

Abstract
X-ray crystallography, the workhorse of structural biology, has been revolutionized by the advent of serial femtosecond crystallography using X-ray free electron lasers. Here, the fast pace and history of discoveries are discussed together with current challenges and the method’s great potential to make new structural discoveries, such as the ability to generate molecular movies of biomolecules at work.
Discovering the structure and mechanisms of the complete collection of biomolecules is one of the grand challenges of chemical biology. A new milestone was reached this year when the number of biomolecule structures solved by X-ray crystallography, NMR and electron microscopy exceeded 100,000. Although this number may seem to imply that structural biology is a mature field, we have only just scratched the surface of the structural diversity of biomolecules. Indeed, fewer than 600 membrane structures are known to date, of which fewer than 50 are human membrane proteins. Serial femtosecond crystallography (SFX) promises to extend the range of protein structures that can be solved by addressing several of the major shortcomings of X-ray crystallography. This Commentary discusses recent SFX advances and also points to challenges that must be resolved to make SFX available to the broad community in structural biology.
One of the major challenges in the structure determination of membrane proteins and large multiprotein complexes is the growth of crystals large enough and of high enough order for conventional X-ray crystallography. It can take years, if not decades, to solve the structures of these difficult-to-crystallize proteins. It would be ideal to determine structures from very small crystals, which can be much more readily obtained and also lack long-range disorder, but such nanocrystals are not suitable for conventional X-ray structure analysis because the dose limit of X-ray damage severely affects data collection of small crystals, even under cryogenic conditions at synchrotron sources.
Another major challenge of structural studies is capturing the dynamics of biomolecules. Most of the current X-ray structures provide only a static picture of the molecule. Time-resolved Laue crystallography, in which reversible photoinduced reactions are studied at synchrotron sources using a nonmonochromatic X-ray (a so-called ‘pink beam’), has unraveled structural changes in proteins but requires very large crystals and so far has mainly been applied to light-driven reactions in small single-domain proteins1.
X-ray free electron lasers (XFELs), which provide short (femtosecond) X-ray pulses that are 1012 stronger than that of a synchrotron, have begun to address these major challenges of X-ray crystallography. In SFX, data are collected in serial fashion, where crystals are delivered to the X-ray beam in their mother liquor at room temperature in a liquid jet (Fig. 1). The femtosecond X-ray pulses from an XFEL are so strong that they destroy any solid material, but the pulses are so short (1 fs = 10−15 s) that the X-ray diffraction snapshots are collected before the molecules and the crystals are destroyed. SFX thereby overcomes the size limitation of crystals and X-ray damage problem of conventional crystallography. The SFX data sets consist of tens of thousands of femtosecond X-ray diffraction snapshots, each collected from one single crystal in random orientation by intersection with a femtosecond X-ray pulse.
Figure 1.
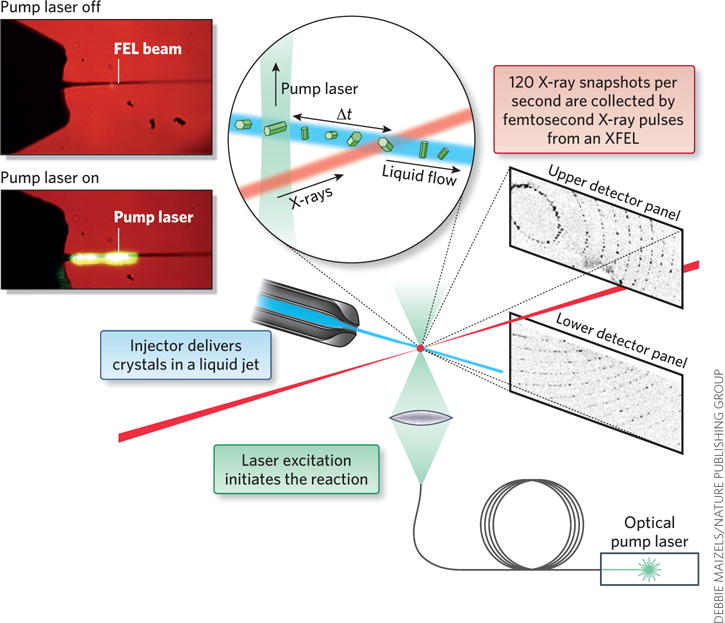
Schematic picture of collection of time-resolved data with SFX crystallography. Crystals are delivered to the X-ray beam in a liquid jet. The structure is probed by femtosecond X-ray pulses from a free electron laser. The time domain is added to the X-ray diffraction experiment by triggering of a reaction by light, where the reaction progression is monitored by variation of the time delay between optical ‘pump’ pulse and the X-ray pulse. Figure modified from ref. 15.
