

Abstract
A few reports suggest combination of ANCA-associated vasculitis (AAV) and neutrophilic dermatoses (ND). We aimed to describe the main characteristics of patients presenting with both AAV and ND in a French cohort and through a systematic literature review, and to discuss the possible common pathogenic process involved.
We conducted a retrospective study of patients with both conditions. Patients were selected via the French Internal Medicine Society (SNFMI) and the French Vasculitis Study Group (FVSG). A literature review focusing on a combination of both conditions, concentrated only on publications with well-established diagnoses and individual detailed data.
Seventeen patients diagnosed with AAV and ND were identified in this cohort. Twelve patients had granulomatosis with polyangiitis (GPA), 4 had microscopic polyangiitis (MPA) and one had eosinophilic GPA (EGPA). Eight patients, all with GPA, displayed pyoderma gangrenosum (PG). Sweet's syndrome was observed in 6 patients (4 with MPA, one with GPA and one with EGPA) and erythema elevatum diutinum in the other three (2 with GPA and 1 with MPA). The literature review identified 33 additional patients with both conditions, including 26 with GPA. Altogether, of the 50 patients (17 from our study and 33 from the literature review), 33 (66%) patients presented with PG associated with GPA in 29 cases (89%). Corticosteroids were the first-line treatment in conjunction with an immunosuppressive agent in most cases. Outcomes were good and a total of 15 patients experienced a relapse. Patients who relapsed were more likely to have ear, nose and throat manifestation than patients who did not [12/15 (80%) relapsing patients vs. 15/35 (43%) non-relapsing patients; p = 0.03)].
In our stud, the most frequent association concerned GPA and PG. ND should be considered and specifically researched within the spectrum of cutaneous manifestations observed in AAV.
INTRODUCTION
Antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis (AAV) is a group of rare and potentially life-threatening diseases comprising 3 main conditions: granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic GPA (EGPA). Pathophysiological mechanisms include dysregulation of neutrophils that are a target of ANCA. Skin lesions are frequent and polymorphic in AAV and can affect 10% to 50% of patients.1–3 Purpura and leukocytoclastic vasculitis are still the most common clinical and histopathologic features, respectively. Neutrophilic dermatoses (ND) are a heterogeneous group of rare inflammatory disorders characterized by sterile infiltration of the skin with neutrophil infiltrates. Pyoderma gangrenosum (PG), Sweet syndrome (SS), and erythema elevatum diutinum (EED) are the 3 most common ND entities. Their association with some other systemic diseases is well known, especially malignant blood disorders, inflammatory bowel diseases, and primary or iatrogenic autoimmune disorders.4,5 A combination of ND and AAV has only been described in a few case reports. However, in the absence of a perceived need for dermatological expertise when evaluating a skin lesion associated with an established diagnosis of AAV, underdiagnosis of ND is probable and therefore raises the question of the role played by ANCA in both conditions. In this report, we describe a case series of patients with both conditions. We also conducted a comprehensive literature review on the combination of both conditions.
PATIENTS AND METHOD
Study Design and Patients
The patients were selected through an e-mail sent to physicians belonging to the French Internal Medicine Society (SNFMI) and the French Vasculitis Study Group (FVSG). A search was also carried out in the FVSG database to obtain additional patients. Patients had to satisfy the following 2 criteria to take part in the study. First, diagnosis of AAV between 1990 and 2015, and positive ANCA result. We enrolled patients with GPA, MPA, and EGPA, according to the 1990 American College of Rheumatology (ACR) modified criteria.6 Second, diagnosis of histologically proven ND was also a prerequisite. We did not include patients with other types of vasculitis or patients with dubious AAV. Diagnosis of ND was established on the basis of medical expertise and histological evidence.
Physicians who enrolled patient (s) received a standard data collection form.
This study was conducted in compliance with good clinical practices and the Declaration of Helsinki principles. In accordance with French law, formal approval from an ethics committee is not required for this type of study.
Parameters Studied
For each patient, detailed information regarding AAV and ND was recorded. Demographics, medical history, clinical presentation, and date of onset for both conditions were noted. For vasculitis, clinical manifestations as well as ANCA status, laboratory tests (including hemogram, ionogram, and creatinine level), and histological findings (when available) were collected. When available, the Birmingham Vasculitis Activity Score (BVAS) was also noted. For ND, the type of dermatosis was specified as well as the localization and histological findings. For both conditions, treatment and outcomes were recorded. Moreover, diseases or iatrogenic conditions known to be associated with ND and/or secondary vasculitis were targeted in particular (and were thus excluded) when establishing the patients’ medical and current histories, including other autoimmune diseases, spondylarthropathies, inflammatory bowel diseases, Behçet disease, hematological malignancies, or autoinflammatory diseases, and the use of some drugs such as synthesis anti-thyroid or anti-tumor necrosis factor (TNF)-alpha agents, minocycline, d-penicillamine, or allopurinol.
Review of the Literature and Selection
We searched MEDLINE via PubMed for all reports published in English from 1990 to 2015, using the following keywords: “neutrophilic dermatoses” or “Sweet's syndrome” or “pyoderma gangrenosum” or “erythema elevatum diutinum”, which were each linked to “antineutrophil cytoplasmic antibody”, “antineutrophil cytoplasmic antibody associated vasculitis”, “Wegener's granulomatosis”, “granulomatosis with polyangiitis”, “microscopic polyangiitis”, “Churg-Strauss syndrome” and “eosinophilic granulomatosis with polyangiitis”. All relevant articles were retrieved and additional references quoted in these articles were checked. We excluded articles with doubtful diagnosis or inadequate data.
Detailed information was extracted for all of the patients enrolled in the study and pooled for statistical analysis.
Statistical Analysis
Categorical variables are expressed as numbers (percentage) and quantitative variables as median values (range). Categorical variables were compared using the χ2 test, or where appropriate, Fisher exact test. The nonparametric Mann–Whitney test was used to compare median values. Differences were considered significant when P < 0.05. All tests were performed using GraphPad Prism 5.0c.
RESULTS
The French AAV and ND Cohort
Clinical Characteristics of the Patients
Twenty-five patients were reported, but only 17 were eligible for inclusion. Of the 8 patients who were excluded, 6 had ND and dubious vasculitis with negative ANCA, whereas the other 2 patients lacked information in their medical records, including treatments and outcomes.
