+) and intermediate monocytes (CD14+CD16+). D, the population of different subsets of monocyte in nAMD patients and controls. Mean ± SEM, n = 22 in control, n = 27 in nAMD.
Chemokine Receptor Expression in Blood Monocytes in nAMD Patients
L-selectin (CD62L), chemokine receptors CCR2 and CX3CR1 are commonly used to differentiate different subsets of monocytes [17, 48]. The classical monocytes expressed higher levels of CD62L, followed by intermediated and non-classical monocytes (Fig. 2A). There was no significant difference in expression levels of CD62L in different subsets of monocytes between nAMD and controls (Fig. 2B). The classical and intermediate monocytes expressed significantly higher levels of CCR2 compared to the non-classical monocytes (Fig. 2C), and the expression levels did not differ between nAMD and controls in different monocytes (Fig. 2D). The expression level of CX3CR1 was significantly higher in the non-classical and intermediate monocytes compared to that in classical monocytes (Fig. 2E). Interestingly, the intermediate monocytes from nAMD patients expressed significantly higher levels of CX3CR1 than their counterparts from controls, and the difference remained significant after adjustment for age and gender (Fig. 2F). The non-classical monocytes from nAMD also expressed signi-ficantly higher levels of CX3CR1 compared to controls (p = 0.048), however, after adjusting for age and gender, the difference became insignificant (Fig. 2F). There was no significant difference in the expression levels of CX3CR1 in classical monocytes between nAMD and controls (Fig. 2F).
Fig. (2).
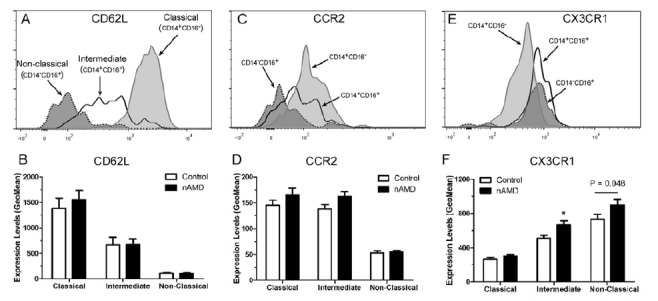
The expression of L-selectin (CD62L), CCR2 and CX3CR1 in different subsets of monocytes in nAMD patients. (A, C, E) Representative histogram from healthy control donor showing CD62L (A), CCR2 (C) and CX3CR1 (E) expression in different subsets of monocytes. (B, D, F), the average expression level of CD62L (B), CCR2 (D) and CX3CR1 (F) in monocytes from nAMD patients and controls. *, P < 0.05 compared to control of the same monocyte subset. Mean ± SEM, n = 22 in control; n = 27 in nAMD.
MHC-II and HLA-DR Expression in Circulating Monocytes in nAMD Patients
To further explore the difference between monocytes from nAMD patients and controls, we examined the activation state of the cells using MHC-II (HLA-DR, DP, DQ) and HLA-DR as markers. The intermediate monocytes expressed the highest levels of MHC-II and HLA-DR, followed by the non-classical monocytes, and then the classical monocytes (Fig. 3A-D). There was no significant difference in the expression levels of MHC-II in different subsets of monocyte between nAMD and controls (Fig. 3B). However, the intermediate monocytes from nAMD patients expressed significantly higher levels of HLA-DR compared to those from controls (Fig. 3D, E), and the difference remain significant after adjusting for age, gender, vitamin intake, diabetes and BMI. Our results suggest that the intermediate monocytes from nAMD patients are more active than those from age-matched controls.
Fig. (3).
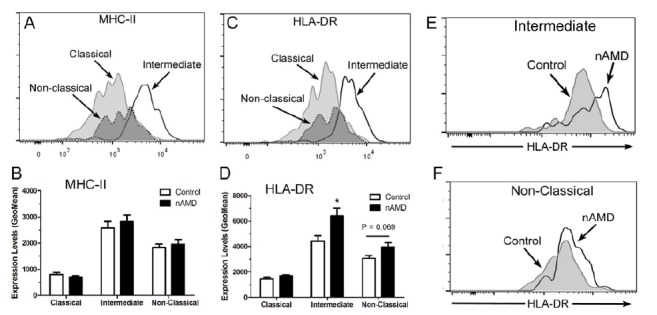
MHC-II and HLA-DR expression in different subsets of monocyte in nAMD patients. (A, C) Representative histogram from healthy control donor showing MHC-II (A) and HLA-DR (C) expression in different subsets of monocytes. (B, D), the average expression level of MHC-II (B) and HLA-DR (D) in monocytes from nAMD patients and controls. *, P < 0.05 compared to control of the same monocyte subset. Mean ± SEM, n = 22 in control; n = 27 in nAMD. (E, F), representative histogram showing HLA-DR expression in intermediate monocytes (E), and non-classical monocytes (F) in nAMD and controls.
Angiogenic Phenotype of Monocytes from nAMD Patients
To further understand if the increased activation of circulating monocyte resulted in an angiogenic phenotype in nAMD patients, VEGF production by PBMCs was measured. PBMCs from nAMD patients produced 3~5 times more VEGF than those from controls in both normoxic and hypoxic conditions (Fig. 4A).
Fig. (4).
