

Abstract
Dystrophin is expressed in differentiated myofibers where it is required for sarcolemmal integrity, and loss-of-function mutations in its gene result in Duchenne Muscular Dystrophy (DMD), a disease characterized by progressive and severe skeletal muscle degeneration. Here we found that dystrophin is also highly expressed in activated muscle stem cells (also known as satellite cells) where it associates with the Ser/Thr kinase Mark2 (also known as Par1b), an important regulator of cell polarity. In the absence of dystrophin, expression of Mark2 protein is downregulated, resulting in the inability to polarize Pard3 to the opposite side of the cell. Consequently, the number of asymmetric divisions is strikingly reduced in dystrophin-deficient satellite cells, while also displaying a loss of polarity, abnormal division patterns including centrosome amplification, impaired mitotic spindle orientation, and prolonged cell divisions. Altogether, these intrinsic defects strongly reduce the generation of myogenic progenitors needed for proper muscle regeneration. Therefore, we conclude that dystrophin has an essential role in the regulation of satellite cell polarity and asymmetric division. Our findings indicate that muscle wasting in DMD is not only caused by myofiber fragility, but is also exacerbated by impaired regeneration due to intrinsic satellite cell dysfunction.
Keywords: Duchenne Muscular Dystrophy, dystrophin, Skeletal muscle, Satellite cell, Stem cell, Asymmetric cell division, Polarity, PAR-complex, Mark2, Mitotic defect
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a devastating genetic muscular disorder of childhood manifested by progressive debilitating muscle weakness and wasting, and ultimately death in the second or third decade of life1. DMD is characterized by the absence of dystrophin, a 3,500 aa rod-shaped protein expressed in differentiated myofibers, that connects the myofiber cytoskeleton to the extracellular matrix through the dystrophin-associated glycoprotein complex (DGC)2,3. In the absence of dystrophin, myofibers are extremely susceptible to injury, which leads to multiple cycles of degeneration and regeneration, that in turn lead to elevated inflammation, fibrosis and eventual progressive loss of muscle mass and function4.
Previous studies have shown that impairment of the DGC by specific deletion of Dystroglycan (Dag1) in myofibers does not mimic the robust muscle degeneration observed in mdx mice (dystrophin-null mice), suggesting that myofiber fragility is not the only mechanism involved in muscle degeneration in DMD patients5. It has been suggested that human DMD progression is exacerbated by reduced function of muscle stem cells due to exhaustion caused by telomere shortening6,7. However, in human and mouse dystrophic skeletal muscles, satellite cell numbers are elevated, even in advanced stages of dystrophy, suggesting that the depletion of satellite cells is not the primary cause for failed regeneration8–10. Importantly, the proportion of myogenin-expressing (Myog) progenitors entering the differentiation program is unusually low in DMD muscle8. Together, these data suggest the hypothesis that the homeostasis between stem cells and committed progenitors within the satellite cell compartment is perturbed in dystrophin-deficient muscle.
A recent study has indicated that the polarity protein MAP/Microtubule affinity-regulating kinase 2 (Mark2, also known as Partitioning-defective 1b; Par1b) binds to the R8–R9 spectrin-repeat domain of dystrophin in differentiated myofibers11. Mark2 has also been shown to be required for the basolateral formation of a functional DGC in epithelial cells12. Importantly, Par1 (homolog of Mark2 in Drosophila) plays an essential role in asymmetric stem cell divisions to regulate self-renewal of the stem cell reserve by establishing cellular polarity through direct phosphorylation of Par3 (or Pard3 in mammals) causing asymmetric distribution of the PAR complex13. For example, in dividing neuroblasts in Drosophila, Par1 is localized on the basal cell surface that will give rise to differentiated ganglion mother cell, while Par3/Par6 proteins accumulate in the apical cell surface and prevents differentiation by inactivating Numb so that it no longer inhibits Notch14. On the other hand, in intestinal stem cells, integrin bound to the basement membrane segregates the Par3/Par6/aPKC complex to the apical daughter cell that will become the differentiated cell15. Consistent with the hypothesis that the PAR complex regulates satellite cell polarity, Pard3 knockdown in satellite cells results in loss of asymmetric divisions and reduced capacity to form myogenic progenitors16.
Here, we demonstrate that dystrophin is expressed in activated satellite cells where it regulates polarity establishment by interacting with Mark2. Dystrophin-deficient satellite cells show impaired polarity establishment, loss of apicobasal asymmetric division, and higher proportion of abnormal division leading to reduced generation of myogenic progenitors and impaired muscle regeneration.
RESULTS
Dystrophin is expressed in satellite cells
Dystrophin is not expressed in myoblasts cultured in vitro, and consequently was generally considered not expressed in satellite cells17,18. However, our RNA-seq and microarray analysis of prospectively isolated satellite cells reveals that DGC-encoding genes such as Dystrophin (Dmd) and Dag1, which are transcribed at low levels in myoblasts and upregulated in differentiated myotubes, are highly transcribed in satellite cells (Fig. 1a and Supplementary Fig. 1a,b)19. Clustering analysis of expression changes in DGC-encoding genes also revealed that distinct complex components, such as ɛ-sarcoglycan (Sgce), β2-syntrophin (Stnb2), and β-dystrobrevin (Dtnb) are exclusively expressed in satellite cells and not myofibers (Fig. 1a and Supplementary Fig. 1c). Validation by qPCR confirmed that Dmd and Dag1 mRNA levels are elevated by 475% and 250%, respectively, in prospectively isolated satellite cells compared to the level found in in vitro differentiated myotubes (Fig. 1b,c and Supplementary Fig. 1d).
Figure 1.
