

Supplemental Digital Content is available in the text
Abstract
von Willebrand disease (VWD) is a genetic bleeding disease due to a defect of von Willebrand factor (VWF), a glycoprotein crucial for platelet adhesion to the subendothelium after vascular injury. VWD include quantitative defects of VWF, either partial (type 1 with VWF levels <50 IU/dL) or virtually total (type 3 with undetectable VWF levels) and also qualitative defects of VWF (type 2 variants with discrepant antigenic and functional VWF levels). The most bleeding forms of VWD usually do not concern type 1 patients with the mildest VWF defects (VWF levels between 30 and 50 IU/dL).
The French reference center for VWD performed a laboratory phenotypic and genotypic analysis in 1167 VWD patients (670 families) selected by their basic biologic phenotype: type 3, type 2, and type 1 with VWF levels <30 IU/dL. In these patients indeed, to achieve an accurate diagnosis of VWD type and subtype is crucial for the management (treatment and genetic counseling).
A phenotype/genotype correlation was present in 99.3% of cases; 323 distinct VWF sequence variations (58% of novel) were identified (missense 67% versus truncating 33%). The distribution of VWD types was: 25% of type 1, 8% of type 3, 66% of type 2 (2A: 18%, 2B: 17%, 2M: 19%, 2N: 12%), and 1% of undetermined type. Type 1 VWD was related either to a defective synthesis/secretion or to an accelerated clearance of VWF. In type 3 VWD, bi-allelic mutations of VWF were found in almost all patients. In type 2A, the most frequent mechanism was a hyper-proteolysis of VWF. Type 2B showed 85% of patients with deleterious mutations (distinct from type 2B New York). Type 2M was linked to a defective binding of VWF to platelet glycoprotein Ib or to collagen. Type 2N VWD included almost half type 2N/3.
This biologic study emphasizes the complex mechanisms for both quantitative and qualitative VWF defects in VWD. In addition, this study provides a new epidemiologic picture of the most bleeding forms of VWD in which qualitative defects are predominant.
INTRODUCTION
von Willebrand factor (VWF) is a large multimeric glycoprotein crucial for platelet-dependent primary hemostasis; it is also the carrier protein for coagulation factor VIII (FVIII).1 VWF is synthesized as a 2813-amino-acid (aa) monomeric protein exhibiting repeated domains in the order D1-D2-D′-D3-A1-A2-A3-D4-C1-C2-C3-C4-C5-C6-CK.2 VWF maturation includes dimerization, multimerization, and cleavage of the 741 aa pro-peptide (D1-D2) producing the 2050 aa mature VWF subunit.3 VWF multimers are secreted into plasma and their size is regulated by a specific cleaving-protease named ADAMTS13 (A Disintegrin and Metalloproteinase with ThromboSpondin type 1 repeats, member 13).3 The VWF gene (VWF) is located at the tip of the short arm of chromosome 12 (178 kb of genomic DNA and 52 exons).4,5
Considering its large size and its structural complexity involving many distinct domains and an original multimeric organization, VWF is prone to both quantitative and qualitative defects. In a very large majority of cases, VWF defects are genetically inherited (related to mutations of VWF gene or of other still unknown genes) and lead to von Willebrand disease (VWD).6 In contrast, few VWF defects are acquired by miscellaneous mechanisms, consisting in an acquired von Willebrand syndrome (AVWS).7 VWF defects induce bleeding manifestations, mainly mucosal, which intensity is usually proportional to the severity of the protein deficiency.8 The revised classification of VWD includes 6 types:9 VWD type 1 (OMIM ID#193400) is defined by low levels (<50 IU/dL) of a functionally normal VWF and a dominant inheritance. Within type 1, a ∼30 IU/dL threshold (plasma VWF levels lower than 30 IU/dL) is likely to distinguish VWF defects related to VWF gene sequence variation(s) from VWF defects potentially related to other genetic causes/contributors such as gene modifiers.10,11 VWD type 3 defined as severe VWD (OMIM ID#277480) is characterized by undetectable (<1 IU/dL) plasma and platelet VWF levels and a recessive inheritance.12 VWD type 2 (OMIM ID #613554) consisting in qualitative functional VWF defects is very heterogeneous and includes 4 main types:13 type 2A is defined by a decreased binding of VWF to platelet GPIb due to a significant reduction or absence of the high-molecular-weight (HMW) multimers of VWF; type 2M is characterized by a decreased binding of VWF to platelet GPIb or to collagen with normal or subnormal multimeric distribution; type 2B is defined by an increased binding of VWF to platelet GPIb; type 2A, 2B and 2M have in common to affect primary hemostasis and a usually dominant inheritance. In contrast, type 2N VWD is defined by an impaired binding of VWF to FVIII and a recessive inheritance. A supplementary level of classification is used to distinguish specific mechanisms within type 2A (IIA, IIC, IID, IIE),14 within type 2B (i.e. “New York” type),15 or within type 1 (i.e. “Vicenza” rapid VWF clearance type).16
The diagnosis of VWD is based on clinical and biological information.17 The laboratory phenotypic investigation for VWD includes many tests ranked in 3 levels.18 Screening assays include classically activated partial thromboplastin time (aPTT), platelet count, and closure time (PFA-100 analyzer). Second-level-specific VWF assays are crucial to diagnose VWF deficiency and they include the measurement of FVIII activity (FVIII:C), VWF antigen (VWF:Ag), and VWF ristocetin cofactor activity (VWF:RCo) allowing the calculation of ratios (VWF:RCo/VWF:Ag and FVIII:C/VWF:Ag) and the measurement of ristocetin-induced platelet aggregation (RIPA). Third-level VWF assays are devoted to a better characterization of VWD types and they include structural assays (VWF multimers analysis, VWF propeptide [VWFpp]) and functional assays (VWF binding to platelet GPIb, to collagen (VWF:CB) and to FVIII [VWF:FVIIIB]).19,20 Some of these assays are available only in some hospital laboratories specialized in the management of inherited bleeding disorders. The genotypic investigation for VWD, also performed in expert laboratories, is considered as a final step to definitely confirm the diagnosis of VWD-specific type.21,22 The diagnosis of VWD type and subtype is crucial for many purposes: to make a differential diagnosis between VWD 2N and hemophilia A or between VWD 2B and a platelet disorder named “pseudo VWD or platelet type VWD,”23 to predict the response to desmopressin24 or the kinetic of VWF during pregnancy, to anticipate the risk of allo-immunization in patients with type 3 VWD,12 and to help for genetic counseling.5
In terms of prevalence, VWD is usually reported to be “the most common human inherited bleeding disorder.” Actually, the 1% prevalence reported to support this observation includes all subjects with either qualitative or any quantitative VWF defects including the mildest quantitative defects (values ranging between 30 and 50 IU/dL), the latter subjects being usually pauci- or nonsymptomatic.25 In contrast, when excluding these milder forms and thus, only considering usually “bleeder” patients (who overlap VWD type 3, type 2, and type 1 with VWF levels usually <30 IU/dL), VWD prevalence is reported to be only ∼1/10,000.10,26 This low prevalence, lower than the 1/2000 threshold defining rare diseases, thus allows to consider the most bleeding forms of VWD as a rare disease. This new epidemiologic point of view prompted our group (hemostasis departments from Paris Lariboisière, Lille, Nantes, Bicêtre and Caen university hospitals) to candidate for a national certification for rare diseases provided by the National Plan for Rare Disease (NPRD) on behalf of the French Health Ministry. Our group has been qualified as the national reference center for VWD (CRMW for “Centre de Référence de la Maladie de Willebrand”) since 2006 and we have been collaborating with the main hemostasis departments of France (from about 50 university hospitals) as a national network. The CRMW provides to patients with VWD diagnosed in these latter hemostasis departments, a unique national biologic platform for both the highly specialized phenotype and the genotype of VWD.
The current paper reports the biologic analysis (focus on the phenotypic/genotypic correlation) of 1167 French patients with VWD (from 670 families) enrolled during the first 6 years of functioning of the CRMW. All patients were extensively investigated by the CRMW biologic platform to achieve an accurate diagnosis of VWD type. Thanks to stringent laboratory inclusion criteria required by the definition of rare diseases (limitation to VWD type 3, type 2 and type 1 with VWF levels <30 IU/dL), this cohort brings new insights in the epidemiology of VWD specifically related to mutation(s) of VWF gene.