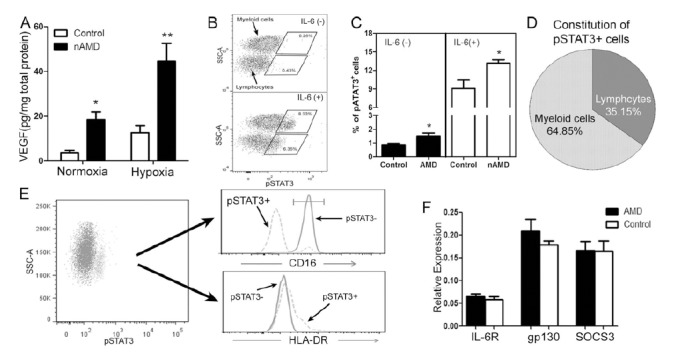
Angiogenic phenotype of circulating monocyte in nAMD patients. A, VEGF production by PBMC from nAMD patients and controls cultured under normoxia or hypoxia conditions, * P < 0.05; **, P < 0.01 compared to controls; B, flow cytometry analysis of pSTAT3 expression in blood cells with/without IL-6 stimulation; C, the percentage of pSTAT3+ circulating immune cells in nAMD and controls with/without IL-6 stimulation; *, P < 0.05 compared to controls. D, the constitution of pSTAT3+ immune cells; E, the phenotype of pSTAT3+ cells; F, mRNA expression of IL6R, gp130 and SOCS3 in PBMC from nAMD patients and controls. Mean ± SEM, n = 13 in controls, n = 12 in nAMD.
The JAK2/STAT3 pathway critically regulates vegf gene expression [49]. To understand whether this cytokine pathway is involved in the angiogenic phenotype of monocytes in nAMD patients, p-STAT3 expression in blood cells from nAMD and controls was examined by flow cytometry. In the absence of any stimulation a small population of leukocytes (0.5-2%) expressed p-STAT3+ (Fig. 4B) and the population of p-STAT3+ cells was significantly higher in nAMD patients than that in controls (Fig. 4C). A brief (20min) treatment with IL-6 resulted in a significant increase in pSTAT3 expression in blood cells, including lymphocytes and myeloid cells (Fig. 4B). Significantly more pSTAT3+ cells were detected in nAMD patients compared to that in controls following IL-6 stimulation (Fig. 4C). 64.85% of pSTAT3+ leukocytes were myeloid cells and 35.15% were lymphocytes (Fig. 4B, D). Approximately 90% of pSTAT3+ myeloid cells were CD16- (Fig. 4E) and they express higher levels of HLA-DR (Fig. 4E), suggesting that the majority of them are likely to be monocytes rather than neutrophils. Due to lack of anti-CD14 antibodies that are suitable for the intracellular pSTAT3 staining protocol (none available commercially), we were unable to further identify the subset(s) of pSTAT3-expressing monocytes.
The JAK2/STAT3 pathway is activated through the IL-6R/gp130 receptor, and the pathway is negatively regul-ated by the Suppressor Of Cytokine Signalling 3 (SOCS3) [50]. Real-time RT-PCR showed that PBMC from nAMD patients and controls expressed comparable levels of IL-6R, gp130 and SOCS3 mRNAs (Fig. 4F), suggesting that increased pSTAT3 expression in nAMD patients was not caused by abnormalities of the IL-6R/gp130 or SOCS3.
Taken together, our results suggest that monocytes from nAMD patients are more active and exhibit an angiogenic phenotype compared to monocytes from age-matched controls. The angiogenic phenotype is related to increased STAT3 activation in these cells.
Laser-Induced Choroidal Neovascularization in the LysM-Cre+/-:SOCS3fl/fl Mice
To understand whether increased STAT3 activation in circulating monocytes could promote retinal angiogenesis, we conducted the laser-induced CNV in the LysM-Cre+/-:SOCS3fl/fl mice, in which SOCS3 was deleted in myeloid cells resulting in spontaneous STAT3 activation [51, 52]. Although the expression of non-phosphorylated STAT3 did not differ between the KO and WT mice (Fig. 5A, B), significantly increased p-STAT3 expression was detected in the retina (Fig. 5C) and RPE/choroid (Fig. 5D) in the LysM-Cre+/-:SOCS3fl/fl mice, suggesting a spontaneous activation of STAT3 in ocular myeloid cells (e.g., retinal microglia, choroidal macrophages and dendritic cells) in the KO mice.
Fig. (5).
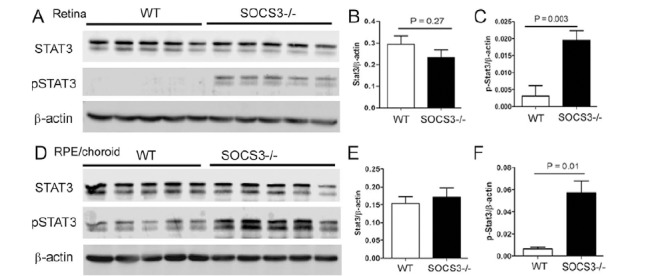
Western blot of STAT3 and pSTAT3 in the retina, RPE/choroid of WT and SOCS3 KO mice. Retinas (A-C) and RPE/choroidal tissues (D-F) were collected from 10-12 week old WT and LysM-Cre:SOCS3fl/fl (SOCS3-/-) mice for Western blotting analysis of inactivated STAT3 and activated pSTAT3. β-actin was used as housekeeping reference protein for semi-quantitative analysis. B, relative STAT3 expression in the retina of WT and SOCS3-/- mice. C, relative pSTAT3 expression in the retina of WT and SOCS3-/- mice. E, relative STAT3 expression in the RPE/choroid of WT and SOCS3-/- mice. F, relative pSTAT3 expression in the RPE/choroid of WT and SOCS3-/- mice. Mean ± SD, n = 5.