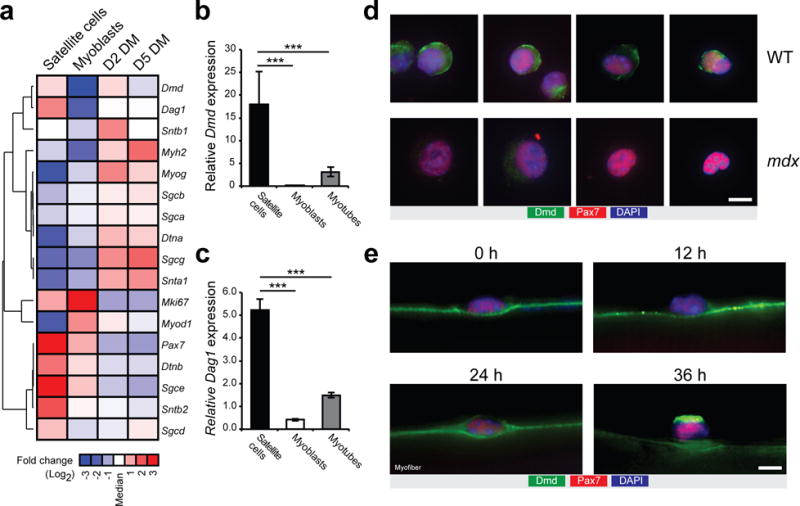
Dystrophin expression in satellite cells. (a) Microarray heatmap representing genes from the DGC from prospectively isolated satellite cells, proliferating myoblasts cultured in vitro, and 2- and 5-day-differentiated myotubes. Signal intensities represent the average of n = 3 microarrays for myoblasts and myotubes, and n = 1 microarray for satellite cells obtained from pooled freshly isolated satellite cells of nine mice. (b,c) Quantitative Real-time PCR for Dmd and Dag1 expression in satellite cells, myoblasts and myotubes. Error bars represent means ± SEM. ***P < 0.005. Statistical significance was calculated by Student’s t test. (d) Representative pictures (n > 20 pictures per condition) of immunostaining for Pax7 (red), Dmd N-terminal (green) and DAPI (blue) of satellite cells isolated by FACS from cardiotoxin-injured WT and mdx mice 2 days post-injury. n = 3 mice. (e) Representative pictures (n > 50 pictures per condition) of immunostaining for Pax7 (red), Dmd C-terminal (green) and DAPI (blue) of satellite cells from cultured myofiber at 0, 12, 24, or 36 h. n = 3 mice. Scale bars, 5 μm.
In sections from normal muscle, dystrophin protein expression in satellite cell is not easily discernable from dystrophin expression of the myofiber due to their close juxtaposition. Therefore, we isolated satellite cells by FACS from cardiotoxin-injured Pax7-zsGreen reporter mice, and we cytospun and immunostained the sorted satellite cells. We observed dystrophin protein expression in satellite cells from wild type (WT) but not mdx mice (Fig. 1d). To examine the dystrophin expression pattern during satellite cell activation, we isolated myofibers from extensor digitorum longus (EDL) muscle and cultured them ex vivo for 0, 12, 24, and 36 h. We found that high level of dystrophin protein is expressed 24 h after satellite cell activation and is polarized on one side of the cell by 36 h (Fig. 1e). Immunostaining of myofibers cultured for 72 h revealed expression of dystrophin with both N-terminal and C-terminal antibodies in a subset of WT satellite cells, whereas a small subset of mdx satellite cells were stained with the C-terminal antibody (only observed at the 72 h time point) (Supplementary Fig. 1e).
Dystrophin regulates generation of myogenic progenitors
We next examined the developmental program of WT versus dystrophin-deficient satellite cells following activation in myofiber cultures (Fig. 2 and Supplementary Fig. 2). We observed that the number of Pax7-expressing satellite cells per myofiber was 175% higher in freshly isolated myofibers (time 0) from mdx mice relative to WT mice (Fig. 2a). However, after 72 h of culture the number of satellite cells in myofibers from WT mice increased by about 3.4-fold, while the number of satellite cells only increased by 1.4-fold in myofibers from mdx mice (Fig. 2a).
Figure 2.
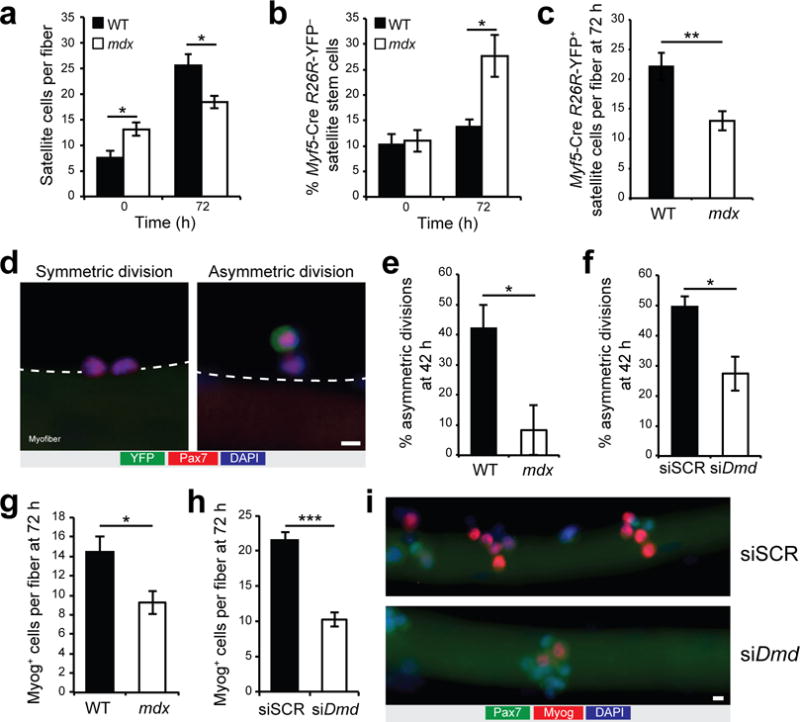
Impaired satellite stem cell asymmetric divisions and reduced generation of myogenic progenitors in absence of dystrophin. (a) Quantification of Pax7-expressing satellite cells per myofiber cultured for 0 or 72 h from WT and mdx Myf5-Cre:R26R-YFP mice. (b) Proportion of Pax7-positive YFP-negative satellite stem cells on myofibers cultured for 0 h or 72 h. (c) Quantification of Pax7-positive YFP-positive committed satellite cells per fiber. (d) Representative micrographs (n > 20 micrographs) of a planar symmetric stem cell division (left) and an apicobasal asymmetric division (right). (d–f) Myofibers from Myf5-Cre:R26R-YFP mice immunostained for YFP (green), Pax7 (red), and DAPI (blue) after 42 h of culture. (e,f) Quantification of asymmetric divisions relative to total satellite stem cell divisions in (e) WT and mdx myofibers and (f) WT myofibers knockdown of dystrophin (siDmd) or scramble control (siSCR). (g,h) Myog-expressing cells per myofiber cultured for 72 h from (g) WT and mdx mice and (h) WT myofibers treated with siDmd or siSCR. Error bars represent means ± SEM. n = 4 mice for every panel except for f (n = 5) and h (n = 3); 30–50 myofibers per mice. *P < 0.05, **P < 0.01, ***P < 0.005. Statistical significance was calculated by Student’s t test or Wilcoxon Rank-sum test for e. (i) Representative micrographs (n = 10 micrographs per condition) of cultured myofibers from WT mice at 72 h and treated with siSCR or siDmd and stained for Pax7 (green), Myog (red), and DAPI (blue), n = 3 mice. Scale bar, 5 μm.