Detailed characteristics of the 17 patients are shown in Tables 1 and 2. With regard to the patients’ current status or medical history, 1 patient who developed GPA along with PG suffered from ulcerative colitis for 4 years. This condition had not been apparent at the time of GPA and PG diagnosis. Another patient had thyroid papillary cancer 2 years before GPA developed, which was considered cured at the time of GPA diagnosis. No drug exposure or other diseases was cited as a possible cause of secondary vasculitis or ND recorded in our cohort, including alpha-1-antitrypsin deficiency, the typical manifestations of which (including emphysema, bronchiectasis, chronic hepatic disorders, panniculitis and pancreatitis) were not observed in our cohort. However, this test is not performed routinely and was only carried out in 5 of our AAV patients, all of whom exhibited normal values.
TABLE 1.
Main Patient Characteristics at Diagnosis in the French Cohort
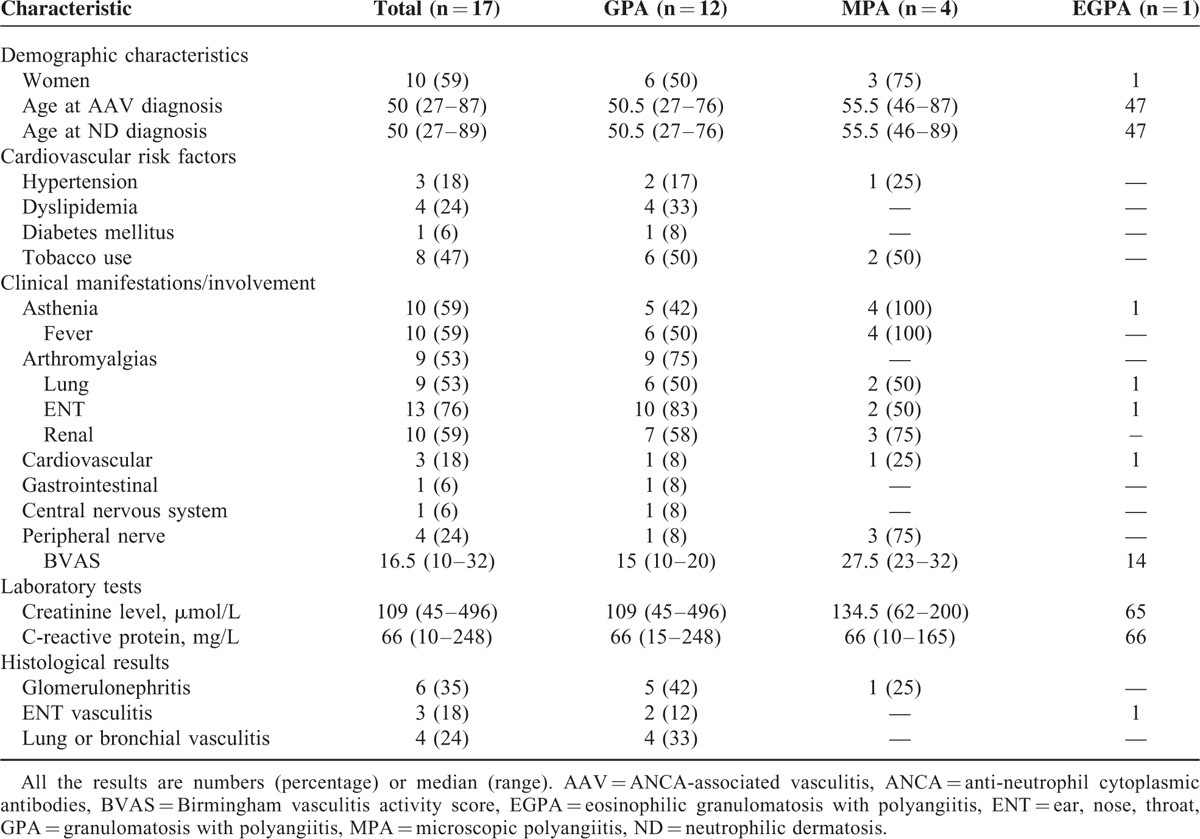
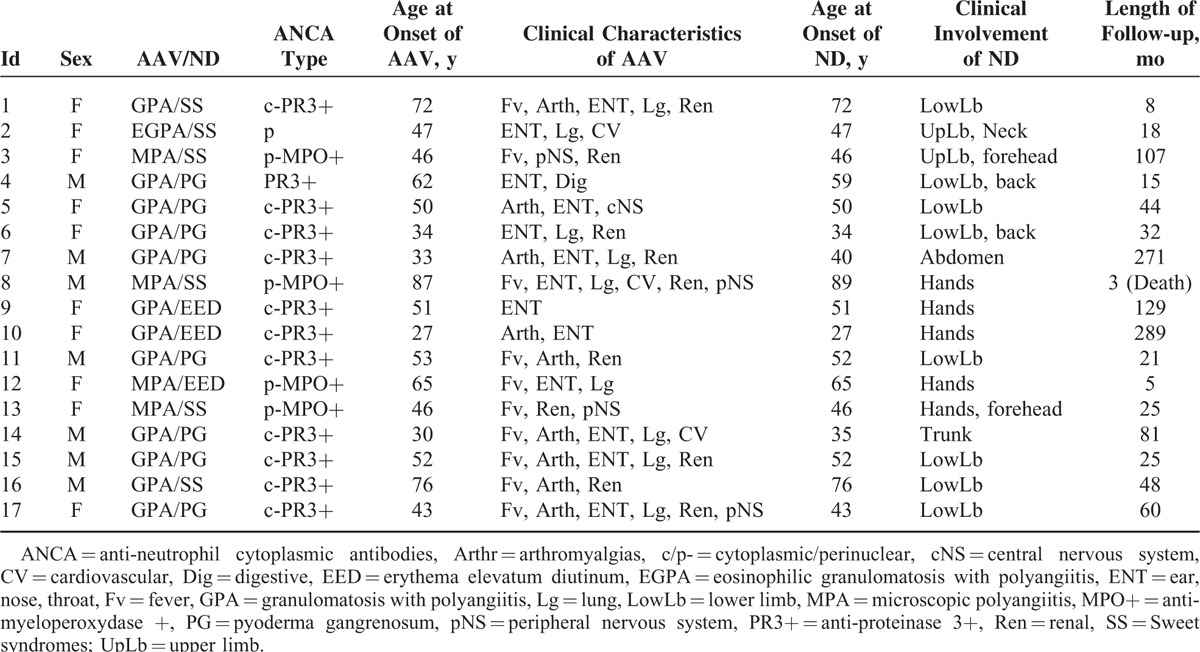
TABLE 2.