At 5 and 7 days after CNV induction, the LysM-Cre+/-:SOCS3fl/fl mice developed more severe lesion compared to WT mice (Fig. 6A-D, I). The CNV lesion regressed from day 10-14 and the lesion size was similar between the LysM-Cre+/-:SOCS3fl/fl mice and WT mice at this stage (Fig. 6E-I). This result suggests that increased STAT3 activation in myeloid cells promoted retinal angiogenesis.
Fig. (6).
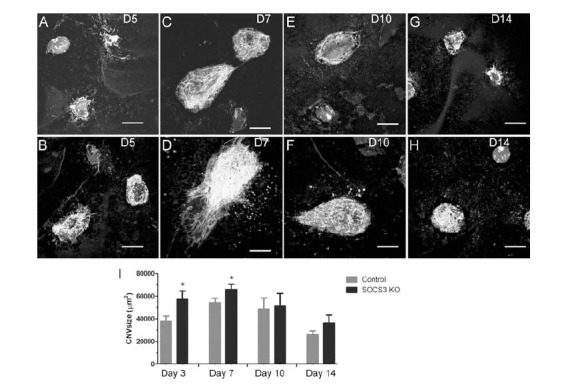
Laser-induced CNV in WT and SOCS3 KO mice. Three laser burns were applied to each eye in WT (A, C, E, G) and LysM-Cre+/-:SOCS3fl/fl (SOCS3-/-) (B, D, F, H) mice. Samples were collected at day 5 (A, B), 7 (C, D), 10 (E, F), and 14 (G, H) post-CNV induction. RPE/choroidal flatmounts were stained for Isolectin B4, and imaged by confocal microscopy. Scale bar – 100 µm. I, histogram showing the average size of CNV in each group from different time points. *, P < 0.05 compared to controls of the same time point. Mean ±SEM, N = 5 mice. Two-way ANOVA, followed by Bonferroni Post Test.
The Effect of STAT3 Inhibition in Laser-Induced Choroidal Neovascularization
To further confirm the role of myeloid cell STAT3 activation in retinal angiogenesis, the expression level of pSTAT3 in the laser-induced CNV was examined. The non-phosphorylated STAT3 was detected in both the retina (Fig. 7A) and RPE/choroid (Fig. 7B) in control and laser-treated eyes, and the expression levels increased significantly in the retina 72h post-
Fig. (7).
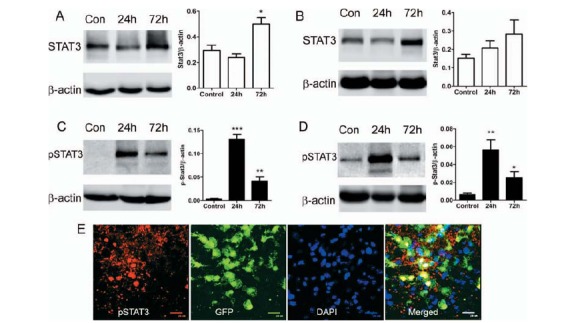
The expression of STAT3, pSTAT3 in the retina and RPE/choroid in laser-induced CNV. Laser-induced CNV was conducted in WT C57BL/6 mice. Eyes were collected at 24h and 72h after and retina (A, C) and RPE/choroid (B, D) were processed for Western Blotting analysis of STAT3 (A, B) and pSTAT3 (C, D). β-actin was used as housekeeping reference protein. Mean ± SD, n = 5. *, P < 0.05, **, P < 0.01, ***, P < 0.001 compared to non-CNV controls. One-Way ANOVA followed by Tukey’s Multiple Comparison Test. E, Laser-induced CNV was conducted in CX3CR1gfp/+ mice. 24h later, eyes were collected for confocal flatmount investigation of pSTAT3 expression. A confocal image of RPE/choroidal flatmount showing pSTAT3 expression in CNV lesion and infiltrating CX3CR1+ cells.
CNV induction (Fig. 7A). The expression of pSTAT3 was markedly increased at 24h post-CNV induction in both the retina (Fig. 7C) and RPE/choroid (Fig. 7D), which then reduced (but was still significantly higher than in non-CNV tissues) at 72h (Fig. 7C, D). RPE/ choroidal whole mount staining showed that 49.24 ± 3.98% of infiltrating CX3CR1+ macrophages expressed pSTAT3 at 24h after CNV induction (Fig. 7E).
Administration of a STAT3 inhibitor LLL12 significantly reduced fluorescence leakage at day 7 post-CNV induction (Fig. 8A-C). Confocal microscopy of RPE/choroid flatmount stained for isolectin B4 revealed significantly reduced size of CNV following LLL12 but not vehicle treatment (Fig. 8D-G).
Fig. (8).