Studies from different laboratories demonstrate that satellite cells are heterogeneous with a subpopulation of satellite cells that can self-renew and another subpopulation that is more prone to differentiate10,20–22. Using Myf5-Cre and R26R-YFP alleles, we previously observed that about 10% of satellite cells, which have never expressed Myf5-Cre (Pax7-positive YFP-negative satellite stem cells), are able to self-renew and extensively contribute to the satellite cell pool after transplantation into host skeletal muscle20. By contrast, satellite cells that have expressed Myf5-Cre during development (Pax7-positive YFP-positive committed satellite cells) are committed to undergo differentiation and do not efficiently contribute to the satellite cell pool following transplantation20. The proportion of YFP-negative satellite stem cells was about 10% in freshly isolated myofibers (time 0) from both WT and mdx mice (Fig. 2b). While this proportion remained relatively stable after 72 h of culture in myofibers from WT mice, it increased by 2.5-fold in myofibers from mdx mice (Fig. 2b). Accordingly, the generation of committed YFP-expressing satellite cells per fiber was substantially lower in myofibers isolated from mdx mice compared to myofibers from WT mice after 72 h of culture (Fig. 2c).
Changes in the proportions of YFP-negative satellite stem cells could be explained by their ability to undergo apicobasal asymmetric cell division to generate one YFP-negative and one YFP-expressing daughter cell (Fig. 2d)20. Therefore, we examined satellite stem cells immediately after the first round of cell division on isolated mdx myofibers cultured for 42 h and observed a reduction of 80% in the proportion of asymmetric satellite stem cell divisions compared to WT myofibers cultured for 42h (Fig. 2e and Supplementary Fig. 2a). We also observed an unusually low proportion of asymmetric cell divisions in 2-week-old mdx mice, prior to the major regenerative and inflammatory phase that occurs in mdx mice at 4–5 weeks of age (Supplementary Fig. 2b,c). By performing knockdown experiments with siRNA targeting Dmd (siDmd) or scramble control (siSCR) on myofibers from WT mice, we confirmed that the low proportion of asymmetric division is directly a consequence of the loss of dystrophin expression (Fig. 2f and Supplementary Fig. 2d–f). Consistent with this hypothesis, we observed a reduced proportion of asymmetric divisions when the dystrophin binding transmembrane protein Dag1 was deleted specifically in satellite cells using mice carrying floxed alleles of Dag1 and the satellite cell-specific Pax7-CreER driver of Cre recombinase (Pax7CreER:Dag1fl/fl) compared to Dag1fl/fl mice without Cre recombinase activity (Supplementary Fig. 2g,h)5,20,23. Notably, we did not observe any change in the proportion of asymmetric divisions using myofibers from adult α-sarcoglycan-deficient mice (Sgca−/−), in which DGC assembly is not dysregulated, compared to littermate control mice (Sgca+/−) (Supplementary Fig. 2i).
To examine the consequences of reduced asymmetric division on the generation of myogenic progenitors, we cultured myofibers for 72 h and enumerated the number of differentiating cells based on immunostaining for Myog. Notably, we observed a reduction of 36% in the number of Myog-expressing cells in myofibers isolated from mdx mice compared to WT mice (Fig. 2g). Similarly, we observed a reduction of 52% in the number of Myog-expressing cells in myofibers isolated from WT mice treated with siDmd compared to siSCR (Fig. 2h,i).
Dystrophin regulates polarity by interacting with Mark2
Using cultured myofibers, we detected that dystrophin expression is polarized (that is, asymmetrically distributed on one side of the cell) in activated satellite cells that are about to divide after 36 h of culture (Fig. 3a,b and Supplementary movie 1). We performed an in situ proximity ligation assay (PLA) on isolated myofibers to investigate whether Mark2 associates with dystrophin in satellite cells as in myofibers24 (Fig. 3c–f and Supplementary Fig. 3). PLA detection of endogenous Mark2 and dystrophin generated a signal in activated satellite cells on cultured EDL myofibers and on prospectively isolated satellite cells from WT mice, whereas the signal was absent in satellite cells from mdx mice (Fig. 3c and Supplementary Fig. 3e,f)11. We also found Mark2 interacted with Dag1, however no signal was visible for Pard3 with dystrophin or Dag1 (Fig. 3d–f and Supplementary Fig. 3c,d)12. Notably, PLA also revealed that dystrophin and Dag1 interact with α7-integrin (Itga7), another laminin-binding receptor that plays an important role in polarity, although this interaction is temporally restricted and not exclusive (Fig. 3a and Supplementary Fig. 3a,b)15.
Figure 3.