Details of Each Patient's Characteristics in the French Cohort
GPA was the most frequently observed AAV in 12 (71%) patients, followed by MPA in 4 (23%) and EGPA in 1 (6%). We observed PG in 8 (47%) patients, Sweet syndrome in 6 (35%), and erythema elevatum diutinum in 3 (18%) (Figure 1 a).
FIGURE 1.
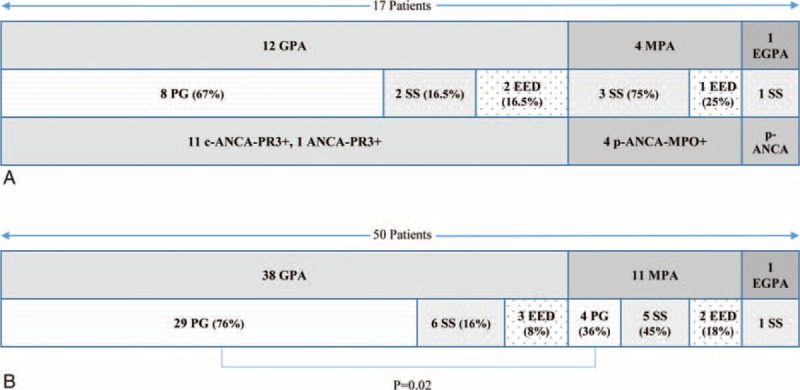
Distribution of vasculitis, neutrophilic dermatosis, and ANCA type assays in a French cohort of 17 patients (A) and in the whole cohort of 50 patients including 33 additional patients from a literature review (B). There were significantly more PG cases in GPA patients than in other forms of vasculitis (76% vs 36%; P = 0.02). ANCA = anti-neutrophil cytoplasmic antibodies, C = cytoplasmic fluorescence, EED = erythema elevatum diutinum, EGPA = eosinophilic GPA, GPA = granulomatosis with polyangiitis, MPA = microscopic polyangiitis, MPO = myeloperoxidase, P = perinuclear fluorescence, PG = pyoderma gangrenosum, PR3 = proteinase 3, SS = Sweet syndrome.
PG was described as large ulcers beginning with inflamed nodules or papules, and developing into large painful ulcers with clear-cut margins, deep, irregular, and necrotic centres and surrounding inflammation. Two patients presented with a solitary lesion, whereas the others displayed multiple lesions that could fuse together. Lesions advanced within a few days or weeks. The lower limbs, trunk, and both sites were involved in 6, 4, and 2 patients, respectively.
The 6 patients with SS presented painful erythematous plaques, papules, or nodules on limb extremities. Two of the patients displayed facial manifestations and 1 patient presented with neck lesions. Some lesions were tender.
The hands were affected in the 3 patients with EED whose lesions took the form of elevated erythematous papular or nodular lesions involving the extensor surfaces of the extremities.
AAV and ND occurred concomitantly in 12 (70%) patients. In 3 (18%) patients, AAV diagnosis predated ND diagnosis (2, 5, and 7 years before; patients 8, 14, and 7, respectively; Table 2). In 2 (12%) patients with PG, ND occurred before AAV (1 and 3 years before, patients 11 and 4, respectively).
At onset, the patients with MPA had a more severe presentation than those with GPA (median BVAS of 27.5 [23–32] vs 15 [10–20], P = 0.01). All 12 patients with GPA had anti-PR3 ANCA, including 11 with cytoplasmic fluorescence. The 4 patients with MPA presented with anti-MPO ANCA with perinuclear fluorescence. The 1 patient with EGPA had perinuclear ANCA with negative ELISA. The 2 patients (4 and 11) who developed PG before GPA had positive ANCA at the time of PG diagnosis, but no sign of vasculitis.
Figure 1A shows the distribution of AAV, ND, and ANCA. PG occurred only in GPA patients; 3 of the 4 MPA patients exhibited Sweet syndrome.
Histological Features
Histological evidence of extracutaneous vasculitis was obtained in 10 (59%) patients, including 8 with a GPA diagnosis. Three patients were biopsied at 2 sites (Table 1).
As regards ND, all of the patients underwent a skin biopsy that showed common features indicative of ND: intense inflammation of the superficial dermis, dermal neutrophil infiltration with irregular lymphocytes and histiocytes, no granuloma or vasculitis, except for 1 patient with EED whose biopsy confirmed leukocytoclastic vasculitis along with neutrophil aggregates.
Patients with PG showed various histological findings depending on the site of the lesion from which the biopsy was obtained and the stage of evolution of the lesions. Typical histological findings were observed in 6 of our 8 patients: intense neutrophil infiltrate with microabscess or perifollicular neutrophilic infiltrate, along with central necrosis. The epidermis and dermis were ulcerated and surrounded by an intense acute infiltration of inflammatory cells. Two other patients displayed moderate and mixed neutrophil and lymphocyte infiltration. Although neither of these patients displayed typical histological findings, their clinical presentation was very typical. None of our patients with PG presented with intralesional vascular involvement on cutaneous biopsy.
Most patients with SS presented with superficial and deep dermal infiltration of neutrophils with perivascular localization, but without vasculitis. Dermal edema was often observed.
EED presented with perivascular neutrophil infiltration with possible leukocytoclastic vasculitis of capillaries.
Treatments and Outcomes
All of the patients received corticosteroids at the time of AAV diagnosis except 1 patient whose treatment was started on PG diagnosis, 3 years before GPA (patient 4).