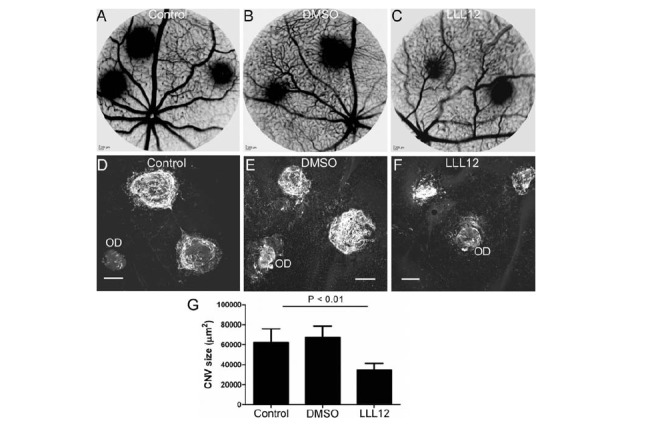
The effect of LLL12 in laser-induced CNV. CNV was induced in WT mice by applying laser ruptures in the Bruch’s membrane. Immediately after CNV induction, animals were untreated (A, D) or treated with LLL12 (B, E) or Vehicle (DMSO) (C, F) daily (see Materials & Methods). On day 7 post CNV induction fluorescence angiography (A-C) was conducted, and the eyes were then collected. RPE/choroidal flatmounts were stained for Isolectin B4 and examined by confocal microscopy (D-F). OD, optic disc. G, the average size of CNV in each group. Mean ± SEM, N = 6 mice. Tukey’s Multiple Comparison Test.
Discussion
We show in this study that PBMCs from nAMD patients expressed significantly high levels of pSTAT3 and produced 3~5 times more VEGF compared to those from controls. We also show that circulating monocytes, in particular the CD14+CD16+ intermediate subset, from nAMD patients were more active and expressed significantly higher levels of CX3CR1 and HLA-DR compared to those from controls. Using a mouse model of laser-induced CNV, we further show that STAT3 activation in monocytes/macrophages contributes significantly to the development of choroidal neovascularization. Our results suggest that activation of circulating monocytes, in particular the CD14+CD16+ intermediate subset may contribute to macular lesion development in nAMD.
Compelling evidence suggests that monocytes and macrophages are involved in the pathogenesis of
nAMD. Infiltrating macrophages have been detected in CNV lesions in nAMD patients [39]. Animal studies have shown that circulating CCR2+ monocytes are critically involved in retinal angiogenesis in the laser-induced CNV [41, 53, 54], and depletion of monocytes reduces CNV lesion size [41, 53]. However, exactly how monocytes contribute to CNV in nAMD remains poorly defined. After monocytes are recruited to the lesion site, they can be differentiated into either macrophages or dendritic cells - both can argument choroidal neovascularization [41, 55]. In this study, we found that increased STAT3 activation in monocytes is related to an angiogenic phenotype of PBMC in nAMD patients. STAT3 is a signal transducer as well as a gene transcription activator that is critically involved in VEGF production and angiogenesis under both physiological (e.g. development) and pathological conditions [56-58]. STAT3 can be activated by a number of growth factors, including IL-6, IL-10, IL-17, GM-CSF, IGF, and EPO [52, 58], and activation of this pathway leads to a diverse transcriptional activation, including vegf expression [58, 59]. Interestingly, STAT3 can also be activated through the VEGF/VEGFR2 pathway [60-62]. STAT3 can modulate HIF-1-mediated VEGF expression in various cells [63-66] and constitutive STAT3 activity in tumour cells can upregulate VEGF expression and induce tumour angiogenesis [67]. The basal levels of pSTAT3 expression and the VEGF production in PBMC are both higher in nAMD patients compared to controls (Fig. 4A, C), suggesting that constitutive STAT3 activation may contribute to higher levels of VEGF production. The constitutive STAT3 activation in nAMD is not caused by the abnormalities of the IL-6R/gp130 receptor or the negative regulator SOCS3 as their expression levels were comparable between nAMD patients and controls. Increased STAT3 activation in PBMC from nAMD patients was also observed in IL-6 treated cells. Since the same amount of IL-6 was used to stimulate cells from nAMD patients and controls, and IL-6R/gp130 was expressed at similar levels, the higher levels of response to IL-6 by PBMC from nAMD patients may suggest that cells in nAMD patients are more sensitive (or primed) for STAT3 activation and VEGF production. A previous study by Jiang et al. has reported increased pSTAT3 expression in aged human eyes, particularly in eyes with large drusen [68], suggesting that the pSTAT3 is related to retinal aging and drusen development.
The role of STAT3 activation in circulating immune cells in retinal angiogenesis was further confirmed by our in vivo study using the LysMCre+/-:SOCS3fl/fl mice, where SOCS3 was deleted specifically in myeloid cells (monocytes and neutrophils) [51, 52], resulting in increased STAT3 activation through the JAK2/STAT3 pathway [50, 57]. In the laser-induced CNV, where macrophages play an important role in retinal angiogenesis [41, 53, 54], the LysMCre+/-:SOCS3fl/fl mice developed more severe CNV at days 5 and 7 post-laser treatment, and inhibition of pSTAT3 suppressed laser-induced CNV. Our results suggest that STAT3 activation in myeloid cells promotes pathogenic retinal angiogenesis.