The first low-energy XFEL began operation in 2005 at the German research center Deutsches Elektronen-Synchrotron (DESY). With this XFEL, known as FLASH, Chapman and colleagues were able to demonstrate the diffract-before-destroy principle with a single-femtosecond X-ray snapshot of an object (a picture of two men and the sun edged into a silicon-nitrate window)2. However, most structural biologists were highly skeptical that the diffract-before-destroy principle could be extended to nanocrystals of biomolecules and that FELs would be of use for structural biology. It was argued that even if diffraction-before-destruction would occur, one would not be able to detect diffraction. The argument was that though the flux (photons/time) is 1012 times higher, the exposure times would be 1015 times shorter than those at synchrotron sources: that is, the number of photons in an femtosecond X-ray pulse would be 1,000 times smaller than that produced by a second of exposure in a synchrotron. The detection problem would be further heightened by the fact that nanocrystals contain only a few hundred unit cells, as compared to the trillions of unit cells in a large single crystal, leading to the conclusion that even if the nanocrystals diffracted, no signal would be detected. Furthermore, even with larger crystals, only one diffraction snapshot could be observed from a given crystal, so all diffraction patterns would be still images of crystals in random orientation. Only partial reflections would be recorded, leading to difficulties in the determination of accurate structure factors.
At the end of 2009, the first high-energy XFEL, the Linac Coherent Light Source (LCLS) at Stanford’s Linear Accelerator Center (SLAC), started user operations (at 2.5 keV). The first X-ray diffraction experiments demonstrating the proof of concept for femtosecond crystallography of proteins were conducted in December 2009 by our team (a large international group of scientists from 24 institutions including DESY, Arizona State University, the University of Uppsala and the Max Planck Institute in Heidelberg), focusing on photosystem I as the model system3. Consisting of 36 proteins and 381 cofactors, photosystem I is the most complex membrane protein crystallized thus far, with a molecular weight of over 1 million Da. In this first proof-of-principle study, the crystals were delivered to the XFEL beam at room temperature in their mother liquor using an injector with gas-dynamic virtual nozzles4, which replenished the sample between the then 30-Hz X-ray shots (LCLS now runs at a frequency of 120 Hz). The first results exceeded all expectations: millions of diffraction images were collected, and the crystals showed diffraction to the edge of the detector at 8-Å resolution3.
In 1952, Sayre proposed that if it were ever possible to get a diffraction pattern from a nanocrystal where one could count the number of unit cells (<20), then the patterns should show shape transforms5, also known as the Fourier transforms, of the object. Indeed, the diffraction images on the back detector showed strong shape transforms and fringes between the Bragg peaks3. Simply counting the fringes provides the number of unit cells in the corresponding direction. The shape transforms may be used for direct phasing in the future (further discussion on phasing methods are discussed below).
Since the publication of the first proof-of-principle study of SFX just 4 years ago3, the technique has progressed at a breathtakingly fast pace on all fronts, from nanocrystal growth and high-throughput identification and characterization to advances in sample delivery and data evaluation. Structural biology milestones include the application of SFX at atomic resolution6, crystals grown inside living insect cells7,8 and bacteria9, structure determination of human membrane proteins of high medical importance in a membrane environment10–14 and the first time-resolved SFX studies on large photosynthetic proteins at medium resolution15–17. Very recently, the first high-resolution time-resolved SFX (TR-SFX) study was reported18.