All of the patients received another combined immunosuppressant. Cyclophosphamide (CYC) was administered to 10 patients, including 8 GPA and 2 MPA patients. Eight patients received azathioprine, including 4 who had previously received intravenous CYC. One patient with a concomitant diagnosis of GPA and Sweet syndrome received colchicine with corticosteroids (patient 16). One patient, with both refractory GPA and PG, remained unresponsive to 18 boluses of CYC; intravenous immunoglobulin combined with azathioprine finally led to a clear improvement in both conditions with mycophenolate mofetil (patient 7). Another patient with GPA and PG was successfully treated with cyclosporine following a poor response to CYC in both entities (patient 11).
Overall, all of the patients recorded favorable outcomes, except for 1 patient (patient 8, 89 years’ old), who died from a myocardial infarction at the time of the third CYC bolus. The other 16 patients achieved remission for both AAV and ND 4 (1–33) months (median) after introducing first-line treatment. However, 7 patients relapsed, including 6 patients with GPA and PG (patients 5, 6, 7, 10, 14, and 17, 5 of whom were first treated with CYC) and 1 patient with EGPA and Sweet syndrome (patient 2, initially treated with corticosteroids and azathioprine). ANCA was positive at the time of relapse in 6 of the 7 relapsing patients. Relapses corresponded to vasculitis in only 2 patients (patients 5 and 6) and to both conditions in 5 others (patients 2, 7, 10, 14, and 17).
All 7 relapsing patients initially presented with ENT manifestations. Therefore, all exhibited sustained complete remission with an increased dose of corticosteroids combined with an immunosuppressant in three cases, or with rituximab in 3 other cases (patients 5, 6, 10). One patient (patient 7, suffering from GPA and PG), was treated with rituximab for a 3rd relapse after CYC failed for the second time.
The latest findings show that the patients were monitored for a median period of 32 (3–289) months.
Published Data
We initially identified 45 articles in the field, mostly single case reports or short series of 2 to 3 patients; we excluded 17 of them because of doubtful diagnosis regarding AAV or ND in 10, a lack of cardinal information in 6, and 1 letter that reported on our “patient 3.”7
Thus, 28 relevant articles were analyzed in our systematic review; data on 33 patients (median age 54 [7–78] years) were retrieved and are described in Table 3 .8–35
TABLE 3.
Literature Review on ANCA-associated Vasculitis With Neutrophilic Dermatosis
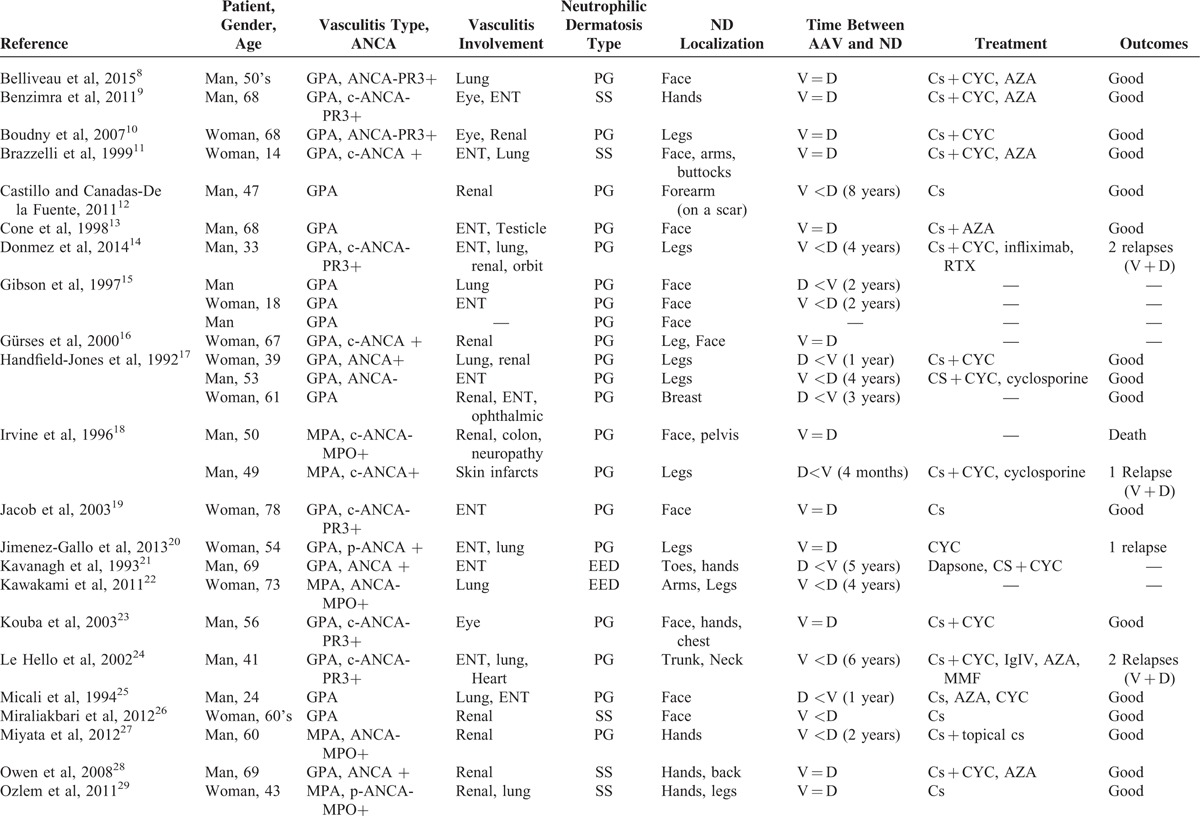
TABLE 3 (Continued).
Literature Review on ANCA-associated Vasculitis With Neutrophilic Dermatosis
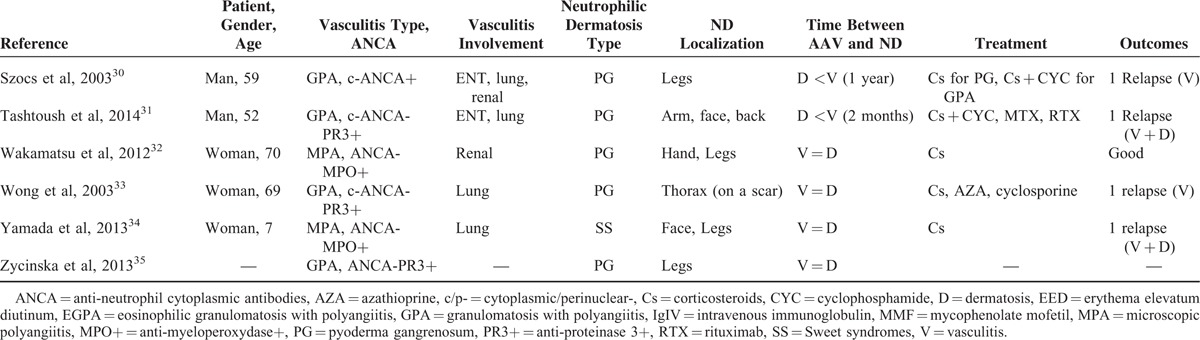
In these articles, GPA was also the most frequent AAV associated with ND in 26 (79%) patients, followed by MPA observed in 7 (31%) others, whereas no case of EGPA was found in the literature review.