The intermediate monocytes express higher levels of MHC-II, HLA-DR, and CCR5 [17] and are linked to antigen presentation, inflammation and angiogenesis [15]. They are known to be involved in atherosclerosis [69, 70], in particular, activation of intermediate monocyte increases the cardiovascular risk in patients with chronic kidney disease [71]. We found that the intermediate monocytes from nAMD patients expressed higher levels of HLA-DR and CX3CR1 than those from controls. A previous study has reported increased CCR2 expression in CD14+CD16+ intermediate monocytes in nAMD patients [72]. In our study, although the expression of CCR2 was slightly increased in the CD14+CD16- and CD14+CD16+ monocytes in nAMD patients compared to those from controls, the increment did not reach statistical significance (Fig. 2C, D). The discrepancy may be related to differences in patient populations and gate strategies in flow cytometry data analysis. Although the link between increased CX3CR1 and HLA-DR expression and pSTAT3 expression/VEGF production in circulating monocytes is unknown, genetic studies have shown that polymorphisms of cx3cr1 [28] and HLA [73] are associated with AMD. CX3CR1 is the receptor for chemokine CX3CL1, and is known to be involved in monocyte adhesion and migration [74, 75]. More recently, it has been shown that the CX3CR1-CX3CL1 pathway also plays a major role in monocyte survival [76]. In the laser-induced CNV, CX3CR1+ cells were recruited to the lesion site prior to angiogenesis and many of them expression pSTAT3 (Fig. 7E). HLA-DR is a subtype of the MHC-II molecule encoded by the human leukocyte antigen complex on chromosome 6p21.31, and is constitutively expressed by antigen presenting cells (e.g., dendritic cells, monocytes, etc.). The primary function of HLA-DR is to present peptide antigens to CD4 T cells. The expression levels of MHC-II and its subtypes such as HLA-DR can be upregulated upon pathogen or cytokine (e.g., interferon) stimulation. Whether the increased HLA-DR and CX3CR1 expression in nAMD patients relates to genetic predisposition warrants further investigation. Nevertheless, our results suggest that the intermediate monocytes may contribute to nAMD pathology, probably through producing inflammatory cytokines and angiogenic growth factors.
Conclusion
Our study suggests that circulating monocytes, in particular the CD14+CD16+ intermediate monocytes in nAMD patients appear to be pre-conditioned to an inflammatory phenotype, and may contribute to CNV development when they are recruited to the damaged macula. Further understanding the role of the intermediate monocytes in CNV pathogenesis may uncover novel targets for therapy.
ACKNOWLEDGEMENTS
This work was supported by the Dunhill Medical Trust (R188/0211), Fight for Sight (1361 / 1362) and Guide Dogs for the Blind Association UK (2008-5a). The authors thank the patients who participated in this study. We also thank the research nurses Rebecca Denham and Georgina Sterrett for their help in patient recruitment.
Abbreviations
- AMD
Age-related macular degeneration
- CNV
Choroidal neovascularization
- nAMD
Neovascular AMD
- PBMC
Peripheral blood mononuclear cell
- pSTAT3
Phosphorylated STAT3
- RPE
Retinal pigment epithelium
- SOCS3
Suppressor of cytokine signalling 3
- STAT3
Signalling transducer and activator of transcription 3
- VEGF
Vascular endothelial growth factor.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.Sunderkotter C., Steinbrink K., Goebeler M., Bhardwaj R., Sorg C. Macrophages and angiogenesis. J. Leukoc. Biol. 1994;55:410–422. doi: 10.1002/jlb.55.3.410. [DOI] [PubMed] [Google Scholar]
- 2.Dirkx A.E., Oude Egbrink M.G., Wagstaff J., Griffioen A.W. Monocyte/macrophage infiltration in tumors: modulators of angiogenesis. J. Leukoc. Biol. 2006;80:1183–1196. doi: 10.1189/jlb.0905495. [DOI] [PubMed] [Google Scholar]
- 3.Szekanecz Z., Besenyei T., Paragh G., Koch A.E. New insights in synovial angiogenesis. Joint Bone Spine. 2010;77:13–19. doi: 10.1016/j.jbspin.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crawford T.N., Alfaro D.V., III, Kerrison J.B., Jablon E.P. Diabetic retinopathy and angiogenesis. Curr. Diabetes Rev. 2009;5:8–13. doi: 10.2174/157339909787314149. [DOI] [PubMed] [Google Scholar]
- 5.Lim L.S., Mitchell P., Seddon J.M., Holz F.G., Wong T.Y. Age-related macular degeneration. Lancet. 2012;379:1728–1738. doi: 10.1016/S0140-6736(12)60282-7. [DOI] [PubMed] [Google Scholar]
- 6.Sica A., Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu W.K., Llewellyn O.P., Bates D.O., Nicholson L.B., Dick A.D. IL-10 regulation of macrophage VEGF production is dependent on macrophage polarisation and hypoxia. Immunobiology. 2010;215:796–803. doi: 10.1016/j.imbio.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 8.Xiong M., Elson G., Legarda D., Leibovich S.J. Production of vascular endothelial growth factor by murine macrophages: regulation by hypoxia, lactate, and the inducible nitric oxide synthase pathway. Am. J. Pathol. 1998;153:587–598. doi: 10.1016/S0002-9440(10)65601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lavin Y., Winter D., Blecher-Gonen R., et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crowther M., Brown N.J., Bishop E.T., Lewis C.E. Microenvironmental influence on macrophage regulation of angiogenesis in wounds and malignant tumors. J. Leukoc. Biol. 2001;70:478–490. [PubMed] [Google Scholar]