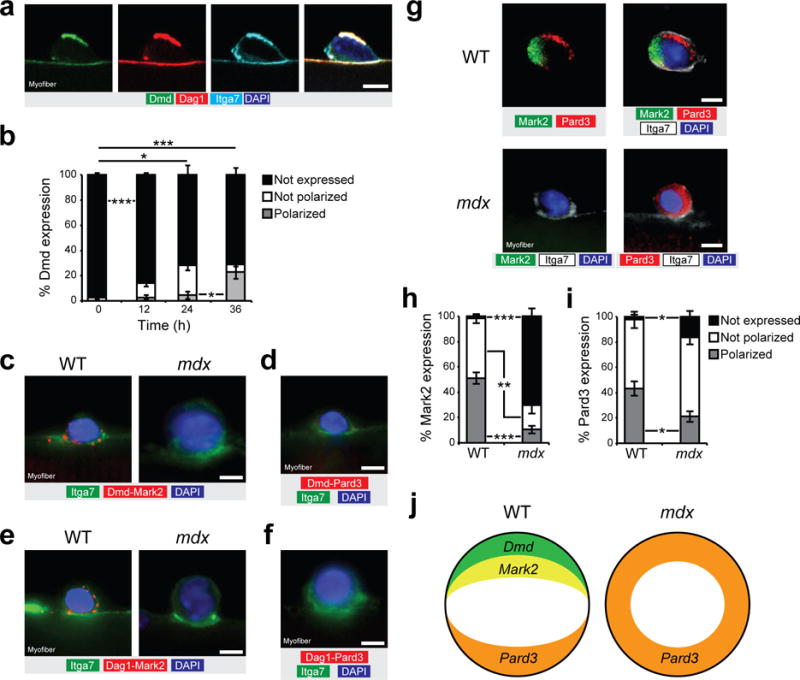
Dystrophin regulates PAR polarity protein localization. (a) Immunostaining for Dmd C-terminal (green), Dag1 (red), itga7 (cyan) and DAPI (blue) of cultured myofiber at 36 h. (b) Quantification of Dmd expression (rod domain) and localization in satellite cells of cultured myofibers of WT mice at 0, 12, 24, and 36 h. Only undivided cells were quantified. n = 3 mice, approx. 50 cells per mice. (c) Representative micrograph (n = 10 micrographs per condition) of proximity ligation assay (PLA) for Dmd (Dy4/6D3 clone) and Mark2 (red) and for (d) Dmd and Pard3 (red), along with immunostaining for itga7 (green) and DAPI (blue) on cultured myofibers from WT and mdx mice at 36 h. n = 3 mice. (e) Representative micrograph (n = 10 micrographs per condition) of proximity ligation assay (PLA) between Dag1 and Mark2 (red), and between (f) Dag1 and Pard3 (red), along with immunostaining for itga7 (green) and DAPI (blue) on cultured myofibers from WT and mdx mice at 36 h. n = 3 mice. (g) Example (n = 10 micrographs per condition) of polarity protein distribution from immunostaining for Mark2 (green), Pard3 (red), itga7 (white) and DAPI (blue) of cultured myofiber from WT and mdx mice at 36 h. n = 3 mice. (h,i) Quantification for (h) Mark2 and (i) Pard3 expression and localization in satellite cells of cultured myofibers of WT and mdx mice at 36 h. n = 4 mice, 30–50 cells per mice. (j) Schematic representation of the polarity establishment in WT and mdx satellite cells. Error bars represent means ± SEM. P* < 0.05, **P < 0.01, ***P < 0.005. Statistical significance was calculated by Student’s t test. Scale bars, 5 μm.
To further interrogate whether the PAR proteins are involved in satellite cell polarity, we analyzed Mark2 and Pard3 expression patterns by immunostaining of satellite cells on myofibers cultured for 36 h prior to the first cell division. We observed polarized localization of Mark2 or Pard3 in about half the satellite cells from WT mice (Fig. 3g–j). By contrast, we observed that the majority of dystrophin-deficient satellite cells expressed low levels of Mark2, while Pard3 was localized around the cell periphery in a non-polarized manner (Fig. 3g–i). These findings are diagrammatically summarized in Fig. 3j.
PAR proteins regulate satellite cell asymmetric division
We isolated EDL myofibers from Myf5-Cre:R26R-YFP mice and cultured them for 42 h (after the first cell division) to measure the asymmetric inheritance of the different polarity effectors in relation to the myogenic fate of the daughter cells. We observed that expression of dystrophin and Mark2 was retained in YFP-negative satellite stem cells after asymmetric division, but was rarely detectable in YFP-positive daughter cells (Fig. 4a and Supplementary Table 1). By contrast, we observed that Pard3 was segregated to the YFP-positive satellite cells and was rarely detectable in YFP-negative daughter cells (Fig. 4a and Supplementary Table 1). Contrary to what we observed in asymmetric cell pairs, dystrophin, Mark2, and Pard3 were equally distributed in both YFP-negative daughter cells after symmetric division (Fig. 4b and Supplementary Table 1). Asymmetric segregation of dystrophin was also observed at 72 h of culture after satellite cells underwent multiple rounds of division, where staining for dystrophin was only observed in a small subset of satellite cells (Supplementary Figs. 1e and 4a).
Figure 4.