Of the 33 patients, 25 (76%), 6 (18%), and 2 (6%) presented with PG, SS, and EED, respectively. PG was by far the most frequent ND seen in GPA, involving 21 of the 26 (81%) patients with vasculitis. As regards ND clinical characteristics, PG affected limb extremities in 13 cases, the face in 12 cases, and the trunk in 6 patients; SS also mostly affected limb extremities in 5 patients, the face in 3 cases, and the chest in 2 patients, whereas EED was only observed on limb extremities.
AAV and ND were concomitantly diagnosed in 16 (48%) patients; vasculitis preceded neutrophilic dermatosis in 8 patients 4 (2–8) years before, whereas ND was diagnosed before vasculitis in 8 other patients, 12 (2–60) months before. In the last patient, there was a lack of information regarding the time of occurrence of both conditions.15 Where discussed, treatment mainly comprised corticosteroids combined with an immunosuppressive agent (mostly CYC). Two-thirds of patients exhibited long-term sustained favorable outcomes, whereas 8 others were reported to have relapsed. Given the frequent lack of data about specific ANCA characteristics, the relationship between the subtypes of this antibody and those of the related AAV and ND could not be analyzed in this study.
French Cohort and Literature Data Compilation
Regarding the 50 patients (17 from our cohort and 33 from the literature review), GPA remained the most frequent AAV, followed by MPA and then EGPA, in 38 (76%), 11 (22%), and 1 (2%) case(s) respectively (Figure 1B). The distribution of the various NDs showed that PG remained the most frequent, followed by SS and finally EED in 33 (66%), 12 (24%), and 5 (10%) patients, respectively.
On analyzing the relationship between the 2 entities, even though all 3 ND subtypes were found in both GPA and MPA, some differences in terms of distribution were detected among these 2 AAVs, as shown in Figure 1B. Indeed, whereas PG was by far the most common ND found in GPA, its frequency in MPA was significantly lower (29/38 [89%] in GPA patients vs 4/11 [36%] in MPA patients; P = 0.02). Overall, 15 patients relapsed (12 with GPA, 2 with MPA, 1 with EGPA). Both conditions relapsed simultaneously in 10 patients, whereas only vasculitis relapsed in 5 patients. Patients who relapsed presented with more ENT manifestations than those who did not (12/15 [80%] relapsing patients vs 15/35 (43%) nonrelapsing patients; P = 0.03).
DISCUSSION
Our study including 50 AAVs associated with ND correlates a possible link between certain AAVs and ND, especially between GPA and PG. This type of association has only been suggested in case reports or in very small series to date.
Indeed, considering the overall population studied, the patients included here exhibited systemic forms of AAV with multiple organ involvement and clinical presentation that was concordant with conventionally described findings. GPA was the most common AAV, with the usual clinical and biological patterns. Although we found the 3 main subtypes of ND, that is, PG, SS, and EED in this study, PG was present in half and two-thirds of the patients, in the French cohort and literature review, respectively. Furthermore, PG was found in all the GPA patients in the French cohort and in more than three-quarters of the GPA patients in the literature review. Even though PG was also found in MPA, the second most common AAV in both the French cohort and data compilation, its frequency of occurrence in only about one-third of the patients appeared significantly lower than that observed in patients with GPA. SS, which is the second most common ND, was not overobserved in a vasculitis group. Finally, EGPA seems to be seldom associated with ND, as only 1 case of this AVV was found among the 50 patients in the data set, namely in the French cohort. In more than two-thirds of our patients, both conditions occurred concomitantly and treatment of vasculitis improved ND in almost all of the patients.
Cutaneous involvement in AAV has been reported in 10% to 69% of patients. In various studies, palpable purpura on the lower extremities was the most frequent finding observed in 47% to 76% of cases, followed by ulcers (14%–27%), papules (14%–20%), and subcutaneous nodules (13%–18%). Biopsy, when performed, mostly highlighted leukocytoclastic vasculitis in 80% of cases, but granulomatous inflammation or mixed inflammatory patterns can be seen in addition to the typical findings of ND reported here.1–3,6
PG is definitely another possible manifestation of AAV. An association of GPA and PG has been suggested for 50 years, especially in a particular form of the disease, called “malignant pyoderma” that often involves the face. Malignant pyoderma may indicate a severe inflammatory pyoderma, which progresses to exaggerated destruction of ulcerative tissue frequently accompanied by systemic illness.15 Some authors observed that most patients with an initial diagnosis of malignant pyoderma were eventually diagnosed with GPA.1 Our literature review identified 11 patients with PG involving the face. Of these, 10 suffered from GPA. We did not include enough patients with MPA to suggest a pattern of association with ND. Of the 11 patients with MPA (4 in our cohort and 7 in the literature review), 5 (45%) had SS, 4 PG, and 2 EED.