- 11.Passlick B., Flieger D., Ziegler-Heitbrock H.W. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. 1989;74:2527–2534. [PubMed] [Google Scholar]
- 12.Ancuta P., Rao R., Moses A., et al. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J. Exp. Med. 2003;197:1701–1707. doi: 10.1084/jem.20022156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ziegler-Heitbrock H.W. Heterogeneity of human blood monocytes: the CD14+ CD16+ subpopulation. Immunol. Today. 1996;17:424–428. doi: 10.1016/0167-5699(96)10029-3. [DOI] [PubMed] [Google Scholar]
- 14.Wong K.L., Tai J.J., Wong W.C., et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood. 2011;118:e16–e31. doi: 10.1182/blood-2010-12-326355. [DOI] [PubMed] [Google Scholar]
- 15.Zawada A.M., Rogacev K.S., Rotter B., et al. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood. 2011;118:e50–e61. doi: 10.1182/blood-2011-01-326827. [DOI] [PubMed] [Google Scholar]
- 16.Frankenberger M., Sternsdorf T., Pechumer H., Pforte A., Ziegler-Heitbrock H.W. Differential cytokine expression in human blood monocyte subpopulations: a polymerase chain reaction analysis. Blood. 1996;87:373–377. [PubMed] [Google Scholar]
- 17.Stansfield B.K., Ingram D.A. Clinical significance of monocyte heterogeneity. Clin. Transl. Med. 2015;4:5. doi: 10.1186/s40169-014-0040-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaipersad A.S., Lip G.Y., Silverman S., Shantsila E. The role of monocytes in angiogenesis and atherosclerosis. J. Am. Coll. Cardiol. 2014;63:1–11. doi: 10.1016/j.jacc.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 19.Anbazhagan K., Duroux-Richard I., Jorgensen C., Apparailly F. Transcriptomic network support distinct roles of classical and non-classical monocytes in human. Int. Rev. Immunol. 2014;33:470–489. doi: 10.3109/08830185.2014.902453. [DOI] [PubMed] [Google Scholar]
- 20.Cros J., Cagnard N., Woollard K., et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33:375–386. doi: 10.1016/j.immuni.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Venneri M.A., De Palma M., Ponzoni M., et al. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood. 2007;109:5276–5285. doi: 10.1182/blood-2006-10-053504. [DOI] [PubMed] [Google Scholar]
- 22.De Palma M., Murdoch C., Venneri M.A., Naldini L., Lewis C.E. Tie2-expressing monocytes: regulation of tumor angiogenesis and therapeutic implications. Trends Immunol. 2007;28:519–524. doi: 10.1016/j.it.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 23.Lewis C.E., De Palma M., Naldini L. Tie2-expressing monocytes and tumor angiogenesis: regulation by hypoxia and angiopoietin-2. Cancer Res. 2007;67:8429–8432. doi: 10.1158/0008-5472.CAN-07-1684. [DOI] [PubMed] [Google Scholar]
- 24.Lu W., Zhang Z., Fu C., Ma G. Intermediate monocytes lead to enhanced myocardial remodelling in STEMI patients with diabetes. Int. Heart J. 2015;56:22–28. doi: 10.1536/ihj.14-174. [DOI] [PubMed] [Google Scholar]
- 25.Schauer D., Starlinger P., Zajc P., et al. Monocytes with angiogenic potential are selectively induced by liver resection and accumulate near the site of liver regeneration. BMC Immunol. 2014;15:50. doi: 10.1186/s12865-014-0050-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lotery A., Xu X., Zlatava G., Loftus J. Burden of illness, visual impairment and health resource utilisation of patients with neovascular age-related macular degeneration: results from the UK cohort of a five-country cross-sectional study. Br. J. Ophthalmol. 2007;91:1303–1307. doi: 10.1136/bjo.2007.116939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Owen C.G., Fletcher A.E., Donoghue M., Rudnicka A.R. How big is the burden of visual loss caused by age related macular degeneration in the United Kingdom? Br. J. Ophthalmol. 2003;87:312–317. doi: 10.1136/bjo.87.3.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tuo J., Smith B.C., Bojanowski C.M., et al. The involvement of sequence variation and expression of CX3CR1 in the pathogenesis of age-related macular degeneration. FASEB J. 2004;18:1297–1299. doi: 10.1096/fj.04-1862fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tuo J., Grob S., Zhang K., Chan C.C. Genetics of immunological and inflammatory components in age-related macular degeneration. Ocul. Immunol. Inflamm. 2012;20:27–36. doi: 10.3109/09273948.2011.628432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katta S., Kaur I., Chakrabarti S. The molecular genetic basis of age-related macular degeneration: an overview. J. Genet. 2009;88:425–449. doi: 10.1007/s12041-009-0064-4. [DOI] [PubMed] [Google Scholar]
- 31.Seddon J.M., Gensler G., Milton R.C., Klein M.L., Rifai N. Association between C-reactive protein and age-related macular degeneration. JAMA. 2004;291:704–710. doi: 10.1001/jama.291.6.704. [DOI] [PubMed] [Google Scholar]