PATIENTS, MATERIALS, AND METHODS
Patients
Among the missions of the CRMW, one is to help, on their request, all the physicians of France to make a precise diagnosis of VWD in their patients thanks to a unique biologic platform devoted to an exhaustive phenotypic and genotypic analysis of VWF. On purpose, only patients with the following laboratory inclusion criteria (defined by the CRMW but performed by the enrolling hemostasis departments from at least 2 distinct samples) could beneficiate from further laboratory testing by the CRMW biologic platform: for type 1 phenotype, VWF levels <30IU/dL (together with VWF:RCo/VWF:Ag and FVIII:C/VWF:Ag ratios >0.6); for type 2 phenotype, a decreased or normal VWF levels with a discrepancy between the antigenic and the functional levels (VWF:RCo/VWF:Ag or FVIII:C/VWF:Ag ratios < or = 0.6); and for type 3 phenotype, VWF plasma levels <5 IU/dL. Only inherited VWF defects were considered: any patient with a clinical context potentially responsible for an AVWS was thus excluded. Affected family members of index cases (IC) were also eligible. The inclusion period of patients spanned from January 1st, 2007, to December 31, 2012.
Blood Collection, Ethical Issues
Venous blood was collected at the time of enrolment in the study, into 1:10 final volume of 3.2% or 3.8% sodium citrate; platelet-poor plasma was obtained as described.27 Blood was also collected on EDTA for molecular genetic analysis. An informed consent, specific for the CRMW and explaining that both the phenotypic and genotypic assays were performed for medical diagnosis purposes, was obtained from each patient according to the Declaration of Helsinki. The study was approved by the Ethics Committee of Lille University Hospital. The CRMW database and biobank (plasma and DNA) were declared to the French data protection authority.
Phenotypic Assays
Phenotypic assays performed in VWD patients were as follows. First-level assays performed locally by the enrolling hemostasis departments, consisted in VWF:Ag, VWF:RCo, FVIII:C, platelet count, aPTT, Ristocetin-Induced Platelet Aggregation (RIPA), and PFA-100R Occlusion Time (ADP and Epinephrine).27 The second-level VWF assays were performed by the CRMW biologic platform as previously described: VWF multimeric distribution (1.5% SDS-agarose gel electrophoresis);28 VWF binding to platelet glycoprotein Ib,29 VWF:CB,30 VWF:FVIIIB,31 VWF pp.32 For each patient, the first-line test panel included VWF multimers and either VWF binding to platelet glycoprotein Ib (phenotype 2A, 2B, 2M) or VWF:FVIIIB (phenotype 2N). Other assays were added as a function of specific phenotypes (VWF:CB to document some type 2M or type 1, VWF pp to document VWF clearance in type 1).
Genetic Analysis
The strategy for VWF gene analysis in IC patients was performed as a function of VWD phenotype previously documented by second-level VWF assays (suppl. Material, suppl. Figure 1). Patients genomic DNA was screened for sequence variations by direct sequencing of VWF gene and in some cases, by multiplex ligation-dependent probe amplification (MLPA). HGVS nomenclature (URL: http://www.hgvs.org/mutnomen/) was used for the sequence variations. To analyze conservation across evolution and prediction on structure and functional effect of protein changes, each novel sequence variation was checked on miscellaneous software (suppl. Material).
Phenotype/Genotype Correlation
For each patient, the results of the phenotypic tests panel were compared to the results of the genetic analysis by a specific committee including some expert members of the CRMW. The VWF gene mutations in our patients were screened in both the international literature (www.pubmed.com) and the databases for VWF gene variations (Sheffield mutations database: http://www.ragtimedesign.com/vwf/mutation/ and LOVD VWF database: https://grenada.lumc.nl/LOVD2/VWF/home.php?select_db=VWF) to determine their deleterious or candidate status. The mutation(s) was (were) qualified to support the phenotype if already reported as deleterious or if candidate but located within a VWF domain likely to explain the phenotype.
RESULTS
Study Population and Demographic Features
According to the inclusion criteria, 1856 subjects were eligible for a phenotypic/genotypic analysis by the CRMW. Six hundred and eighty-nine subjects could not be tested because their blood samples were finally not sent to the CRMW biologic platform. Among the 1167 subjects tested for both phenotype and genotype, 670 were IC (consisting in VWD propositi from 670 unrelated families) whereas 497 were their affected family members. The cohort of 1167 VWD patients exhibited a sex ratio of 1.46F/1M) and an age distribution ranging from 6 months to 90 years-old with a 34 y.o. median. The proportion of O blood group was 56%, and in those patients, mean VWF:Ag levels were 19 IU/dL lower than in patients with non-O blood group. The main ethnic groups were White (90% including 15% of people from North Africa) and Afro-Caribbean (10%).
Phenotype/Genotype Correlation
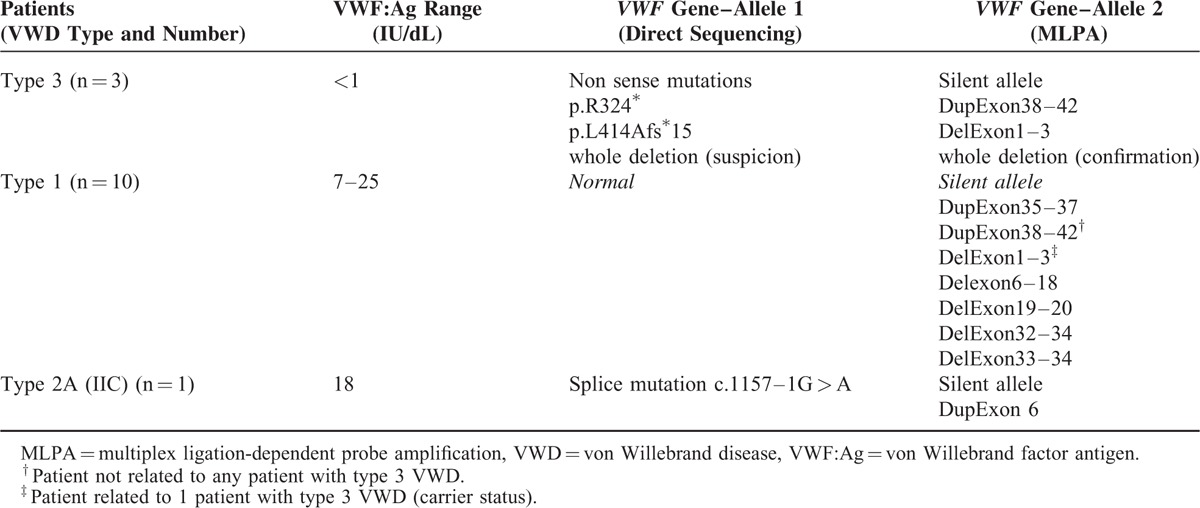
The proportions of the miscellaneous VWD types and subtypes were similar between the 670 IC and the global cohort of 1167 subjects (Figure 1). Among 670 IC, after second-level phenotypic assays and direct sequencing of VWF gene, 648 patients exhibited a genotype supporting their phenotype (1 mutation of VWF gene in 87% of cases, several mutations in 13% of cases) (Figure 2). In contrast, the genotype did not support the phenotype in 22 patients after direct sequencing who thus underwent MLPA analysis (Figure 2). Large deletions, insertions, and duplications were identified in 14 patients (Table 1) but 8 patients remained with a genotype either not explaining or only partially supporting their phenotype (Figure 2).
FIGURE 1.