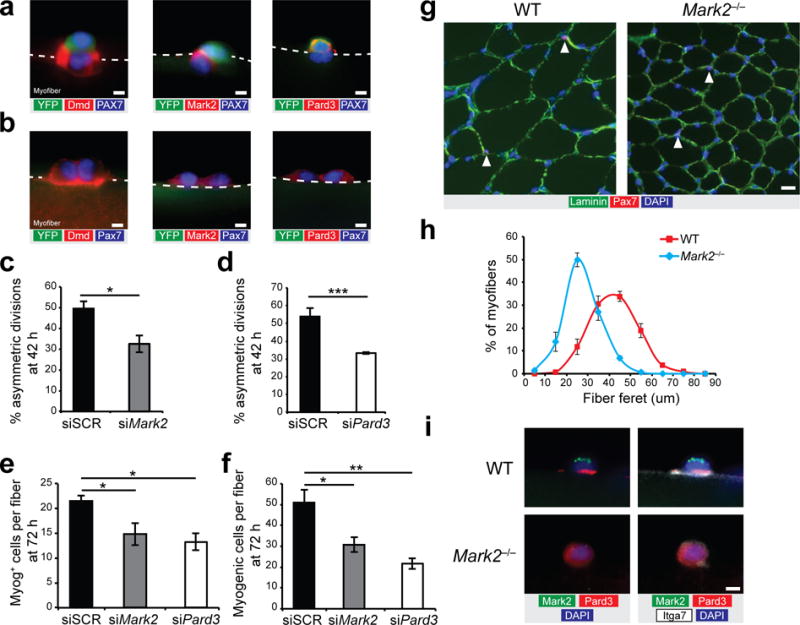
PAR polarity proteins are required for muscle stem cell asymmetric divisions. (a,b) Myofibers from Myf5-Cre:R26R-YFP mice cultured for 42 h and immunostained for YFP (green), Pax7 (blue) and Dmd (Dy4/6D3, red; left panel), or Mark2 (red; middle panel), or Pard3 (red; right panel) in (a) asymmetric cell pairs (YFP-positive/YFP-negative pairs) and (b) symmetric YFP-negative cell pairs (YFP-negative/YFP-negative pairs). Representative pictures from n = 10–20 pictures per condition. (c,d) Quantification of asymmetric divisions relative to total satellite stem cell divisions in cultured myofibers of Myf5-Cre:R26R-YFP mice following knockdown of (c) Mark2 (siMark2) or (d) Pard3 (siPard3) compared to scramble siRNA (siSCR). (e) Quantification of Myog-expressing cells per fiber and (f) total myogenic cells (Pax7-expressing or Myog-expressing cells) per fiber from WT myofibers cultured 72 h treated with siMark2, siPard3, or siSCR. (c–f) n = 3 mice except for c where n = 5 and d where n = 4, 30–40 myofibers per mice. (g) Representative pictures (n > 10 pictures) from WT and Mark2−/− section from TA muscle immunostained for laminin (green), Pax7 (red), and DAPI (blue). n = 3 mice. (h) Distribution of minimal fiber feret size from the TA muscle of Mark2−/− mice (blue) and WT littermate (red). n = 3 mice, >300 fibers counted per mouse. (i) Representative pictures (n = 5-10 pictures per condition) of immunostaining for Mark2 (green), Pard3 (red), itga7 (white), and DAPI (blue) of cultured myofiber from WT and Mark2−/− mice at 36 h. n = 3 mice. Error bars represent means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.005. Statistical significance was calculated by Student’s t test. (a,b,i) Scale bars, 5 μm and (g)10 μm.
To investigate the specific function of PAR proteins on satellite cell fate decision, we performed siRNA knockdowns for Mark2 (siMark2) or Pard3 (siPard3) on myofibers isolated from Myf5-Cre:R26R-YFP mice and cultured them for 42 h (Supplementary Fig. 4b–e). We found that siMark2 and siPard3 both resulted in a reduction of approximately 35% in the proportion of asymmetric divisions compared to siSCR-treated myofibers cultured for 42 h (Fig. 4c,d and Supplementary Fig. 4f). Moreover, we observed that siMark2 and siPard3 treatment on myofibers cultured for 72 h resulted in a reduction of 30% and 38% in the number of Myog-expressing cells, and approximately 50% in the number of total myogenic cells (Pax7-expressing or Myog-expressing cells) compared to siSCR-treated myofibers cultured for 72 h (Fig. 4e,f). Double knockdown with siMark2 and siDmd did not exacerbate the reduction in the number of Myog-expressing cells compared to siMark2 alone (Supplementary Fig. 4g,h).
To further analyze Mark2 function, we investigated the muscle phenotype of mice carrying null alleles of Mark2 (Mark2−/− mice)25. We observed that myofiber size of muscle from Mark2−/− mice was strikingly lower compared to muscle from WT mice (Fig. 4g,h). Moreover, we detected asymmetric distribution of polarity proteins in satellite cells from WT but not in Mark2−/− mice, where the absence of Mark2 expression resulted in non-polarized localization of Pard3 (Fig. 4i). However, polarization of dystrophin was still observed in satellite cells from Mark2−/− mice (Supplementary Fig. 4i).
Impaired mitotic spindle orientation
Centrosomes specify the alignment of the mitotic spindle (that is, apical-basal versus planar orientations)26. Therefore, to determine the axis of cell division, we immunostained mitotic centrosomes with antibody reactive with phosphorylated forms of Aurora kinases (p-Aurk)27. In pro-metaphase and metaphase satellite cells, immunostaining for p-Aurk detects two centrosomes per cell localized at opposite cell poles28. In telophase and cytokinesis, p-Aurk dissociates from the centrosome and accumulates at the midbody (Fig. 5a)28.
Figure 5.