Nanocrystal growth and delivery
With the ability to collect data on crystals <1 μm, SFX allows the structural determination of challenging proteins from which no large crystals can be grown. However, with the crystals’ small size comes the challenges in detecting them as they are smaller than a wavelength of light and cannot be detected by optical microscopy. Second-order nonlinear imaging of chiral crystals (SONICC) can detect nanocrystals as small as 100 nm in high throughput19. Proteins that contain cofactors are ideally suited for SONICC detection as they enhance second harmonics generation and thereby increase the signal strength. We have had success with nanocrystal growth and used SONICC to detect over 100 conditions where nanocrystals are observed for proteins from which no large crystals have been detected under traditional crystallization conditions. Further methods to detect nanocrystals are powder diffraction and electron microscopy.
SFX poses a logistical problem with sample amounts and handling, as the liquid jet uses large quantities (tens of milligrams) of sample. Furthermore, data collection at room temperature and the dynamics of nanocrystals necessitate on-site crystallization. Nanocrystals have a larger surface/volume ratio compared to large crystals. The surface of a crystal is dynamic, with molecules attaching and deattaching from the crystal surface. Shaking of crystals during transport leads to the growth of larger, disordered crystals at the expense of the small, well-ordered nanocrystals. New methods have been developed to grow nanocrystals of high quality and quantity by ultrafiltration20 and modified free interface diffusion21, and the crystals can be sorted by size22.
Experiments are currently underway to explore the mechanism of in vivo crystal growth in more detail with the goal of enhancing these pathways during in-cell crystal growth. Nanocrystals suitable for SFX have been observed in vivo by overexpression of proteins in living insect cells8 and in a bacterium9. However, inclusion bodies, which are observed frequently during protein overexpression in Escherichia coli, have so far not shown either SONICC signals or powder diffraction in our lab and therefore are assumed not to contain nanocrystals.
One of the most exciting developments of SFX has been its development for membrane protein crystals grown in lipidic cubic phases (LCPs)10. LCPs mimic the natural membrane environment and are ideally suited to crystallize human membrane proteins. However, although nano- and microcrystals (<5 μm) are often observed in the first screens, it can take months or years to grow larger crystals in LCP for conventional X-ray crystallography. SFX also avoids freezing and X-ray damage, making it ideally suited for structure determination of crystals grown in LCP. LCP sample delivery for SFX was initially a challenge as it has the consistency of toothpaste. A special new type of injector has been developed for delivery of crystals in highly viscous media (the LCP injector) that delivers the crystals at a reduced flow rate, leading to a reduction in sample amount by a factor of 100 (ref. 11). An agarose-based delivery medium with very low background scattering has been developed that allows pregrown crystals of soluble proteins, protein complexes, membrane proteins and even virus crystals to be delivered to the FEL beam with low sample consumption23.
Recently, the method of serial crystallography has been extended to data collection at synchrotron sources, where crystals were delivered to the synchrotron beam at the European Synchrotron Radiation Facility (ESRF) in the LCP jet24. Single-snapshot X-ray diffraction patterns can be collected on macrocrystals at room temperature in the jet in a serial fashion using fast detector readouts. Although the method is not damage free, the short exposure times limit secondary X-ray damage. Also, as no mounting or freezing is involved, serial macro-X-ray crystallography may become a premier method to determine X-ray structures at a synchrotron source in the future, especially at the new high-brilliance synchrotron beamlines, which have such high flux that X-ray damage is severe even under cryogenic conditions.
An alternate concept to SFX for data collection at the FEL is based on the more conventional approach, in which data are collected from hundreds of large frozen crystals individually mounted and shifted on a goniometer or fixed target holder during data collection. This concept has been successfully used to determine an undamaged high-resolution structure of the cytochrome oxidase25 and the dark state of photosystem II26. Although this concept allows more conventional methods of data evaluation to be used, it requires large crystals, crystal freezing and huge amounts of precious FEL beamtime for data collection. It is therefore mainly useful for determining high-resolution X-ray structures of proteins that are strongly susceptible to X-ray damage, such as those containing metal centers.