- 32.Wu K.H., Tan A.G., Rochtchina E., et al. Circulating inflammatory markers and hemostatic factors in age-related maculopathy: a population-based case-control study. Invest. Ophthalmol. Vis. Sci. 2007;48:1983–1988. doi: 10.1167/iovs.06-0223. [DOI] [PubMed] [Google Scholar]
- 33.Scholl H.P., Charbel Issa P., Walier M., et al. Systemic complement activation in age-related macular degeneration. PLoS One. 2008;3:e2593. doi: 10.1371/journal.pone.0002593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lechner J., Chen M., Hogg R.E., et al. Higher plasma levels of complement C3a, C4a and C5a increase the risk of subretinal fibrosis in neovascular age-related macular degeneration: Complement activation in AMD. Immun. Ageing. 2016;13:4. doi: 10.1186/s12979-016-0060-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reynolds R., Hartnett M.E., Atkinson J.P., et al. Plasma complement components and activation fragments: associations with age-related macular degeneration genotypes and phenotypes. Invest. Ophthalmol. Vis. Sci. 2009;50:5818–5827. doi: 10.1167/iovs.09-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nassar K., Grisanti S., Elfar E., et al. Serum cytokines as biomarkers for age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 2015;253:699–704. doi: 10.1007/s00417-014-2738-8. [DOI] [PubMed] [Google Scholar]
- 37.Lechner J., Chen M., Hogg R.E., et al. Alterations in Circulating Immune Cells in Neovascular Age-Related Macular Degeneration. Sci. Rep. 2015;5:16754. doi: 10.1038/srep16754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ilhan N., Daglioglu M.C., Ilhan O., et al. Assessment of Neutrophil/Lymphocyte Ratio in Patients with Age-related Macular Degeneration. Ocul. Immunol. Inflamm. 2014:1–4. doi: 10.3109/09273948.2014.921715. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 39.Cao X., Shen D., Patel M.M., et al. Macrophage polarization in the maculae of age-related macular degeneration: a pilot study. Pathol. Int. 2011;61:528–535. doi: 10.1111/j.1440-1827.2011.02695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan C.C., Ardeljan D. Molecular pathology of macrophages and interleukin-17 in age-related macular degeneration. Adv. Exp. Med. Biol. 2014;801:193–198. doi: 10.1007/978-1-4614-3209-8_25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakurai E., Anand A., Ambati B.K., van Rooijen N., Ambati J. Macrophage depletion inhibits experimental choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 2003;44:3578–3585. doi: 10.1167/iovs.03-0097. [DOI] [PubMed] [Google Scholar]
- 42.Dheda K., Huggett J.F., Bustin S.A., et al. Validation of housekeeping genes for normalizing RNA expression in real-time PCR. Biotechniques. 2004;37:112–114, 116, 118-119. doi: 10.2144/04371RR03. [DOI] [PubMed] [Google Scholar]
- 43.Chen M., Glenn J.V., Dasari S., et al. RAGE regulates immune cell infiltration and angiogenesis in choroidal neovascularization. PLoS One. 2014;9:e89548. doi: 10.1371/journal.pone.0089548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin L., Hutzen B., Zuo M., et al. Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer Res. 2010;70:2445–2454. doi: 10.1158/0008-5472.CAN-09-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao J., Chen M., Xu H. Experimental autoimmune uveoretinitis (EAU)-related tissue damage and angiogenesis is reduced in CCL2(-)/(-)CX(3)CR1gfp/gfp mice. Invest. Ophthalmol. Vis. Sci. 2014;55:7572–7582. doi: 10.1167/iovs.14-15495. [DOI] [PubMed] [Google Scholar]
- 46.Heimbeck I., Hofer T.P., Eder C., et al. Standardized single-platform assay for human monocyte subpopulations: Lower CD14+CD16++ monocytes in females. Cytometry A. 2010;77:823–830. doi: 10.1002/cyto.a.20942. [DOI] [PubMed] [Google Scholar]
- 47.Abeles R.D., McPhail M.J., Sowter D., et al. CD14, CD16 and HLA-DR reliably identifies human monocytes and their subsets in the context of pathologically reduced HLA-DR expression by CD14(hi) /CD16(neg) monocytes: Expansion of CD14(hi) /CD16(pos) and contraction of CD14(lo) /CD16(pos) monocytes in acute liver failure. Cytometry A. 2012;81:823–834. doi: 10.1002/cyto.a.22104. [DOI] [PubMed] [Google Scholar]
- 48.Geissmann F., Jung S., Littman D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 49.O'Shea J.J., Holland S.M., Staudt L.M. JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med. 2013;368:161–170. doi: 10.1056/NEJMra1202117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alexander W.S., Starr R., Metcalf D., et al. Suppressors of cytokine signaling (SOCS): negative regulators of signal transduction. J. Leukoc. Biol. 1999;66:588–592. doi: 10.1002/jlb.66.4.588. [DOI] [PubMed] [Google Scholar]
- 51.Qin H., Yeh W.I., De Sarno P., et al. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc. Natl. Acad. Sci. USA. 2012;109:5004–5009. doi: 10.1073/pnas.1117218109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qin H., Holdbrooks A.T., Liu Y., et al. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J. Immunol. 2012;189:3439–3448. doi: 10.4049/jimmunol.1201168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Espinosa-Heidmann D.G., Suner I.J., Hernandez E.P., et al. Macrophage depletion diminishes lesion size and severity in experimental choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 2003;44:3586–3592. doi: 10.1167/iovs.03-0038. [DOI] [PubMed] [Google Scholar]