Neutrophilic dermatoses have been reported in association with other systemic diseases or with treatment or drug consumption.5 We did not include these patients in this cohort, except for 2 who have medical histories of cured and nonactive ulcerative colitis and thyroid papillary cancer, respectively. The literature review identified only 1 patient with a history of Crohn disease.19
AAV and ND are likely to share some common pathophysiological features as neutrophil dysregulation appears to be the main pathological and immunohistochemical substratum in both conditions. In AAV, neutrophils adhere to the vessel walls and release lytic enzymes and toxic oxygen radicals that promote invasive inflammation and fibrinoid necrosis of the arterial wall. ANCA target neutrophils and may induce leukocyte activation.36 Neutrophil fragmentation seems to be essential and leads to the exposure of intracytoplasmic molecules (such as myeloperoxidase or proteinase 3) that are normally sequestered inside cells, reaching the immune system. In vitro, it has been shown that ANCA induces neutrophil activation and degranulation. MPO released from activated neutrophils can bind to nonactivated neutrophils and render these cells susceptible to anti-MPO antibodies.37,38 NDs are part of and/or share many features of monogenic inflammatory disorders such as fever, arthralgias, and neutrophilic infiltrates, as witnessed in Crohn disease, PAPA syndrome (pyogenic sterile arthritis, PG, acne), and other autoinflammatory diseases.4 The role of ANCA in ND is not well established, and there are no fundamental data. ANCA may be directly or indirectly pathogenic in ND and/or may be generated because of neutrophil activation and apoptosis in these dermatoses, resulting in an autoamplifying loop. Moreover, some clinical features in the literature review argue in favor of ANCA as a credible causal factor or at least as a surrogate marker of some ND. Indeed, several clinical conditions such as inflammatory bowel diseases (IBDs), including Crohn disease and ulcerative colitis can exhibit both ND and ANCA, especially their anti-MPO subtypes.20,29,32,39 Because of the high prevalence of ANCA in IBD (60%–80% and 10%–30% in ulcerative colitis and Crohn disease, respectively),40 and their negativity in Behcet disease, Filik et al41 propose an ANCA, but also an ASCA assay as an aid to differential diagnosis of both entities. Although ANCA is negative in Behçet disease, ND and especially PG have been reported in this condition, but less frequently than in IBD.4 Drug-induced vasculitis, including a link to cocaine use, anti-TNF-alpha, or synthesis of anti-thyroid agents, sometimes exhibited both ND and ANCA associated with typical necrotizing systemic vasculitis.20,29,32,39 Cases of SS or PG associated with Sneddon-Wilkinson syndrome, which is another subtype of ND, have been described with propylthiouracil-induced p-ANCA.42–44 Considering primary ND, apart from several isolated case reports,45–48 Kemmet et al49 published a series on 6 of 7 patients with SS displaying positive ANCA. Thus, von den Driesch et al and Ozlem et al queried ANCA's role as a serological marker/diagnostic tool for SS.29,50 In 2004, Ayoub et al51 showed the positivity of IgA ANCA in 10 patients with EED. However, the specific target of these antibodies has not been identified to date.
Overall, these data suggest that ANCA may be tested for in ND, and when positive, careful attention should be paid to detect signs suggestive of systemic vasculitis. Interestingly, our 2 patients who developed PG before GPA were already ANCA-positive at the time of PG diagnosis since the case reported by Hunt et al47 showed ANCA-related ND associated with pauci-immune glomerulonephritis and arthritis. The close relationship between ND and AAV is also reinforced by the works of Darné et al and Kawakami et al supporting the paradigm of the proliferative effect of ANCA on neutrophil activation and their pathogenic and clinical extension beyond the realm of vasculitis alone. Indeed specific histopathological analysis demonstrated necrotizing vasculitis with moderate neutrophilic infiltrations and/or palisading granulomas in the papillary to middle dermis in 2 MPA patients.37,52
Corticosteroids remain the mainstay treatment for ND and, in most cases, are sufficient. However, some refractory cases exist and may require immunosuppressive agents, such as azathioprine, cyclosporine, or methotrexate. Targeted therapies including interleukin-1 or TNF blockade agents seem to be another effective option.14,53 In our cases, immunosuppressive drugs were administered for vasculitis with concomitant improvement of the dermatological disease. Overall, outcomes seem to be good under treatment, and relapses, which often occur in parallel for both entities, were reported in one-third of patients.
Here we report the largest series of patients with AAV and ND, implemented with a literature review. The retrospective design of our study is a limitation, especially given the potential lack of certain clinical, biological, or outcome information. However, a standard registration form was used and physicians were contacted as necessary to obtain any missing information. Moreover, the French AAV network is committed to providing complete and consistent clinical, biological, histological, and outcome data.
CONCLUSION
Overall, our study acts as a reminder that AAV is a systemic disease in which cutaneous manifestations are not rare. ND should probably be considered part of the cutaneous spectrum of AAV, especially the association of GPA and PG. We presume that the various clinical forms of ND are underdiagnosed in AAV through lack of systematic cutaneous biopsy and/or dermatologist expertise. Treatment and outcomes seem to be good and may not differ from those in patients with isolated vasculitis. The complex relationships and pathogenic mechanisms shared by AAV and ND are far from clear and require further prospective clinical and fundamental research, which will probably allow the real frequency of this association to be assessed.
Acknowledgments
None.
Footnotes
Abbreviations: AAV = ANCA-associated vasculitis, ANCA = antineutrophil cytoplasmic antibody, EED = erythema elevatum diutinum, EGPA = eosinophilic granulomatosis with polyangiitis, FVSG = french vasculitis study group, GPA = granulomatosis with polyangiitis, IBD = intestinal bowel disease, MPA = microscopic polyangiitis, ND = neutrophilic dermatosis, PG = pyoderma gangrenosum, SNFMI = société nationale française de médecine interne, SS = Sweet's syndrome, TNF = tumor necrosis factor
The authors report no conflicts of interest.