Structural biology highlights from FELs
It was recently found that nanocrystals of CatB, the protease that degrades red blood cells in patients suffering from sleeping sickness, can be grown by overexpression of proteins in living insect cells8. CatB is a potential drug target against sleeping sickness, which affects 3 million patients each year worldwide. The structure of CatB determined at 2.1 Å by SFX with its N-terminal inhibitor peptide could form the basis for new drugs against this serious disease.
The high impact of structures determined by SFX on bioenergy conversion and medicine can be seen from their effect on the structure determination of photosynthetic proteins (Fig. 2a,b) and the recent structural discoveries of G protein– coupled receptors (Fig. 2c–g). GPCRs are one of the most important classes of human membrane proteins, with 40% of all current drugs in the US targeting GPCRs. Although all GPCRs consist of seven transmembrane helices, they show large variations in function and substrate binding and have key roles in various pathways, including nerve function, signaling, taste perception, hormone signaling, mood control, blood pressure control and cancer development. GPCR nano- or microcrystals can now be grown in LCP in syringes from which they can be directly introduced into the sample chamber of the LCP injector.
Figure 2.
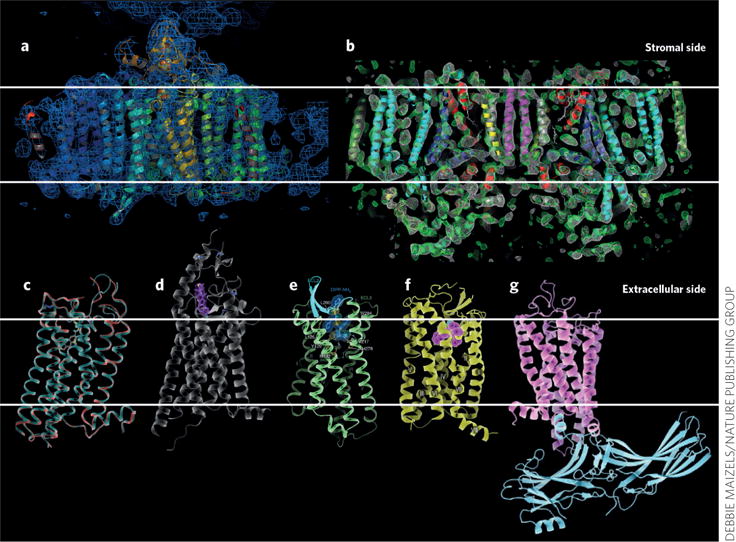
Highlights of structures of membrane proteins solved by SFX. (a) Photosystem I3. (b) Photosystem II16. (c) β-adrenergic receptor10. (d) Smoothened receptor11. (e) Opinoid receptor13. (f) Angiotensin receptor12. (g) Rhodopsin–arrestin complex14. Images reproduced from refs. 3 (a), 16 (b), 11 (d) 13 (e) and 14 (g) and, with permission, from ref. 10 (c) and ref. 12 (f).
The first GPCR whose structure was determined by SFX was the β-adrenergic receptor10 (Fig. 2c). Comparing the room-temperature SFX structure and the previously determined synchrotron structure showed improved electron density based on the SFX data sets, where missing side chains became visible, new salt bridges were identified and even the backbone showed differences in some parts of the protein. Since this breakthrough, five more GPCR structures have been solved by SFX, including that of the Smoothened receptor bound with its inhibitor cyclopamine11 (Fig. 2d). This structure revealed that cyclopamine does not bind the receptor in the membrane intrinsic region of the receptor, as originally assumed; it is instead bound closer to the extracellular loop regions, whose conformations are thereby significantly altered. In February 2015, co-crystal structures of the δ-opinoid receptor, which controls emotions and pain, were solved in complex with several peptide inhibitors27 (Fig. 2e).