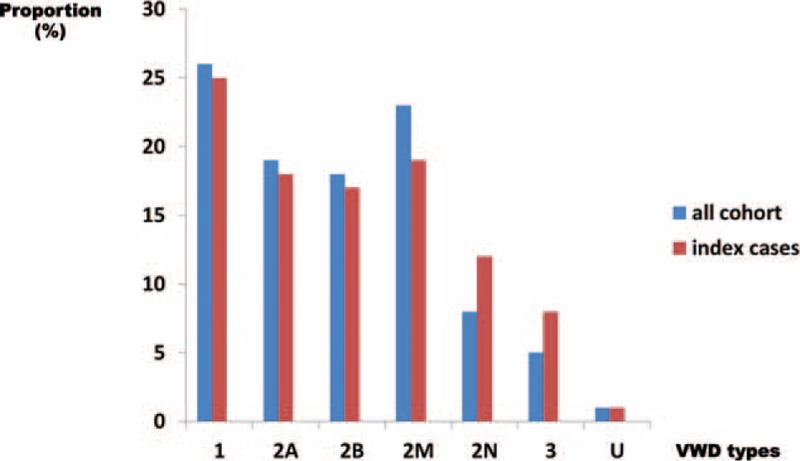
Distribution of von Willebrand disease (VWD) types in the global cohort (n = 1167 patients) and in the index cases (n = 670 cases). The proportion of each VWD type (1, 2A, 2B, 2M, 2N, and 3) is represented with blue histograms for the global cohort of 1167 patients and with red histograms for the 670 index cases (IC). These proportions are similar for both groups of patients within each VWD type. IC = index cases, VWD = von Willebrand disease.
FIGURE 2.
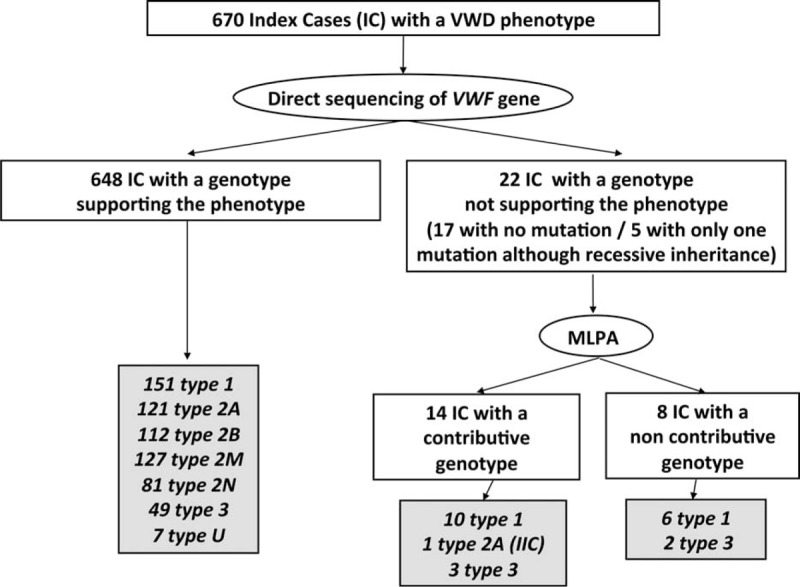
Phenotype/genotype correlation in 670 index cases (IC) patients with von Willebrand disease (VWD). Among 670 IC, after second level phenotypic assays and direct sequencing of VWF gene, 648 patients exhibited a genotype supporting their phenotype: 151 patients had type 1 VWD, 121 patients type 2A, 112 patients type 2B, 127 patients type 2M, 81 patients type 2N, 49 patients type 3, and 7 patients undetermined (U) VWD. In contrast, after direct sequencing, the genotype was not supporting the phenotype in 22 patients (17 patients with no mutation and 5 patients with only 1 mutation although recessive inheritance) who thus underwent Multiplex Ligation-dependent Probe Amplification (MLPA) analysis. MLPA helped identifying large deletions, insertions, and duplications in 14 patients consisting in 10 type 1, 1 type 2A (IIC), and 3 type 3. Finally, 8 patients remained with a genotype either not explaining their phenotype (6 type 1 patients with no mutation found) or only partially supporting their phenotype (2 type 3 patients with a single mutation found).IC = index cases, MLPA = multiplex ligation-dependent probe amplification, VWD = von Willebrand disease.
TABLE 1.
von Willebrand Factor Antigen (VWF:Ag) Levels and Genotype of 14 Index Cases Patients With von Willebrand Disease (VWD) in Whom MLPA (multiplex ligation-dependent probe amplification) Was Used to Document the Presence of a Deletion or Duplication
One hundred and sixty-seven (25%) IC patients were classified as type 1 VWD: normal or subnormal VWF multimeric pattern, presence of 1 heterozygous VWF gene mutation previously described or candidate in type 1 or type 3 VWD (n = 161), or no mutation identified (n = 6) (Figures 2 and 3). Type 1 mutations associated with a decreased synthesis/secretion of VWF were present in 88 patients (Figure 3). Interestingly, 33 patients exhibited mutations inducing an accelerated VWF clearance with a corresponding phenotype combining plasma VWF levels <15 IU/dL, increased VWF propeptide and sometimes ultralarge VWF multimers. Six patients had mutations localized within the propeptide-cleavage site. Also, 34 patients with type 1 exhibited heterozygous mutations also described in type 3 VWD (Figure 3) and were thus considered as type 3 carriers. In 6 patients with type 1 VWD, no mutation was found.
FIGURE 3.
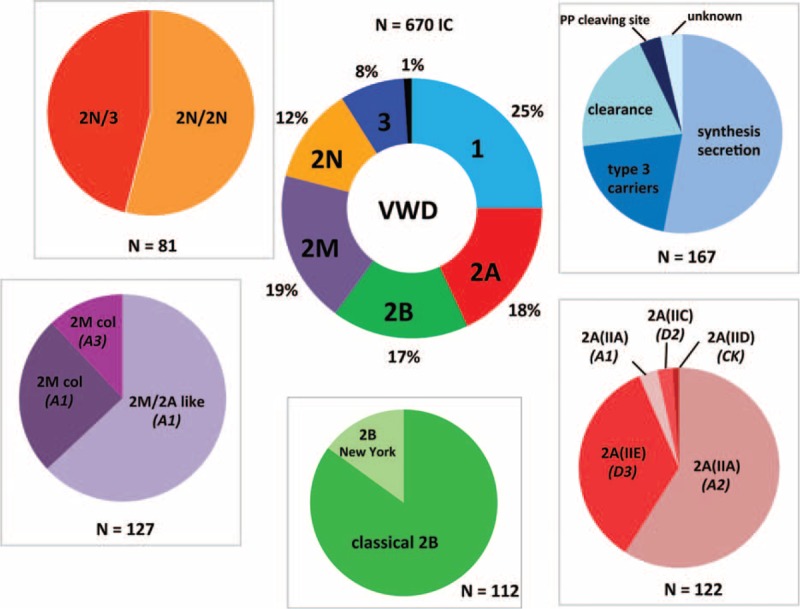
Distribution of von Willebrand disease (VWD) types and subtypes in 670 index cases patients. The central ring represents the proportion of VWD types in the 670 IC patients: 25% of type 1, 18% of type 2A, 17% of type 2B, 19% of type 2M, 12% of type 2N, 8% of type 3, and 1% of undetermined (U) VWD. The peripheral disks indicate the proportion of the miscellaneous mechanisms responsible for some VWD type. For type 1 VWD, the main mechanism was classical dominant negative mutations of VWF gene leading to synthesis/secretion defects (53%), whereas mutations responsible for an accelerated VWF clearance represented 20% of cases; heterozygous mutations usually described in type 3 VWD accounted for 20% of patients (type 3 carriers); mutations within VWF propeptide cleavage site were rare (3.5%) and the mechanism remained unknown in 3.5% of cases (no mutation found). In patients with type 2A VWD, the most common subtype was 2A(IIA) (62%) including a large majority of mutations within the A2 VWF domain inducing an increased VWF proteolysis by ADAMTS13 (59%), whereas mutations within the A1 VWF domain were rare (4%); subtype 2A(IIE) was frequent, accounting for 34.5% of type 2A, whereas subtypes 2A(IIC) and 2A(IID) were rare, representing 3% and 1%, respectively. Type 2B VWD included either a majority of classical 2B mutations located within the A1 VWF domain (85%) or some New York mutations within the D3 VWF domain (15%). In patients with type 2M VWD, the most common mechanism was mutations within the A1 VWF domain inducing a 2M/2A-like phenotype (63%) related to a defective binding of VWF to GPIb; the 2M phenotype consisted in a decreased binding of VWF to collagen (37%) related either to mutations within the A1 VWF domain or the A3 VWF domain. Type 2N VWD included patients with either bi-allelic 2N mutations (2N/2N, 54%) or a combination of a mono-allelic 2N mutation (expressed allele) with a mutation inducing a silent allele (2N/3, 46%).IC = index cases, VWD = von Willebrand disease, VWF = von Willebrand factor.