REFERENCES
- 1.Comfere NI, Macaron NC, Gibson LE. Cutaneous manifestations of Wegener’granulomatosis: a clinicopathologic study of 17 patients and correlation to antineutrophil cytoplasmic antibody status. J Cutan Pathol 2007; 34:739–747. [DOI] [PubMed] [Google Scholar]
- 2.Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener's granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol 1994; 31:605–612. [DOI] [PubMed] [Google Scholar]
- 3.Frances C, Du LT, Piette JC, et al. Wegener's granulomatosis. Dermatological manifestations in 75 cases with clinicopathologic correlation. Arch Dermatol 1994; 130:861–867. [PubMed] [Google Scholar]
- 4.Prat L, Bouaziz JD, Wallach D, et al. Neutrophilic dermatoses as systemic diseases. Clin Dermatol 2014; 32:376–388. [DOI] [PubMed] [Google Scholar]
- 5.Wallach D, Vignon-Pennamen MD. From acute febrile neutrophilic dermatosis to neutrophilic disease: forty years of clinical research. J Am Acad Dermatol 2006; 55:1066–1071. [DOI] [PubMed] [Google Scholar]
- 6.Grayson PC, Cuthbertson D, Carette S, et al. New features of disease after diagnosis in 6 forms of systemic vasculitis. J Rheumatol 2013; 40:1905–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saussine A, Gueguen A, de Menthon M, et al. Sweet syndrome revealing microscopic polyangiitis. Rheumatology (Oxford) 2012; 51:1916–1917. [DOI] [PubMed] [Google Scholar]
- 8.Belliveau MJ, Jakubovic BD, Mahendira D, et al. Necrotizing eyelid inflammation heralding granulomatosis with polyangiitis. Am J Med 2015; 129: e7–8. [DOI] [PubMed] [Google Scholar]
- 9.Benzimra J, Low-Beer J, Twomey J. A case of peripheral ulcerative keratitis associated with neutrophilic dermatosis of the dorsal hand. Int Ophthalmol 2011; 31:149–151. [DOI] [PubMed] [Google Scholar]
- 10.Boudny C, Nievergelt H, Braathen LR, et al. Wegener's granulomatosis presenting as pyoderma gangrenosum. J Dtsch Dermatol Ges 2008; 6:477–479. [DOI] [PubMed] [Google Scholar]
- 11.Brazzelli V, Vassallo C, Baldini F, et al. Wegener granulomatosis in a child: cutaneous findings as the presenting signs. Pediatr Dermatol 1999; 16:277–280. [DOI] [PubMed] [Google Scholar]
- 12.Castillo RF, Canadas-De la Fuente GA. Pyoderma gangrenosum developing over an arteriovenous fistula scar. Intern Med J 2011; 41:436–437. [DOI] [PubMed] [Google Scholar]
- 13.Cone LA, Annunziata GM, Gebhart RN. Malignant pyoderma and Wegener's granulomatosis. Mayo Clin Proc 1998; 73:390. [DOI] [PubMed] [Google Scholar]
- 14.Donmez S, Pamuk ON, Gedik M, et al. A case of granulomatosis with polyangiitis and pyoderma gangrenosum successfully treated with infliximab and rituximab. Int J Rheum Dis 2014; 17:471–475. [DOI] [PubMed] [Google Scholar]
- 15.Gibson LE, Daoud MS, Muller SA, et al. Malignant pyodermas revisited. Mayo Clin Proc 1997; 72:734–736. [DOI] [PubMed] [Google Scholar]
- 16.Gurses L, Yucelten D, Comert A, et al. Wegener's granulomatosis presenting as neutrophilic dermatosis: a case report. Br J Dermatol 2000; 143:207–209. [DOI] [PubMed] [Google Scholar]
- 17.Handfield-Jones SE, Parker SC, Fenton DA, et al. Wegener's granulomatosis presenting as pyoderma gangrenosum. Clin Exp Dermatol 1992; 17:197–200. [DOI] [PubMed] [Google Scholar]
- 18.Irvine AD, Bruce IN, Walsh M, et al. Dermatological presentation of disease associated with antineutrophil cytoplasmic antibodies: a report of two contrasting cases and a review of the literature. Br J Dermatol 1996; 134:924–928. [PubMed] [Google Scholar]
- 19.Jacob SE, Martin LK, Kerdel FA. Cutaneous Wegener's granulomatosis (malignant pyoderma) in a patient with Crohn's disease. Int J Dermatol 2003; 42:896–898. [DOI] [PubMed] [Google Scholar]
- 20.Jimenez-Gallo D, Albarran-Planelles C, Linares-Barrios M, et al. Pyoderma gangrenosum and Wegener granulomatosis-like syndrome induced by cocaine. Clin Exp Dermatol 2013; 38:878–882. [DOI] [PubMed] [Google Scholar]
- 21.Kavanagh GM, Colaco CB, Bradfield JW, et al. Erythema elevatum diutinum associated with Wegener's granulomatosis and IgA paraproteinemia. J Am Acad Dermatol 1993; 28:846–849. [DOI] [PubMed] [Google Scholar]
- 22.Kawakami T, Kyoya M, Matsuoka S, et al. Acceleration of pulmonary interstitial fibrosis in a patient with myeloperoxidase-antineutrophil cytoplasmic antibody-positive erythema elevatum diutinum. J Am Acad Dermatol 2011; 65:674–675. [DOI] [PubMed] [Google Scholar]
- 23.Kouba DJ, Mimouni D, Ha CT, et al. Limited Wegener's granulomatosis manifesting as malignant pyoderma with corneal melt. Int J Dermatol 2003; 42:902–904. [DOI] [PubMed] [Google Scholar]
- 24.Le Hello C, Bonte I, Mora JJ, et al. Pyoderma gangrenosum associated with Wegener's granulomatosis: partial response to mycophenolate mofetil. Rheumatology (Oxford) 2002; 41:236–237. [DOI] [PubMed] [Google Scholar]
- 25.Micali G, Cook B, Ronan S, et al. Cephalic pyoderma gangrenosum (PG)-like lesions as a presenting sign of Wegener's granulomatosis. Int J Dermatol 1994; 33:477–480. [DOI] [PubMed] [Google Scholar]
- 26.Miraliakbari HM, McEarchen J, Prasad B. Sweet's syndrome in a patient with Wegener's granulomatosis and ESRD. BMJ Case Rep 2012; 2012: bcr0820103231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyata T, Yashiro M, Hayashi M, et al. Bullous pyoderma gangrenosum of the bilateral dorsal hands. J Dermatol 2012; 39:1006–1009. [DOI] [PubMed] [Google Scholar]
- 28.Owen CE, Malone JC, Callen JP. Sweet-like dermatosis in 2 patients with clinical features of dermatomyositis and underlying autoimmune disease. Arch Dermatol 2008; 144:1486–1490. [DOI] [PubMed] [Google Scholar]