Recently, SFX provided the first structure of the angiotensin receptor12 (Fig. 2f), which is highly medically relevant as it controls blood pressure. Its structure was solved in complex with the small-molecule drug candidate ZD7155, which is more potent and longer lasting in the treatment of hypertension than the first clinically used angiotension receptor blockers. A further breakthrough in the field of structural biology with FELs was the first structure of a GPCR in complex with arrestin (Fig. 2g). The rhodopsin-arrestin structure provided new insights into GPCR activation14.
Toward molecular movies of proteins
TR-SFX holds the potential to view a reaction happening in real time, creating a ‘molecular movie’. TR-SFX is one of the most exciting advances toward gaining new biological insights with FELs. The first TR-SFX experiments used laser excitation to trigger the light reactions in crystals of a complex between photosystem I and its natural electron acceptor ferredoxin15 (Fig. 1). Light excitation leads to charge separation in photosystem I and electron transfer to ferredoxin, which then undocks from photosystem I to bring electrons to ferredoxin NADP+ reductase, where they are used for the reduction of NADP+ and H+ to NADPH. The electron transfer and undocking of ferredoxin is irreversible and leads to destruction of the crystals as the docked ferredoxin is involved in crystal contacts. Therefore, this reaction can only be studied by SFX. By varying the time delay between the optical laser (‘pump pulse’) and the femtosecond X-ray pulse (probe-pulse), different time points of electron transfer and undocking can be obtained. Large changes in the structure factors were observed in the time frame of the electron transfer rates between photosystem I and ferredoxin.
Another major target of time-resolved SFX studies is photosystem II, which changed Earth by evolving essentially all the oxygen in the atmosphere. In four subsequent charge separation events, two water molecules are ‘split’ into oxygen, four electrons and four protons. Two teams have published TR-SFX studies of PSII16,17 performed with setups that differ in crystal size, sample delivery methods, triggering of laser excitation and data evaluation. Large conformational changes between the dark and double-excited state of photosystem II have been detected in the oxygen-evolving complex and at the acceptor site in our study16. These first ‘snapshots’ of the reaction cycle in photosystem II at medium resolution mark the beginning of the route toward molecular movies of water splitting. However, much more work must be done before high-resolution structures of all five states will be determined by SFX.
In December 2014, the first high-resolution time-resolved structure was published. Tenboer et al. used a setup similar to that developed for PSII16, determining the structure of photoactive yellow protein (PYP) in the dark and at time points of 1 μs and 10 ns after initiation of the photocycle by laser excitation. Because of the small size of the crystals, the photoreaction was induced in up to 40% of the PYP molecules in the crystals in the SFX experiment, whereas previous Laue experiments with larger crystals maximally achieved 10–12% photoconversion (Fig. 3). The SFX structure was solved at 1.9-Å resolution18. This study will pave the way for using SFX to image ultrafast reactions at atomic resolution.
Figure 3.
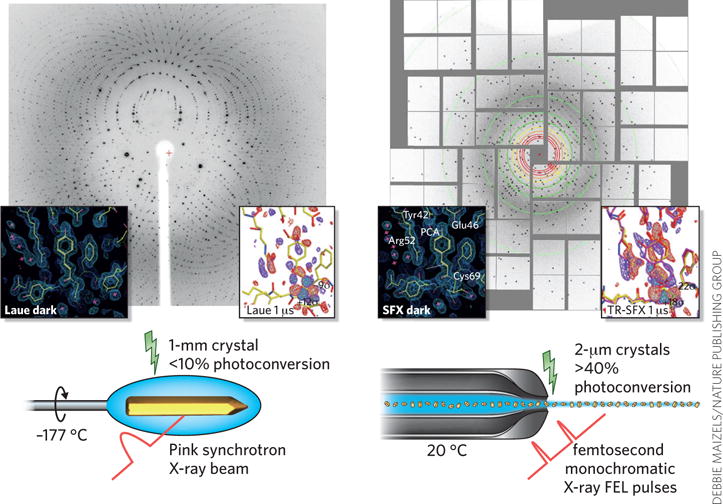
Comparison of time-resolved Laue crystallography and time-resolved SFX crystallography. Left, Laue crystallography; right, time-resolved SFX. Examples are shown here for PYP. The diffraction patterns and the crystal photo were kindly provided by M. Schmidt. Electron density data are reproduced with permission from ref. 18.