Four hundred and forty-two IC patients (66%) were diagnosed with type 2 VWD (Figure 2). One hundred and twenty-two patients (18%) exhibited type 2A VWD: 76 patients exhibited type 2A(IIA) VWD consisting in defect of the HMW and intermediate MW VWF multimers, decreased VWF binding to platelet GPIb and previously reported or candidate mutations within VWF A2 domain (n = 72) or VWF A1 domain (n = 4); 42 patients had type 2A(IIE), 3 patients type 2A(IIC), and 1 patient type 2A(IID) (Figure 3). One hundred and twelve patients (17%) exhibited a type 2B VWD including 95 patients with a “classical” type 2B (increased VWF binding to platelet GPIb, variable loss of the high-molecular-weight VWF multimers, moderate or severe thrombocytopenia and previously reported or candidate mutations within VWF A1 domain) and 17 patients with a type 2B “New York” (always normal VWF multimeric pattern, no thrombocytopenia) (Figure 3).
One hundred and twenty-seven patients (19%) were diagnosed with a type 2M VWD: most often smeary VWF multimeric pattern, decreased VWF binding to platelet GPIb in 80 cases or decreased VWF:CB in 47 cases (Figure 3). Eighty-one patients (12%) were identified with a type 2N VWD: most often normal VWF multimeric pattern, markedly decreased VWF binding to FVIII and previously reported or candidate mutations within D′- or D3 VWF domains. Among type 2N patients, 37 patients exhibited a type 2N/3 phenotype confirmed by the genetic analysis (1 allele with a 2N VWD mutation and 1 nil allele) (Figure 3).
Fifty-four IC patients (8%) were diagnosed with type 3 VWD: VWF levels <5 IU/dL, no VWF multimer detectable by electrophoresis, presence of 2 VWF gene mutations previously reported or candidate in type 3 VWD (n = 52) or only 1 mutation heterozygous VWF gene mutation identified (n = 2) (Figure 2, Figure 3).
Seven IC patients (1%) remained with a type “undetermined” (U) VWD combining either a type 1 or a type 2 phenotype and new VWF gene mutations (Figure 2, Figure 3 and Table 2).
TABLE 2.
Main Phenotypic and Genotypic Features of 7 Index Cases Patients With Undetermined von Willebrand Disease
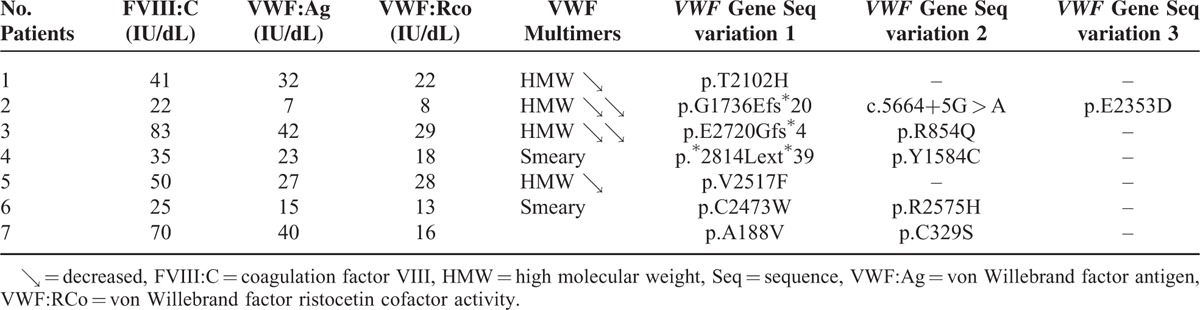
Focus on VWF Gene Mutations
A total of 323 distinct VWF gene mutations were identified in our VWD cohort including 189 (58%) new mutations. Missense mutations were the most frequent (67%), truncating sequence variations including nonsense mutations, small deletions/duplications and frameshift/splice mutations were 31% of all mutations, whereas large deletions/duplications were 2% of all mutations.
In type 1 VWD, 105 distinct sequence variations were identified (two-third of them being novel): they were spread all over VWF gene and they included a similar proportion of missense (51%) and truncating (49%) mutations (Figure 4).
FIGURE 4.
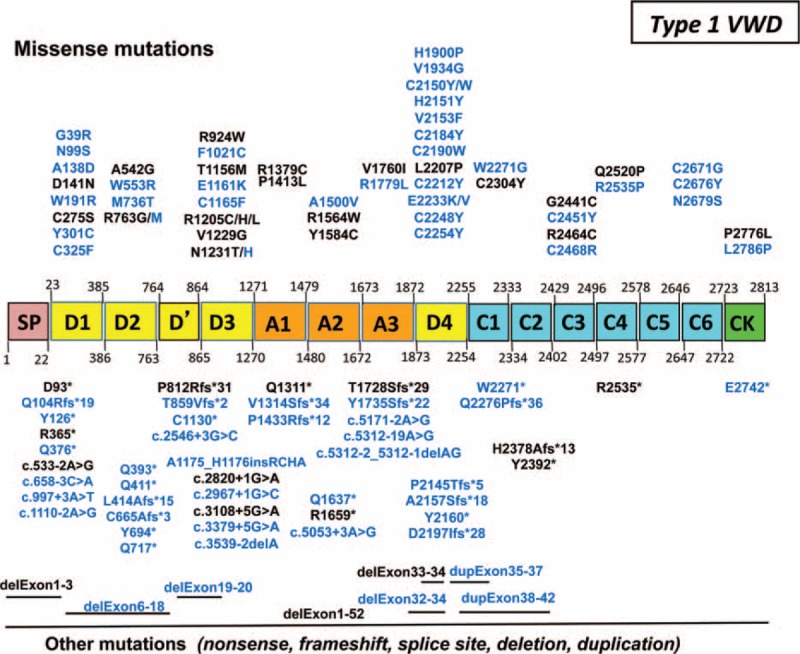
Sequences variations of VWF gene in 161 patients with type 1 von Willebrand disease (VWD). In our 161 patients with type 1 VWD, 105 distinct sequence variations spread all over VWF gene were identified. Novel mutations are indicated in blue. Missense mutations (51%) are presented on the top and truncating sequence variations (49%) are indicated on the bottom. In type 1 “IC” (clearance), the most frequent mutations were either the Vicenza mutation p.Arg1205His or others like p.Arg1205Cys, p.Arg1205Leu, p.Cys1165Phe, and dup.exon35–37. Six patients had mutations localized within the propeptide-cleavage site (p.Arg763Gly or p.Arg763Met). Interestingly, almost half of the truncating mutations found at the heterozygous state in our patients with type 1 VWD were also found in association with another mutation in our patients with type 3 VWD. IC = index cases, VWD = von Willebrand disease.
In type 3 VWD, 61 distinct sequence variations were identified, including two-third of novel mutations (Figure 5). These sequence variations were spread all over VWF gene with however, a hot spot on the N-terminal part of VWF (D domains); they consisted mainly in truncating mutations leading to silent alleles (82%) whereas missense mutations were rare (18%) (Figure 5).
FIGURE 5.