- 54.Tsutsumi C., Sonoda K.H., Egashira K., et al. The critical role of ocular-infiltrating macrophages in the development of choroidal neovascularization. J. Leukoc. Biol. 2003;74:25–32. doi: 10.1189/jlb.0902436. [DOI] [PubMed] [Google Scholar]
- 55.Nakai K., Fainaru O., Bazinet L., et al. Dendritic cells augment choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 2008;49:3666–3670. doi: 10.1167/iovs.07-1640. [DOI] [PubMed] [Google Scholar]
- 56.Wang M., Zhang W., Crisostomo P., et al. STAT3 mediates bone marrow mesenchymal stem cell VEGF production. J. Mol. Cell. Cardiol. 2007;42:1009–1015. doi: 10.1016/j.yjmcc.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Z., Han Z.C. STAT3: a critical transcription activator in angiogenesis. Med. Res. Rev. 2008;28:185–200. doi: 10.1002/med.20101. [DOI] [PubMed] [Google Scholar]
- 58.Miklossy G., Hilliard T.S., Turkson J. Therapeutic modulators of STAT signalling for human diseases. Nat. Rev. Drug Discov. 2013;12:611–629. doi: 10.1038/nrd4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao M., Gao F.H., Wang J.Y., et al. JAK2/STAT3 signaling pathway activation mediates tumor angiogenesis by upregulation of VEGF and bFGF in non-small-cell lung cancer. Lung Cancer. 2011;73:366–374. doi: 10.1016/j.lungcan.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 60.Zhao D., Pan C., Sun J., et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene. 2015;34:3107–3119. doi: 10.1038/onc.2014.257. [DOI] [PubMed] [Google Scholar]
- 61.Okazaki H., Tokumaru S., Hanakawa Y., et al. Nuclear translocation of phosphorylated STAT3 regulates VEGF-A-induced lymphatic endothelial cell migration and tube formation. Biochem. Biophys. Res. Commun. 2011;412:441–445. doi: 10.1016/j.bbrc.2011.07.111. [DOI] [PubMed] [Google Scholar]
- 62.Lu W., Chen H., Yel F., Wang F., Xie X. VEGF induces phosphorylation of STAT3 through binding VEGFR2 in ovarian carcinoma cells in vitro. Eur. J. Gynaecol. Oncol. 2006;27:363–369. [PubMed] [Google Scholar]
- 63.Xu Q., Briggs J., Park S., et al. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene. 2005;24:5552–5560. doi: 10.1038/sj.onc.1208719. [DOI] [PubMed] [Google Scholar]
- 64.Jung J.E., Lee H.G., Cho I.H., et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 2005;19:1296–1298. doi: 10.1096/fj.04-3099fje. [DOI] [PubMed] [Google Scholar]
- 65.Gray M.J., Zhang J., Ellis L.M., et al. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene. 2005;24:3110–3120. doi: 10.1038/sj.onc.1208513. [DOI] [PubMed] [Google Scholar]
- 66.Rathinavelu A., Narasimhan M., Muthumani P. A novel regulation of VEGF expression by HIF-1alpha and STAT3 in HDM2 transfected prostate cancer cells. J. Cell. Mol. Med. 2012;16:1750–1757. doi: 10.1111/j.1582-4934.2011.01472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Niu G., Wright K.L., Huang M., et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000–2008. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 68.Jiang K., To E., Cui J.Z., et al. Drusen and Pro-inflammatory Mediators in the Post-Mortem Human Eye. J. Clin. Exp. Ophthalmol. 2012;3:208. doi: 10.4172/2155-9570.1000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zawada A.M., Rogacev K.S., Schirmer S.H., et al. Monocyte heterogeneity in human cardiovascular disease. Immunobiology. 2012;217:1273–1284. doi: 10.1016/j.imbio.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 70.Rogacev K.S., Seiler S., Zawada A.M., et al. CD14++CD16+ monocytes and cardiovascular outcome in patients with chronic kidney disease. Eur. Heart J. 2011;32:84–92. doi: 10.1093/eurheartj/ehq371. [DOI] [PubMed] [Google Scholar]
- 71.Heine G.H., Ortiz A., Massy Z.A., et al. Monocyte subpopulations and cardiovascular risk in chronic kidney disease. Nat. Rev. Nephrol. 2012;8:362–369. doi: 10.1038/nrneph.2012.41. [DOI] [PubMed] [Google Scholar]
- 72.Grunin M., Burstyn-Cohen T., Hagbi-Levi S., Peled A., Chowers I. Chemokine receptor expression in peripheral blood monocytes from patients with neovascular age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2012;53:5292–5300. doi: 10.1167/iovs.11-9165. [DOI] [PubMed] [Google Scholar]
- 73.Goverdhan S.V., Howell M.W., Mullins R.F., et al. Association of HLA class I and class II polymorphisms with age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2005;46:1726–1734. doi: 10.1167/iovs.04-0928. [DOI] [PubMed] [Google Scholar]
- 74.Fong A.M., Robinson L.A., Steeber D.A., et al. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J. Exp. Med. 1998;188:1413–1419. doi: 10.1084/jem.188.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Umehara H., Goda S., Imai T., et al. Fractalkine, a CX3C-chemokine, functions predominantly as an adhesion molecule in monocytic cell line THP-1. Immunol. Cell Biol. 2001;79:298–302. doi: 10.1046/j.1440-1711.2001.01004.x. [DOI] [PubMed] [Google Scholar]
- 76.White G.E., McNeill E., Channon K.M., Greaves D.R. Fractalkine promotes human monocyte survival via a reduction in oxidative stress. Arterioscler. Thromb. Vasc. Biol. 2014;34:2554–2562. doi: 10.1161/ATVBAHA.114.304717. [DOI] [PMC free article] [PubMed] [Google Scholar]