So far, published TR-SFX work has been applied to reactions that can be triggered by light, but new methods have been developed that will allow for studies of reactions that are induced by rapid mixing ‘on the fly’. These include the use of microfluidic devices that are coupled with the sample injector as well as the development of a double-focusing mixing jet in which the stream of crystals interfaces with the substrate solution directly in the jet28.
In an alternate approach, reactions that are originally not light-driven could be triggered by the so-called ‘caged substrates’ method, in which caged compounds are used to release the substrate molecule of interest by a UV-light pulse. Overall, the full potential for time-resolved molecular movies is underway with the advent of SFX and, as the field progresses, could change the paradigm of structural biology from static pictures toward molecular movies, leading to breakthroughs in our understanding of the mechanistic dynamics of biomolecules and allowing unprecedented progress in fields such as drug design and alternative energy.
Current challenges and outlook
Although biology with FELs has made an exciting entry into structural biology in the last four years, major challenges must still be resolved before SFX becomes a routine method in structural biology. One of the major scientific challenges is the phasing. In standard X-ray crystallography, anomalous scattering methods such as multiple anomalous diffraction (MAD) and single anomalous diffraction (SAD) are the current workhorses for experimental phasing, which is required to solve novel structures for which no homologous structures exist. However, they require the presence of natural or introduced heavy atoms and depend on high-resolution data to be collected with high accuracy as very small differences in the measured intensities must be resolved. At the same time, the large variations in the shot-by-shot X-ray intensity and energy make MAD or SAD phasing of XFEL data very challenging. The first proof-of-principle studies on experimental phasing of SFX data29 were done using gadolinium lysozyme as a model system and required very high multiplicity of data sets for phase determination. New direct methods for phasing SFX data look very promising30 but have not yet been experimentally applied owing to current limitations in the dynamic range and size of the detectors. Because of limitations in the detector that is currently used, it is very difficult to collect data at high resolution as the XFEL beam must currently be attenuated to avoid overheating of pixels from strong reflections at low resolution for strongly diffracting crystals. This limitation can be alleviated with new detectors that allow for true atomic-resolution structures (<1.5 Å) to be determined. New detector developments for SFX, which allow for data collected with a high dynamic range, multiple gain settings and high repletion rate (such as the Jungfrau or adaptive gain integrating pixel detector) will be very important for further progress of SFX not only for phasing but also for the extension of the current resolution range of SFX data.
An ongoing challenge is access to beamtime at FELs. Currently there are only two high-energy FELs in the world (LCLS at Stanford and SACLA (Spring-8 Angstrom Compact Free Electron Laser) in Japan) where experiments can be conducted. Because of the linear nature of the accelerators, only one experiment can be conducted at a time at each FEL (one exception at LCLS allows diffraction experiments at beamline CXI and spectroscopy experiments at beamline XPP to be performed concurrently). With all the breakthrough discoveries and the excitement within the scientific community, many more groups seek access to SFX for structure determination. With three more FELs being currently built (the XFEL at Pohang Accelerator Laboratory in Korea will start lasing in 2016, followed by the European XFEL and the Swiss FEL in 2017), the capacity of experiments that can be performed concurrently worldwide will increase from two to five. This will be a great improvement, but it still will not even remotely meet the needs of the user community, which is not limited to structural biology but also includes chemistry, material science, physics and geology. The high costs of building FELs ($500 million to $1.5 billion) and of their operation are prohibitive. One solution to this serious problem could be the development of compact FELs, which may in the future be able to ‘shrink’ the FEL from 1 km to 1 m and reduce the cost to $20 million.
By providing atomic-level views of chemical reactions, FELs have initiated a new era in structural and chemical biology, opening a pathway for the determination of molecular movies of biomolecules at work. They have the potential for huge impacts in medicine, as these dynamic structures will form the basis for structure-based drug design. The energy field will also benefit as the unraveling of the molecular mechanisms of natural photosynthesis and biocatalysis will open the way for the development of clean energy.