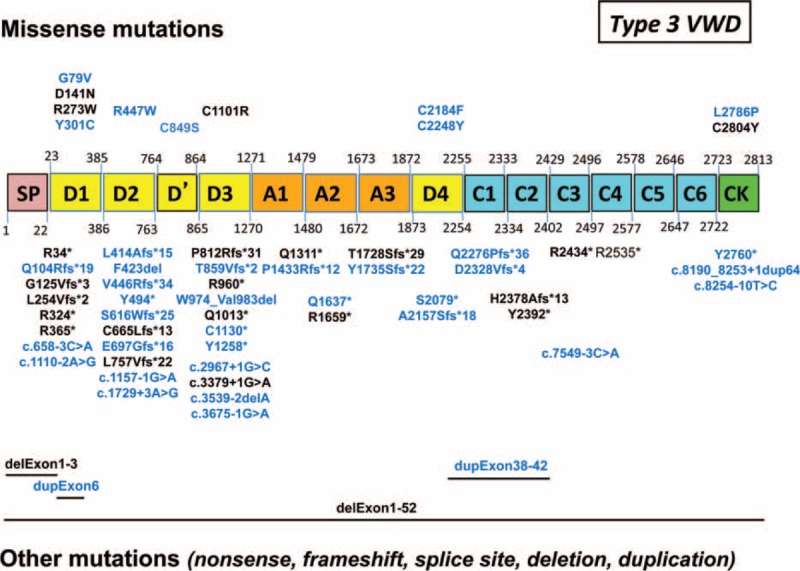
Sequences variations of VWF gene in 54 patients with type 3 von Willebrand disease (VWD). In our 54 patients with type 3 VWD, 61 distinct sequence variations were identified. Novel mutations are indicated in blue. They were spread all over VWF gene with a hot spot on the N-terminal part of VWF (D domains). Truncating mutations (82%) are indicated at the bottom of the figure; missense mutations (18%) are indicated at the top of the figure.VWD = von Willebrand disease, VWF = von Willebrand factor.
In type 2 VWD, a total of 118 distinct sequence variations were identified, including one-third of new mutations (Figure 6). These sequence variations were clustered in the A domains of VWF (types 2A, 2B, and 2M) and in the D′-D3 domains of VWF (type 2N); they consisted in missense mutations in a large majority of cases (95%). Interestingly, in type 2N, 22 truncating mutations leading to a silent allele were also found (type 2N/3 patients) including one-third also found in our type 3 VWD patients (Figures 5 and 6).
FIGURE 6.
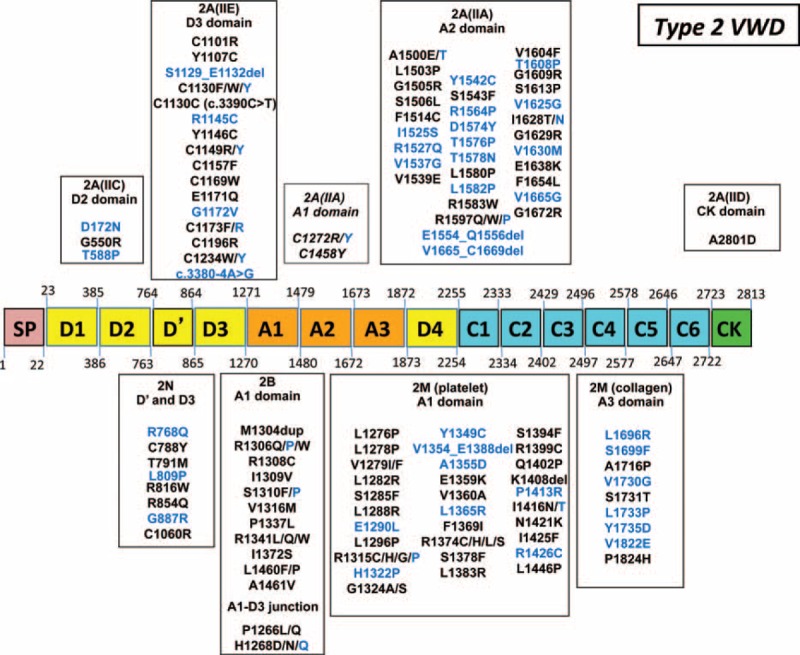
Mutations of VWF gene in 442 patients with type 2 von Willebrand disease (VWD). In our 442 patients with type 2 VWD, 118 distinct mutations were identified. Novel mutations are indicated in blue. In type 2A VWD (56 distinct mutations), most mutations (59%) were located in the A2 domain consisting in subtype 2A(IIA) leading to an increased VWF proteolysis (mainly mutations p.Arg1597Trp, p.Gly1609Arg, and p. Ile1626Thr); 30.5% of mutations were located in the D3 domain consisting in subtype 2A(IIE) (mainly mutations p.Cys1101Arg, p.Tyr1146Cys, and p.Cys1157Phe). Mutations located in either the D2 domain consisting in subtype 2A(IIC), or the A1 domain consisting in subtype 2A(IIA) leading to a decreased VWF secretion (mainly mutations p.Cys1272Arg, p.Cys1272Tyr, and p.Cys1458Tyr) or the CK domain (subtype 2A[IID]) were few (5%, 3.5%, and 2%, respectively). In type 2B VWD, 13 distinct mutations were described, most of them (85%) being located in the A1 domain (most frequently p.Arg1306Trp, p.Arg1308Cys, p.Pro1337Leu, and p.Arg1341Gln); some other mutations responsible for the specific type 2B “New York” (15%) were located in the D3-A1 junction (p.Pro1266Leu or p.Pro1266Gln). In type 2M VWD (40 distinct mutations), most mutations (78%) were located in the A1 domain (mainly p.Arg1315Cys and p.Arg1374Cys), whereas the other mutations (22%) were located within the A3 domain (p.Leu1696Arg was the most frequent mutation). In type 2N VWD, 8 distinct 2N mutations located in the D′-D3 domains were found (the p.Arg854Gln being the most frequent mutation as present in 90% of patients). VWD = von Willebrand disease, VWF = von Willebrand factor.
DISCUSSION
The present study was focused on the laboratory analysis of a prospective cohort of 1167 patients with VWD, selected on the basis of a biologic phenotype including type 3, type 2, and type 1 with VWF levels <30 IU/dL. Thanks to the support of the French NPRD from 2007 to 2012, we performed an exhaustive deciphering of both the biologic phenotype and genotype of these patients who constitute one of the largest VWD cohort enrolled and characterized at a national scale.11,33–38 The most recent and very interesting study dedicated to a cohort of VWD patients was published by Battle et al and it was mostly focused on a laboratory investigation involving next-generation sequencing.38 Almost 100% of our patients (99.3% exactly) carried 1 or several sequence variations of VWF gene, which is obviously linked to our inclusion criteria: 323 distinct mutations (including 58% of novel mutations) were identified. Also, the laboratory phenotype/genotype correlation performed individually in all our patients showed that, with our inclusion criteria, two-third of them exhibited a qualitative VWF deficiency (type 2 VWD, 66%) whereas one-third of them had a quantitative VWF deficiency, either partial (type 1 VWD, 25%) or total (type 3 VWD, 8%). The type of VWD remained undetermined in only 1% of our patients. Also, using a second level of laboratory phenotype/genotype analysis, mechanisms for VWF deficiency were further elucidated.