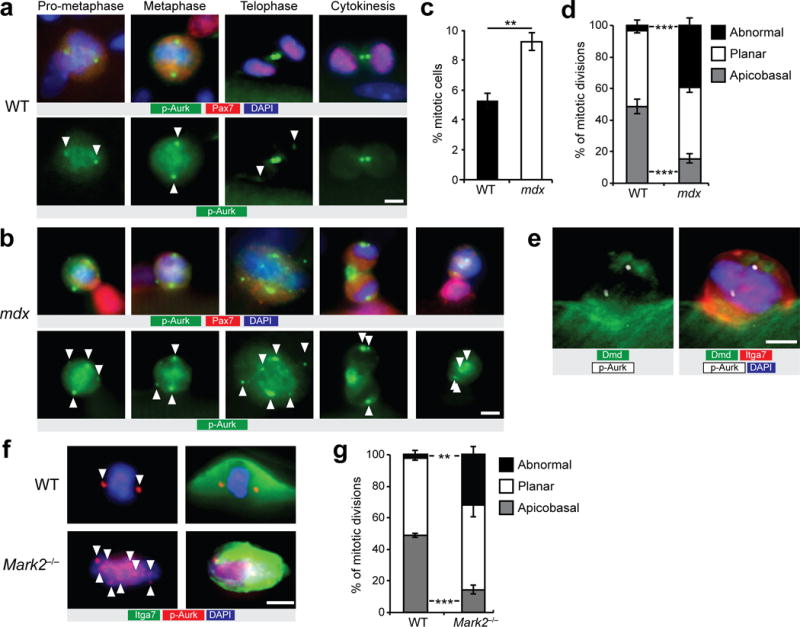
Dystrophin-deficient satellite cells display impaired mitotic spindle orientation and loss of apicobasal division. (a) Representative pictures (n > 50 pictures) of immunostaining for phospho-Aurora kinase (p-Aurk; green), Pax7 (red), and DAPI (blue) of cultured myofibers from WT mice at 36 h. (b) Representative images (n > 20 pictures) of abnormal mitotic events of cultured myofibers from mdx mice at 36 h immunostained for p-Aurk (green), Pax7 (red), and DAPI (blue). (a,b) Mitotic centrosomes (arrowheads) are stained with anti-p-Aurk antibody in prophase and metaphase cells. (c) Proportion of mitotic satellite cells on cultured myofibers from WT and mdx mice at 36 h. (d) Quantification of abnormal, versus planar, and apicobasal orientated mitotic spindles in satellite cells on myofibers from WT and mdx mice after 36 h of culture. (c,d) n = 3 WT mice and n = 4 mdx mice, approximately 100 cells total per condition. (e) Example of immunostaining (n = 10 pictures) for Dmd rod domain (green), itga7 (red), p-Aurk (white) and DAPI (blue) in a mitotic satellite cell undergoing an apicobasal division. (f) Examples of mitotic satellite cells (n > 10 pictures) from immunostaining for itga7 (green), p-Aurk (red), and DAPI (blue) of cultured myofiber from Mark2−/− mice at 36 h. n = 3 mice. (g) Quantification of abnormal, versus planar, and apicobasal orientated mitotic spindles in satellite cells on myofibers from WT and Mark2−/− mice after 36 h of culture. n = 3 mice, >20 fibers counted per condition. (c,d,g) Error bars represent means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.005. Statistical significance was calculated by Student’s t test. Scale bars, 5 μm.
We observed in myofibers from mdx mice, an unexpectedly high number of abnormal mitotic divisions, both in YFP-negative and YFP-positive satellite cells, compared to myofibers from WT mice (Fig. 5b). We characterized abnormal divisions as any events that do not correspond to the classical staining for p-Aurk; for instance, abnormally high numbers of centrosomes, or abnormal kinetics of p-Aurk staining such as persistence of the midbody from previous cell divisions. Paradoxically, we also observed higher proportion of mitotic cells on myofibers from mdx mice at 36 h even though the number of successful cell divisions was lower at 72 h compared to myofibers from WT mice (Figs. 2a and 5c). Notably, the high proportion of abnormal divisions in dystrophin-deficient satellite cells corresponds to the specific loss of apicobasal divisions (Fig. 5d and Supplementary Fig. 5a,b). Taken together, our experiments suggest that dystrophin expression is required to establish the apicobasal mitotic axis required for asymmetric cell division (Fig. 5e). Similar to dystrophin-deficient satellite cells, we observed in cultured myofibers from Mark2−/− mice a higher proportion of satellite cells with abnormal p-Aurk staining pattern together with a specific reduction in apicobasal divisions compared to WT mice (Fig. 5f,g).
DGC-deficient satellite cells display impaired regeneration
To investigate the progression of the dystrophin-deficient satellite cell myogenic program in vivo, we injured muscles from Myf5-Cre:R26R-YFP and mdx:Myf5-Cre:R26R-YFP with cardiotoxin and analyzed satellite cells by flow cytometry 3 days after injury (Supplementary Fig. 6a). We assessed satellite cell activation state by side scatter analysis to measure cell internal complexity, and assessed cell cycle status by Hoechst 33342 staining to measure DNA content. In muscles from WT mice, cardiotoxin injury induced satellite cells to activate, exit quiescence (G0 in Q4), and become proliferative (S-G2M in Q2) (Fig. 6a–d). In the contralateral muscle from mdx mice, the proportion of activated satellite cells was higher than in contralateral muscle from WT mice likely reflecting ongoing repair, which was consistent with the presence of Myod1-expressing and Myog-expressing cells in these samples (Fig. 6a–d and Supplementary Fig. 6b–d). Compared to injured muscle from WT mice, cardiotoxin injury did not provoke a strong proliferative response of satellite cells from mdx mice, with a high-proportion of myogenic cells that remained in G1 (in Q1) (Fig. 6a–d).
Figure 6.