- 29.Ozlem C, Deram B, Mustafa S, et al. Propylthiouracil-induced anti-neutrophil cytoplasmic antibodies and agranulocytosis together with granulocyte colony-stimulating factor induced Sweet's syndrome in a patient with Graves’ disease. Intern Med 2011; 50:1973–1976. [DOI] [PubMed] [Google Scholar]
- 30.Szocs HI, Torma K, Petrovicz E, et al. Wegener's granulomatosis presenting as pyoderma gangrenosum. Int J Dermatol 2003; 42:898–902. [DOI] [PubMed] [Google Scholar]
- 31.Tashtoush B, Memarpour R, Johnston Y, et al. Large pyoderma gangrenosum-like ulcers: a rare presentation of granulomatosis with polyangiitis. Case Rep Rheumatol 2014; 2014:850364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wakamatsu K, Mitsuhashi Y, Yamamoto T, et al. Propylthiouracil-induced antineutrophil cytoplasmic antibody positive vasculitis clinically mimicking pyoderma gangrenosum. J Dermatol 2012; 39:736–738. [DOI] [PubMed] [Google Scholar]
- 33.Wong YW, Lyon CC, Benbow EW, et al. Pyoderma gangrenosum in a thoracotomy wound associated with a pulmonary cavitating lesion. Clin Exp Dermatol 2003; 28:274–276. [DOI] [PubMed] [Google Scholar]
- 34.Yamada Y, Kitagawa C, Kamioka I, et al. A case of microscopic polyangiitis with skin manifestations in a seven-year-old girl. Dermatol Online J 2013; 19:19624. [PubMed] [Google Scholar]
- 35.Zycinska K, Wardyn K, Zielonka TM, et al. Cutaneous changes: an initial manifestation of pulmonary Wegener's granulomatosis. Adv Exp Med Biol 2013; 755:307–310. [DOI] [PubMed] [Google Scholar]
- 36.Schönermarck U, Csemok E, Gross WL. Pathogenesis of anti-neutrophil cytoplasmic antibody-associated vasculitis: challenges and solutions 2014. Nephrol Dial Transplant 2015; Suppl 1:46–52. [DOI] [PubMed] [Google Scholar]
- 37.Darne S, Natarajan S, Blasdale C. Do antineutrophil cytoplasmic antibodies (ANCA) play a key role in neutrophilic dermatoses? A case of propylthiouracil-induced neutrophilic dermatosis with positive perinuclear ANCA. Clin Exp Dermatol 2010; 35:406–408. [DOI] [PubMed] [Google Scholar]
- 38.Reumaux D, Duthilleul P, Roos D. Pathogenesis of diseases associated with antineutrophil cytoplasm autoantibodies. Hum Immunol 2004; 65:1–12. [DOI] [PubMed] [Google Scholar]
- 39.Elkadri AA, Stempak JM, Walters TD, et al. Serum antibodies associated with complex inflammatory bowel disease. Inflamm Bowel Dis 2013; 19:1499–1505. [DOI] [PubMed] [Google Scholar]
- 40.Papp M, Norman GL, Altorjay I, et al. Utility of serological markers in inflammatory bowel diseases: gadget or magic? World J Gastroenterol 2007; 13:2028–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Filik L, Biyikoglu I. Differentiation of Behcet's disease from inflammatory bowel diseases: anti-Saccharomyces cerevisiae antibody and anti-neutrophilic cytoplasmic antibody. World J Gastroenterol 2008; 14:7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boulenger-Vazel A, Kupfer-Bessaguet I, Gouedard C, et al. Neutrophilic dermatosis associated with propylthiouracil-induced p-ANCA (p-antineutrophil cytoplasmic antibodies). Ann Dermatol Venereol 2005; 132:27–31. [DOI] [PubMed] [Google Scholar]
- 43.Lund JJ, Stratman EJ, Jose D, et al. Drug-induced bullous sweet syndrome with multiple autoimmune features. Autoimmune Dis 2011; 2011:176749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seo JW, Son HH, Choi JH, et al. A case of p-ANCA-positive propylthiouracil-induced pyoderma gangrenosum. Ann Dermatol 2010; 22:48–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bayle P, Laplanche G, Gorguet B, et al. Neutrophilic dermatosis: a case of overlapping syndrome with monoclonal antineutrophil cytoplasmic autoantibody activity. Dermatology 1994; 189:69–71. [DOI] [PubMed] [Google Scholar]
- 46.Hoffman MD. Pyoderma gangrenosum associated with c-ANCA (h-lamp-2). Int J Dermatol 2001; 40:135–137. [PubMed] [Google Scholar]
- 47.Hunt RD, Hartman RD, Molho-Pessach V, et al. Palisaded neutrophilic and granulomatous dermatitis in an adolescent girl with perinuclear antineutrophil cytoplasmic antibody-positive pauci-immune glomerulonephritis and arthritis. J Am Acad Dermatol 2012; 67:e164–e166. [DOI] [PubMed] [Google Scholar]
- 48.Malik M, Perkins W, Leach I. Anti-neutrophil cytoplasmic antibody-positive neutrophilic dermatosis of the dorsal hands. Clin Exp Dermatol 2012; 37:869–870. [DOI] [PubMed] [Google Scholar]
- 49.Kemmett D, Harrison DJ, Hunter JA. Antibodies to neutrophil cytoplasmic antigens: serologic marker for Sweet's syndrome. J Am Acad Dermatol 1991; 24:967–969. [DOI] [PubMed] [Google Scholar]
- 50.von den Driesch P, Weber MF. Are antibodies to neutrophilic cytoplasmic antigens (ANCA) a serologic marker for Sweet's syndrome? J Am Acad Dermatol 1993; 29:666. [DOI] [PubMed] [Google Scholar]
- 51.Ayoub N, Charuel JL, Diemert MC, et al. Antineutrophil cytoplasmic antibodies of IgA class in neutrophilic dermatoses with emphasis on erythema elevatum diutinum. Arch Dermatol 2004; 140:931–936. [DOI] [PubMed] [Google Scholar]
- 52.Kawakami T, Kawanabe T, Saito C, et al. Clinical and histopathologic features of 8 patients with microscopic polyangiitis including two with a slowly progressive course. J Am Acad Dermatol 2007; 57:840–848. [DOI] [PubMed] [Google Scholar]
- 53.Kluger N, Gil-Bistes D, Guillot B, et al. Efficacy of anti-interleukin-1 receptor antagonist anakinra (Kineret(R)) in a case of refractory Sweet's syndrome. Dermatology 2011; 222:123–127. [DOI] [PubMed] [Google Scholar]