On an epidemiologic point of view, theoretically, the expected cohort of French patients matching our inclusion criteria should include ∼6000 people (1/10,000 prevalence; population in France: 60 million people). During the first 6 years of this study (2007–2012), we prospectively included 1856 eligible patients. Even if we are aware that reaching an exhaustive national enrollment is impossible, our data suggest that the 1/10,000 prevalence estimated for the most hemorrhagic forms of VWD 25 is likely to be close to reality. In addition, our results suggest that the respective proportions of quantitative and qualitative deficiencies of VWF are totally different as a function of the inclusion criteria used to enroll patients with VWF defects.10 Indeed, in our cohort selected on the basis of a “severe” biologic phenotype mostly associated with a high bleeding risk (i.e., excluding the mildest VWF quantitative defects with VWF levels ranging between 30 and 50 IU/dL), qualitative VWF defects (type 2) are, as it could be expected, the most frequent (66%). In contrast, when including all VWF defects (both the “severe” biologic phenotype and the mildest forms), partial quantitative VWF defects (type 1) are reported to be the most frequent (∼75%).5 For VWD type 3, the proportion of patients of our cohort (8%) is similar to those of the literature.11,33–38
In our cohort, the group of quantitative VWF deficiencies, that is, type 1 and type 3 VWD, showed interesting specific features and also, surprisingly, close genetic relationships. Our 54 IC patients with type 3 VWD exhibited 61 distinct mutations, spread all over VWF gene with a hotspot in the N-terminal part of VWF. Most of them (82%) were truncating mutations predicted to induce a silent allele. These data are in agreement with the ∼100 mutations reported in type 3 VWD in the international literature.11,34,39–43 Our 167 IC patients with type 1 VWD showed 111 distinct mutations, spread all over VWF gene with, however, hotspots within both the N-terminal and the C-terminal parts of VWF. These data are in agreement with the most important studies dedicated to type 1 VWD11,12,24,44–46 altogether including ∼300 patients with 85 distinct VWF gene mutations. Interestingly, in our study, half of the sequence variations found in type 1 VWD were truncating mutations which is more important than the 35% to 40% proportion of truncating mutations usually reported in the literature.11,44 In addition, surprisingly, almost half of the truncating mutations identified at a heterozygous state in our patients with type 1 VWD were also found, in association with another mutation, in our patients with type 3 VWD. This genetic overlap between type 1 and type 3 underlines that the penetrance of some type 3 mutations qualified as “recessive” may be variable. In other terms, the genetic basis of some cases of VWD type 1 may not rely on dominant mutations as classically described and these specific patients may be type 3 carriers with a biological type 1 phenotype.47 In our patients with type 1 VWD, missense mutations were present in 50% of cases, which is less than the 55% to 60% frequency usually reported in the literature.5 These missense mutations were mainly responsible for both either a defect of synthesis/secretion of VWF or an accelerated clearance of VWF. Interestingly, in terms of mechanisms for VWF quantitative deficiency in our patients with type 1 VWD, 53% of cases were related to a synthesis/secretion defect, 20% of cases to an accelerated clearance (mainly involving the classical “Vicenza” mutation), and 20% presented a type 3 carrier status. Rare mutations in the PP cleaving-site represented 3.5% of cases. The high frequency of the “clearance” type 1 VWD16 has important clinical consequences for patients in terms of response to desmopressin.24 Also, the important proportion of VWD patients with a type 1 phenotype who genetically have a status of type 3 carriers may have crucial implications on genetic counseling.39,47 Finally, no mutation could be found in 6 patients with type 1 VWD and a single mutation was found in 2 patients with type 3 VWD. We can speculate on several hypothesis to explain this absence of mutation. As the 6 patients with type 1 exhibited “borderline” VWF levels of 30 IU/dL, they may have no mutation in the VWF gene and their decreased VWF levels may be linked to sequence variations of other genetic systems.6 Another hypothesis valuable for both the latter type 1 and type 3 patients is the presence of intronic mutation(s) of VWF gene (potentially leading to a nil allele) undetectable by our gene sequencing methodology. Last but not least, the role of polymorphisms of VWF gene (which combination on the same allele may either be deleterious by themselves or emphasize the deleterious effect of a concomitant mutation) cannot be excluded in these specific cases.
Qualitative VWD molecular variants represented two-third of our cohort (442 IC patients). They exhibit interesting features in terms of both distribution of VWD types (2A, 2B, 2M, and 2N) and mechanisms of VWF deficiency inducing the miscellaneous VWD subtypes. Our current data are in agreement with the international literature dedicated to type 2 VWD48,49 and specifically, with the numerous studies led previously by the French Inserm Network on type 2 VWD, including 308 patients in which 99 distinct mutations of VWF gene were identified.50,51 In 100% of our current patients with type 2 VWD, a sequence variation of VWF gene was identified: 118 distinct mutations including a large majority of missense mutations distributed in various hotspots were identified, as usually described in the literature.48 The proportions of types 2A, 2B, 2M, and 2N were almost equivalent. Also, some specific points of our study may be underlined: (i) the important proportion of subtype 2A(IIE) (34.5%) within type 2A52, (ii) the presence of 15% of subtype “New York” within type 2B53, (iii) the clear identification of 2 mechanisms for type 2M including both subtype 2M “GPIb” (2M/2A like phenotype) and subtype 2M “collagen,”54 (iv) the important proportion of subtype 2N/3 (46%) within type 2N.55
In conclusion, the current study involving a large series of patients emphasizes the high level of matching between the genotype and the specialized phenotype in VWD.56 This data combined to medico-economic analysis may modify the future laboratory practice for VWD medical diagnosis by upgrading the rank of the genetic investigation when compared to highly specialized phenotypic assays.57 A very recent study published by the Spanish group referent for VWD emphasized this new paradigm for VWD laboratory diagnosis.38 Our study also provides a new epidemiologic picture of VWD: indeed, if the definition of VWD is revised and limited to the symptomatic forms of the disease overlapping type 3, type 2, type 1 with VWF levels <30 IU/dL, VWD will switch from the status of “most common inherited bleeding disorder” to a rare bleeding disorder. Considering this point of view, the 30 to 50 IU/dL range of VWF defects which do not match with a VWF gene abnormality should be renamed “a bleeding risk factor” and not a disease as previously suggested.6 Finally, this work opens the way to further studies dedicated to specific subgroups of our cohort, in order to better investigate the relationships between the bleeding score and both the phenotypic and genotypic profiles.
Supplementary Material
Acknowledgments
The authors are grateful to Sophie Capdenat and Sandrine Thouzeau-Benghezal (Hospital Lariboisière, Paris), Catherine Marichez and Sylvie Hermoire (Hospital Cardiologique, Lille), Hélène Tout-Mandard (Hospital de Bicêtre, Le Kremlin Bicêtre), Patricia Talarmain and Marie-Annick Gourlaouen (Hospital Hôtel Dieu, Nantes), Elise Vallée (Hospital de la Côte de Nacre, Caen) for expert assistance.
The authors also thank the collaborators of the French Reference Center for von Willebrand disease who enrolled some patients: Claire Barro, CHU de La Tronche; Sophie Bayart, CHU de Rennes; Eric Beltan, CHU de Pointe-à-Pitre, Guadeloupe; Elisabeth Benz-Lemoine, CHU de Poitiers; Claire Berger, CHU de Saint Etienne; Marie-Anne Bertrand, CHU de Besançon; Philippe Beurrier, CHU d’Angers; Christine Biron-Andreani, CHU de Montpellier; Florence Blanjouvan, CHU d’Annecy; Jeanne-Yvonne Borg, CHU de Rouen; Tewfik Boutekedjiret, CHU de Bicêtre; Julien Bovet, CHU de Dijon; Catherine Boyer-Neumann, CHU Antoine Béclère Clamart; Marie-Elisabeth Briquel, CHU de Nancy; Sabine Castet, CHU de Bordeaux; Hervé Chambost, CHU de Marseille; Axel Chaminade, CHU Saint Denis de la Réunion; Pierre Chamouni, CHU de Rouen; Erwan Choblet, CHU de Nouméa, Nouvelle Calédonie; Sophie Combe, CHU de Bicêtre; Yesim Dargaud, CHU de Lyon; Luc Darnige, CHU Georges Pompidou Paris; Dominique De Prost, CHU Louis Mourier Colombes; Céline Desconclois, CHU de Bicêtre; Antoine Deschamps, CHU Ambroise Paré Boulogne; Dominique Desprez, CHU de Strasbourg; Ségolène Donadel-Claeyssens, CHU de Toulouse; Valérie Eschwege, CHU de Nancy; Céline Falaise, CHU de Lille; Rémi Favier, CHU Trousseau Paris; Albert Faradji, CHU de Starsbourg; Mathieu Fiore, CHU de Bordeaux; Michaela Fontenay, CHU Cochin Paris; Marc Fouassier, CHU de Nantes; Birgit Frotscher, CHU de Nancy; Philippe Gautier, CHU de Caen; Valérie Gay, CHU de Chamberry; Moana Gelu, CHU de Pointe-à-Pitre Gaudeloupe; Stéphane Girault, CHU de Limoges; Vasiliki Gkalea, CHU Tenon Paris; Alban Godon, CHU d’Angers; Jean-Christophe Gris, CHU de Nîmes; Yves Gruel, CHU de Tours; Viviane Guérin, CHU de Bordeaux; Maryse Guicheteau, CHU de Poitiers; Benoit Guillet, CHU de Rennes; Annie Harroche, CHU Neceker Paris; Cécile Henrio, CHU de Caen; Nathalie Hezard, CHU de Reims; Marie-Hélène Horrelou, CHU Cochin Paris; Yoann Huguenin, CHU de Bordeaux; Marie-Geneviève Huisse, CHU Bichat Paris; Marie-Françoise Hurtaud, CHU Robert Debré Paris; Chloé James, CHU de Bordeaux; Lofti Lahjomri, CHU Raymond Poincarré Garches; Cécile Lavenu-Bombled, CHU de Bicêtre; Thierry Lambert, CHU de Bicêtre; Agnès Lequerrec, CHU de Caen; Anne Lienhart, CHU de Lyon; Alain Marques-Verdier, CHU de Clermont-Ferrand; Elisabeth Mazoyer, CHU Avicenne Bobigny; Isabelle Mazurier, CHU de Colmar; Roland Meley, CHU de Saint Etienne; Sandrine Meunier, CHU de Lyon; Claude Négrier, CHU de Lyon; Philippe Nguyen, CHU de Reims; Caroline Oudot, CHU de Limoges; Brigitte Pan-Petsch, CHU de Brest; Edith Peynaud, CHU Louis Mourier Colombes; Serge Pierre-Louis, CHU de la Réunion; Benoit Polack, CHU de Grenoble; Odile Pouille Lievin, CHU de Chartres; Claire Pouplard, CHU de Tours; Katia Pouymayou, CHU de Marseille; Valérie Proulle, CHU de Bicêtre; Anne Rafowicz, CHU de Bicêtre; Antoine Rauch, CHU de Lille; Yohann Repesse, CHU de Caen; Chantal Rothschild, CHU Necker Paris; Valérie Roussel-Robert, CHU Cochin Paris; Lucia Ruggeri, CHU de Lyon; Jean-François Schved, CHU de Montpellier; Pierre Sié, CHU de Toulouse; Marianne Sigaud, CHU de Nantes; Virginie Siguret, CHU Lariboisière Paris, Alain Stepanian, CHU Lariboisière Paris; Natalie Stieljes, CHU Cochin Paris; Brigitte Tardy, CHU de Saint Etienne; Marie-Françoise Thiercelin-Legrand, CHU de Toulouse; Catherine Trichet, CHU Beaujon Clichy; Jean-Baptiste Valentin, CHU de Tours; Marie-Christine Vergnes, CHU de Nimes; Sophie Voisin, CHU de Toulouse; Fabienne Volot, CHU de Dijon; Anne-Lise Voyer, CHU d’Amiens; Martine Wolf, CHU Antoine Béclère Clamart; France.