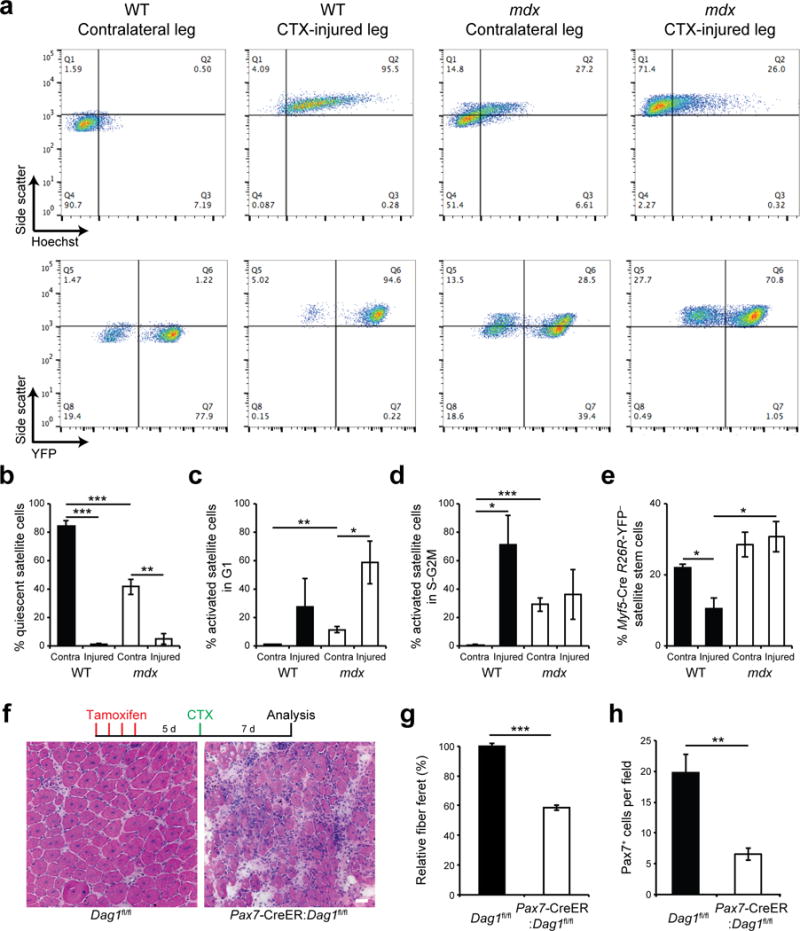
Dystrophin-deficient satellite cells have reduced ability to generate myogenic progenitors in regenerating muscle. (a) Flow cytometric analysis of WT and mdx Myf5-Cre:R26R-YFP mice 3 days after cardiotoxin (CTX) injury. Upper panels display the distribution of myogenic cells by side scatter (SSC, y-axis) and DNA content (x-axis) based on Hoechst 33342 staining, while lower panels profile their side scatter (y-axis) and YFP expression (x-axis). (b–d) Proportions of quiescent (SSC-low, DNA-low), activated G1 (SSC-high, DNA-low), and proliferating S-G2M (SSC-high, DNA-high) myogenic cells from CTX-injured or contralateral (Contra.) muscles of WT and mdx Myf5-Cre:R26R-YFP mice. (e) Proportions of YFP-negative satellite stem cells from CTX-injured or Contra. muscles of WT and mdx Myf5-Cre:R26R-YFP mice. (b–e) n = 3 mice per condition. (f) Representative pictures (n > 10 pictures per condition) from H&E staining of TA muscle sections of tamoxifen-treated Dag1fl/fl and Pax7-CreER:Dag1fl/fl mice 7 days after CTX-injury. n = 3 mice. Scale bar, 20 μm. (g) Relative fiber feret, and (h) satellite cell density (Pax7-expressing cells) from TA muscle sections of tamoxifen-treated Dag1fl/fl and Pax7-CreER:Dag1fl/fl mice 7 days after CTX-injury. n=3 mice per condition. (b–e,g,h) Error bars represent means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.005. Statistical significance was calculated by Student’s t test.
Muscles from WT mice respond to injury through the production of YFP-positive committed satellite cells, and we observed a lower proportion of YFP-negative satellite stem cell in these muscles at 3 days post-injury compared to uninjured muscles (11% and 22%, respectively) (Fig. 6a,e). However, the proportion of YFP-negative satellite stem cells remained unchanged between the contralateral and injured muscle of mdx mice (28% vs 31%, respectively) (Fig. 6a,e). This resulted in lower number of Myog-expressing cells in the injured muscles from mdx mice compared to WT mice (Supplementary Fig. 6c,d).
Previous study showed that myofiber-specific deletion of Dag1 in MCK-Cre:Dag1flfl mice results in a relatively mild regeneration deficit compared to MORE-Cre:Dag1fl/− mice, where Dag1 is deleted in both satellite cells and myofibers5. Therefore, we analyzed muscle regeneration in Pax7-CreER:Dag1fl/fl mice to assess the regenerative capacity of muscle stem cells that specifically lack the DGC. We observed marked delay in muscle regeneration with a lower number of satellite cells and lower caliber of regenerated myofibers in tamoxifen-treated Pax7-CreER:Dag1fl/fl mice compared to tamoxifen-treated Dag1fl/fl littermate control (Fig. 6f–h).
DISCUSSION
Our findings show that Dmd and Dag1 are expressed at the RNA level in prospectively isolated satellite cells, consistent with microarray data from other laboratories29,30. Furthermore, we show that high levels of dystrophin protein are expressed in satellite cells 24 h after their activation, which is consistent with the time predicted to transcribe full-length Dmd31. Immunostaining with antibodies for the N-terminal, C-terminal, and rod domain suggest that full-length dystrophin is expressed in WT satellite cells while shorter isoforms of dystrophin (for example, Dp71/Dp72 that do not interact with Mark2) with transcription start sites after the mdx point mutation may be expressed in satellite cells from mdx mice. Our experiments identify an essential role for dystrophin in regulating the establishment of PAR-mediated polarity in satellite cells. In the absence of dystrophin, the polarity effector Mark2 is dysregulated leading to the failure of Pard3 to be polarized on the pole of the cell that will give rise to the YFP-positive committed progenitor daughter cell (summarized in Fig. 3j and Supplementary Fig. 7). Polarity deficits observed in Mark2−/− mice phenocopy the behavior of dystrophin-deficient satellite cells, supporting the notion that Mark2 is a critical effector of the dystrophin-deficient satellite cell phenotype.
Impaired polarity leads to the dysregulation of mitotic spindle orientation and lower proportion of apicobasal divisions. These results are consistent with the role of Mark2 in epithelial cells to establish apicobasal polarity32. The biased loss of apicobasal divisions suggests that alternative mechanisms exist in satellite cells to determine proper mitotic spindle assembly. Establishment of apicobasal axis is known in various types of stem cells to promote asymmetric distribution of cell fate determinants and lead to asymmetric cell division20,33. Using mdx Myf5-Cre:R26R-YFP mice, we show that Myf5 asymmetric divisions are markedly reduced in the absence of dystrophin and that the generation of YFP-expressing myogenic progenitors is impaired both in vitro and in vivo. Moreover, we observed a high proportion of aberrant divisions in satellite cells from mdx and Mark2−/− mice. Consistent with our results, Par1 has been identified in Drosophila S2 cells as a gene required for centrosome clustering, and knockdown of Par1 leads to centrosome multipolarity34. Notably, abnormal divisions and dystrophin-expressing cells are not exclusive to YFP-negative cells but are also observed in a subset of YFP-positive satellite cells. These results suggest that dystrophin plays a role in other types of asymmetric divisions that have been reported in the progenitor stages of myogenesis35.