Acknowledgments
This work was supported by the Science and Technology Centers Program of the US National Science Foundation through BioXFEL under agreement no. 1231306 and by the US National Institutes of Health awards 617095583 and U54GM094599. Portions of this research were carried out at the LCLS at the SLAC National Accelerator Laboratory. LCLS is an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University.
Footnotes
Competing financial interests
The author declares no competing financial interests.
References
- 1.Neutze R, Moffat K. Curr Opin Struct Biol. 2012;22:651–659. doi: 10.1016/j.sbi.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chapman HN, et al. Nat Phys. 2006;2:839–843. [Google Scholar]
- 3.Chapman HN, et al. Nature. 2011;470:73–77. doi: 10.1038/nature09750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weierstall U, Spence JC, Doak RB. Rev Sci Instrum. 2012;83:035108. doi: 10.1063/1.3693040. [DOI] [PubMed] [Google Scholar]
- 5.Sayre D. Acta Crystallogr. 1952;5:843–843. [Google Scholar]
- 6.Boutet S, et al. Science. 2012;337:362–364. doi: 10.1126/science.1217737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koopmann R, et al. Nat Methods. 2012;9:259–262. doi: 10.1038/nmeth.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Redecke L, et al. Science. 2013;339:227–230. doi: 10.1126/science.1229663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sawaya MR, et al. Proc Natl Acad Sci USA. 2014;111:12769–12774. doi: 10.1073/pnas.1413456111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu W, et al. Science. 2013;342:1521–1524. doi: 10.1126/science.1244142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weierstall U, et al. Nat Commun. 2014;5:3309. doi: 10.1038/ncomms4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H, et al. Cell. 2015;161:833–844. doi: 10.1016/j.cell.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fenalti G, et al. Nat Struct Mol Biol. 2015;22:265–268. doi: 10.1038/nsmb.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang Y, et al. Nature. 2015;523:561–567. doi: 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aquila A, et al. Opt Express. 2012;20:2706–2716. doi: 10.1364/OE.20.002706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kupitz C, et al. Nature. 2014;513:261–265. doi: 10.1038/nature13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kern J, et al. Nat Commun. 2014;5:4371. doi: 10.1038/ncomms5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tenboer J, et al. Science. 2014;346:1242–1246. doi: 10.1126/science.1259357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kissick DJ, Wanapun D, Simpson GJ. Annu Rev Anal Chem. 2011;4:419–437. doi: 10.1146/annurev.anchem.111808.073722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunter MS, Fromme P. Methods. 2011;55:387–404. doi: 10.1016/j.ymeth.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kupitz C, et al. Phil Trans R Soc B Bio Sci. 2014;369:20130316. doi: 10.1098/rstb.2013.0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abdallah BG, Roy-Chowdhury S, Coe J, Fromme P, Ros A. Anal Chem. 2015;87:4159–4167. doi: 10.1021/acs.analchem.5b00589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Conrad CE, et al. IUCrJ. 2015;2:421–430. doi: 10.1107/S2052252515009811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nogly P, et al. IUCrJ. 2015;2:168–176. doi: 10.1107/S2052252514026487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirata K, et al. Nat Methods. 2014;11:734–736. doi: 10.1038/nmeth.2962. [DOI] [PubMed] [Google Scholar]
- 26.Suga M, et al. Nature. 2015;517:99–103. doi: 10.1038/nature13991. [DOI] [PubMed] [Google Scholar]
- 27.Fenalti G, et al. Nature. 2014;506:191–196. doi: 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang D, Weierstall U, Pollack L, Spence J. J Synchrotron Radiat. 2014;21:1364–1366. doi: 10.1107/S160057751401858X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barends TR, et al. Nature. 2014;505:244–247. doi: 10.1038/nature12773. [DOI] [PubMed] [Google Scholar]
- 30.Chen JP, Spence JC, Millane RP. Acta Crystallogr A. 2014;70:154–161. doi: 10.1107/S2053273313032725. [DOI] [PubMed] [Google Scholar]