Footnotes
Abbreviations: aPTT = activated partial thromboplastin time, CRMW = Centre de Référence de la Maladie de Willebrand (French reference center for von Willebrand disease), FVIII = factor VIII, IC = index case, MLPA = multiplex ligation-dependent probe amplification, VWD = von Willebrand disease, VWF = von Willebrand factor, VWF:Ag = von Willebrand factor antigen, VWF:CB = von Willebrand factor collagen binding assay, VWF:pp = von Willebrand factor propeptide, VWF:RCo = von Willebrand factor ristocetin cofactor activity
Funding: this work was supported by the National Plan for Rare Diseases on behalf of the French Ministry of Health.
Authorship contribution: AV wrote the manuscript; AV, PB, EF, CT, CVD, and JG designed the study and analyzed the data; EF analyzed and selected the inclusion requests, specifically performed the phenotype/correlation analysis as well as the cross-sectional analysis of the database; PB, MG, CZ, and SB carried out most of the genotypic analysis and interpreted the results; CC carried out most of the phenotypical analysis and interpreted the results; EF, JG, MT, CT, MD, RD, NIB, and ABD enrolled a large majority of patients, collected clinical and laboratory information, and critically reviewed the manuscript.
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
REFERENCES
- 1.Sadler JE. Von Willebrand factor in its native environment. Blood 2013; 121:2583–2584. [DOI] [PubMed] [Google Scholar]
- 2.Zhou YF, Eng ET, Zhu J, et al. Sequence and structure relationships within von Willebrand factor. Blood 2012; 120:449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lenting PJ, Christophe OD, Denis CV. Von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood 2015; 125:2019–2028. [DOI] [PubMed] [Google Scholar]
- 4.Goodeve AC. The genetic basis of von Willebrand disease. Blood Rev 2010; 24:123–134. [DOI] [PubMed] [Google Scholar]
- 5.James PD, Goodeve AC. Von Willebrand disease. Genet Med 2011; 13:365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sadler JE. New concepts in von Willebrand disease. Annu Rev Med 2005; 56:173–191. [DOI] [PubMed] [Google Scholar]
- 7.Federici AB, Budde U, Castaman G, et al. Current diagnostic and therapeutic approaches to patients with acquired von Willebrand syndrome: a 2013 update. Semin Thromb Haemost 2013; 39:191–201. [DOI] [PubMed] [Google Scholar]
- 8.Laffan MA, Lester W, O’Donnell JS, et al. The diagnosis and management of von Willebrand disease: a United Kingdom Haemophilia Centre Doctors’ Organisation guideline approved by the British Committee for Standards in Haematology. Br J Haematol 2014; 167:453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadler JE, Budde U, Eikenboom JC, et al. Working Party on von Willebrand Disease Classification. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost 2006; 4:2103–2114. [DOI] [PubMed] [Google Scholar]
- 10.Sadler JE. Low von Willebrand factor: sometimes a risk factor and sometimes a disease. Hematology Am Soc Hematol Educ Program 2009; 106–112. [DOI] [PubMed] [Google Scholar]
- 11.Goodeve A, Eikenboom J, Castaman G, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European Study, Molecular and Clinical Markers for the Diagnosis and Management of type 1 von Willebrand disease (MCMDM-1VWD). Blood 2007; 109:112–121. [DOI] [PubMed] [Google Scholar]
- 12.James PD, Lillicrap D, Mannucci PM. Alloantibodies in von Willebrand disease. Blood 2013; 122:636–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tosetto A, Castaman G. How I treat type 2 variant forms of von Willebrand disease. Blood 2015; 125:907–914. [DOI] [PubMed] [Google Scholar]
- 14.Jacobi PM, Gill GC, Flood VH, et al. Intersection of mechanisms of type 2A VWD through defects in VWF multimerization, secretion, ADAMTS13 susceptibility, and regulated storage. Blood 2012; 119:4543–4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baronciani L, Federici AB, Castaman G, et al. Prevalence of type 2B “Malmö/New York” von Willebrand disease in Italy: the role of von Willebrand factor gene conversion. J Thromb Haemost 2008; 6:887–890. [DOI] [PubMed] [Google Scholar]
- 16.Gezsi A, Budde U, Deak I, et al. Accelerated clearance alone explains ultra-large multimers in von Willebrand disease Vicenza. J Thromb Haemost 2010; 8:1273–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ng C, Motto DG, Di Paola J. Diagnostic approach to von Willebrand disease. Blood 2015; 125:2029–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lillicrap D. Von Willebrand disease: advances in pathogenic understanding, diagnosis, and therapy. Hematology Am Soc Hematol Educ Program 2013; 2013:254–260. [DOI] [PubMed] [Google Scholar]
- 19.Eikenboom J, Federici AB, Dirven RJ, et al. MCMDM-1VWD Study Group. VWF propeptide and ratios between VWF, VWF propeptide, and the FVIII in the characterization of type 1 von Willebrand disease. Blood 2013; 121:2336–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flood VH, Lederman CA, Wren JS, et al. Absent collagen binding in a VWF A3 domain mutant: utility of the VWF:CB in diagnosis of VWD. J Thromb Haemost 2010; 8:1431–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Brasi C, El-Maari O, Perry DJ, et al. Genetic testing in bleeding disorders. Haemophilia 2014; 20 (Suppl. 4):54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keeney S, Bowen D, Cumming A, et al. UK Haemophilia Centre Doctors’ Organisation (UKHCDO). The molecular analysis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors’ Organisation Haemophilia Genetics Laboratory Network. Haemophilia 2008; 14:1099–1111. [DOI] [PubMed] [Google Scholar]
- 23.Othman M, Emsley J. Platelet-type von Willebrand disease: toward an improved understanding of the “sticky situation”. Semin Thromb Hemost 2014; 40:146–150. [DOI] [PubMed] [Google Scholar]
- 24.Castaman G, Lethagen S, Federici AB, et al. Response to desmopressin is influenced by the genotype and the phenotype in type 1 von Willebrand disease (VWD): results from the European Study MCMDM-1 VWD. Blood 2008; 111:3531–3539. [DOI] [PubMed] [Google Scholar]
- 25.Rodeghiero F, Castaman G, Dini E. Epidemiological investigation of the prevalence of von Willebrand's disease. Blood 1987; 69:454–459. [PubMed] [Google Scholar]
- 26.Werner EJ, Broxson EH, Tucker EL, et al. Prevalence of von Willebrand disease in children: a multiethnic study. J Pediatr 1993; 123:893–898. [DOI] [PubMed] [Google Scholar]
- 27.Fressinaud E, Veyradier A, Truchaud F, et al. Screening for von Willebrand disease with a new analyzer using high shear stress: a study of 60 cases. Blood 1998; 91:1325–1331. [PubMed] [Google Scholar]
- 28.Budde U, Pieconka A, Will K, et al. Laboratory testing for von Willebrand disease: contribution of multimer analysis to diagnosis and classification. Semin Thromb Hemost 2006; 32:514–521. [DOI] [PubMed] [Google Scholar]
- 29.Caron C, Hilbert L, Vanhoorelbeke K, et al. Measurement of von Willebrand factor binding to a recombinant fragment of glycoprotein Ibalpha in an enzyme-linked immunosorbent assay-based method: performance in patients with type 2B von Willebrand disease. Br J Haematol 2006; 133:655–663. [DOI] [PubMed] [Google Scholar]
- 30.Favaloro EJ. Collagen binding assay for von Willebrand factor (VWF: CBA): detection of von Willebrand disease (VWD) and discrimination of VWD subtypes, depends on collagen source. Thromb Haemost 2000; 83:127–135. [PubMed] [Google Scholar]
- 31.Caron C, Mazurier C, Goudemand J. Large experience with a factor VIII binding assay of plasma von Willebrand factor using commercial reagents. Br J Haematol 2002; 117:716–718. [DOI] [PubMed] [Google Scholar]
- 32.Haberichter SL, Castaman G, Budde U, et al. Identification of type 1 von Willebrand disease with reduced von Willebrand factor survival by assay of the VWF propeptide in the European study: molecular and clinical markers for the diagnosis and management of type 1 VWD (MCMDM-1VWD). Blood 2008; 111:4979–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Federici AB, Bucciarelli P, Castaman G, et al. The bleeding score predicts clinical outcomes and replacement therapy in adults with von Willebrand disease. Blood 2014; 123:4037–4044. [DOI] [PubMed] [Google Scholar]
- 34.James PD, Lillicrap D. The diagnosis and management of von Willebrand disease in Canada. Semin Thromb Haemost 2011; 37:522–527. [DOI] [PubMed] [Google Scholar]
- 35.Flood VH, Gill JC, Bellissimo DB, et al. Von Willebrand disease in the United States: a perspective from Wisconsin. Semin Thromb Haemost 2011; 37:528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yadegari H, Driesen J, Pavlova A, et al. Mutation distribution in the von Willebrand factor gene related to the different von Willebrand disease (VWD) types in a cohort of VWD patients. Thromb Haemost 2012; 108:662–671. [DOI] [PubMed] [Google Scholar]
- 37.Sanders YV, Groeneveld D, Meijer K, et al. WIN study group. Von Willebrand factor propeptide and the phenotypic classification of von Willebrand disease. Blood 2015; 125:3006–3013. [DOI] [PubMed] [Google Scholar]
- 38.Battle J, Perez-Rodriguez A, Corrales I, et al. Molecular and clinical profile of von Willebrand disease in Spain (PCM-EVW-ES): proposal for a new diagnostic paradigm. Thromb Haemost 2015; 114 (6.): Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 39.Bowman M, Tuttle A, Notley C, et al. Association of Hemophilia Clinic Directors of Canada. The genetics of Canadian type 3 von Willebrand disease: further evidence for co-dominant of mutant alleles. J Thromb Haemost 2013; 11:512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sutherland MS, Keeney S, Bolton-Maggs PH, et al. The mutation spectrum associated with type 3 von Willebrand disease in a cohort of patients from the north west of England. Haemophilia 2009; 15:1048–1057. [DOI] [PubMed] [Google Scholar]
- 41.Solimando M, Baronciani L, La Marca S, et al. Molecular characterization, recombinant protein expression, and mRNA analysis of type 3 von Willebrand disease: studies of an Italian cohort of 10 patients. Am J Hematol 2012; 87:870–874. [DOI] [PubMed] [Google Scholar]
- 42.Shahbazi S, Mahdian R, Ala FA, et al. Molecular characterization of Iranian patients with type 3 von Willebrand disease. Haemophilia 2009; 15:1058–1064. [DOI] [PubMed] [Google Scholar]
- 43.Kasatkar P, Shetty S, Ghosh K. Genetic heterogeneity in a large cohort of Indian type 3 von Willebrand disease patients. PLoS One 2014; 9:e92575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.2007; James PD, Notley C, Hegadom C, et al. The mutational spectrum of type 1 von Willebrand disease: results from a Canadian cohort study Blood. 109:145–154. [DOI] [PubMed] [Google Scholar]
- 45.Eikenboom J, Hilbert L, Ribba AS, et al. Expression of 14 von Willebrand factor mutations identified in patients with type 1 von Willebrand disease from the MCMDM-1VWD study. J Thromb Haemost 2009; 7:1304–1312. [DOI] [PubMed] [Google Scholar]
- 46.Peake I, Goodeve A. Type 1 von Willebrand disease. J Thromb Haemost 2007; 5 (Suppl. 1):7–11. [DOI] [PubMed] [Google Scholar]
- 47.Sutherland MS, Cumming AM, Bowman M, et al. A novel deletion mutation is recurrent in von Willebrand disease types 1 and 3. Blood 2009; 114:1091–1098. [DOI] [PubMed] [Google Scholar]
- 48.Goodeve AC. VWF sequence variants: a data goldmine. Blood 2013; 122:471–473. [DOI] [PubMed] [Google Scholar]
- 49.Ahmad F, Jan R, Kannan M, et al. Characterisation of mutations and molecular studies of type 2 von Willebrand disease. Thromb Haemost 2013; 109:39–46. [DOI] [PubMed] [Google Scholar]
- 50.Meyer D, Fressinaud E, Gaucher C, et al. Gene defects in 150 unrelated French cases with type 2 von Willebrand disease: from the patient to the gene. INSERM network on molecular abnormalities in von Willebrand disease. Thromb Haemost 1997; 78:451–456. [PubMed] [Google Scholar]
- 51.Wiley-Blackwell, Meyer D, Fressinaud E, Mazurier C. Federici AB, Lee CA, Berntop E, Lillicrap D, Montgomery RR. Clinical laboratory and molecular markers of type 2 von Willebrand disease. Von Willebrand disease: basic and clinical aspects 2011; 137–147. [Google Scholar]
- 52.Schneppenheim R, Michiels JJ, Obser T, et al. A cluster of mutations in the D3 domain of von Willebrand factor correlates with a distinct subgroup of von Willebrand disease: type 2A/IIE. Blood 2010; 115:4894–4901. [DOI] [PubMed] [Google Scholar]
- 53.Nurden P, Gobbi G, Nurden A, et al. Abnormal VWF modifies megakaryocytopoiesis: studies of platelets and megakaryocyte cultures from patients with von Willebrand disease type 2B. Blood 2010; 115:2649–2656. [DOI] [PubMed] [Google Scholar]
- 54.Castaman G, Federici AB, Tosetto A, et al. Different bleeding risk in type 2A and 2M von Willebrand disease: a 2-year prospective study in 107 patients. J Thromb Haemost 2012; 10:632–638. [DOI] [PubMed] [Google Scholar]
- 55.Mazurier C, Goudemand J, Hilbert L, et al. Type 2N von Willebrand disease: clinical manifestations, pathophysiology, laboratory diagnosis and molecular biology. Best Pract Res Clin Haematol 2001; 14:337–347. [DOI] [PubMed] [Google Scholar]
- 56.Hampshire DJ, Goodeve AC. The molecular basis of von Willebrand disease: the under investigated, the unexpected and the overlooked. Haematologica 2011; 96:798–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Flood VH. New insights into genotype and phenotype of VWD. Hematology Am Soc Hematol Educ Program 2014; 2014:531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.