The loss of polarity in dystrophin-deficient satellite cells ultimately affects the kinetics of cell proliferation. Alterations to the phosphorylation pattern of Aurora kinases are a sign of errors in the mitotic process. Overexpression of Aurora induces abnormal spindle formation and causes cells to bypass the spindle checkpoint leading to apoptosis or senescence36. Notably, a recent study showed that, similar to what we observed in mdx satellite cells, satellite cells deficient in spindle assembly checkpoint effectors displays cell-cycle arrest in G1 and resists differentiation37. The presence of higher numbers of centrosomes could be hypothesized to trigger mitotic catastrophe followed by arrest and cellular senescence38. Moreover, persistence of p-Aurk activity in the midbody is a sign of DNA segregation errors that delay abscission and prevent tetraploization39. Therefore, abnormal p-Aurk staining pattern could also signal the activation of corrective mechanisms that delay division in dystrophin-deficient satellite cells, which is consistent with our observation that there is a higher number of cells in M-phase together with a lower number of successful divisions. Similar defects in satellite cell proliferation has also been observed in Largemyd mice where Dag1 glycosylation is impaired40.
Previous studies documenting impaired muscle regeneration in mdx mice suggest that satellite cell function is perturbed9,41. However, the contribution of satellite cell autonomous defects to the impaired regeneration has not been experimentally addressed. Our results indicate that the failure of regenerative myogenesis to keep pace with disease progression in DMD is not due to muscle stem cell exhaustion, but rather is due to impaired polarity leading to a deficit in cell division, lack of asymmetric divisions and reduced generation of myogenic progenitors (Supplementary Fig. 7). Consistent with this hypothesis, recent results indicate that the granulocyte-colony stimulating factor receptor (G-CSFR) is asymmetrically segregated in activated satellite cells where it plays a role in myogenic progression, and G-CSF treatment improve muscle regeneration in mdx mice42. Our findings explain why the regenerative capacity of muscles from mdx mice or DMD patients is impaired despite high numbers of satellite cells9,41. Moreover, although the total number of satellite cells remains elevated in aged dystrophic muscles, their relative number gradually declines during aging, which is explained by our observations that dystrophin-deficient satellite cells display mitotic errors and a slower division rate.
Different isoforms of dystrophin are expressed in various tissues and a generalized polarity deficit in DMD patients may explain other aspects of the disease such as neurological deficits43,44. Notably, defects in asymmetric stem cell division are associated with tumorigenesis45, and it was recently reported that dystrophin acts as a tumor suppressor gene in rhabdomyosarcoma, consistent with its role in regulating muscle stem cell polarity via its interaction with Mark246.
Overall, our findings demonstrate that the muscle wasting process observed in DMD patients is more complex than anticipated and reveal that in addition to muscle fragility, DMD is also a muscle stem cell disease. These results have important implications for therapeutic interventions such as gene therapy or exon skipping47. For instance, adeno-associated virus (AAV) mini-dystrophin vectors have been shown to partially rescue dystrophin expression in myofibers of mdx mice, however the current mini-dystrophin vectors do not contain the R8–R9 spectrin-repeat domain to which binds Mark248. Gene therapies targeting satellite cells potentially have long-term efficiency due to satellite cell self-renewal. Therefore, in addition to differentiated myofibers, muscle stem cells should be considered as a therapeutic target for restoring muscle function in DMD.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Mice and animal care
We used the following mouse lines: mdx, Dag1fl/fl, Pax7-CreER, R26R-YFP, Mark2−/−, Myf5-Cre, and Myf5-LacZ23,25,49–52. We performed all experiments in accordance with University of Ottawa guidelines for animal handling and animal care determined by the University of Ottawa Animal Care Committee based on the Canadian Council on Animal Care guidelines. If not stated differently, we used 2–3 month old mice for all experiments. Male or female mice were used and always gender-matched for each specific experiment. We delivered tamoxifen as described previously53. We performed cardiotoxin injections (Latoxan, 50 μl of 10 μM solution in saline) under general anaesthesia I.M. through the skin in the right tibialis anterior (TA) muscle (and Gastrocnemius muscle in flow cytometry experiments) 5 days following the last tamoxifen treatment.
Gene Expression Analysis
Microarray analysis of prospectively isolated satellite cells was performed previously19. Briefly, we obtained cells from six-week-old BALB/c mice. We cultured primary myoblasts in HAM-F10 (Wisent) containing 20% FBS (Wisent) and 5 ng/ml bFGF (Cedarlane), and we differentiated myotubes in DMEM/HAM-F10 (Wisent) containing 5% HS (Hyclone). We isolated total RNA from freshly FACS isolated quiescent satellite cells that were pooled from nine mice, or in triplicates from established mouse primary myoblasts and differentiated myotubes using the RNeasy mini kit (Qiagen). The purity of RNA was analyzed by Bioanalyzer (Agilent Technologies). We only used samples with an RNI > 9.0 for subsequent labeling and hybridization with Mouse Gene 1.0 ST Arrays (Affymetrix). Expression data was processed using Gene Expression Consol (Affymetrix). We deposited all microarray data in GEO and are available in the series GSE59272. We performed expression clustering analysis on normalized gene expression fold change with respect to the median expression value using Cluster 3.0 (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm).
RNA-seq
We prospectively isolated satellite cells by FACS from 4 week old Pax7-WT and Pax7-knockout mice as described previously54. A minimum of four mice were pooled together for each experimental condition. RNA-seq libraries were constructed using the Nugen Ovation system from pre-amplifed RNA. We performed single end RNA sequencing on a Solexa GIIx sequencer (Illumina). We used CuffDiff to determine fold change in gene expression between Pax7-WT and Pax7-knockout satellite cells.
Real-time PCR
We isolated total RNA with a commercial kit (NucleoSpin RNA II, Macherey-Nagel). We carried out reverse transcription using a mixture of oligodT and random hexamer primers (iScript cDNA Synthesis Kit, Bio-Rad). We performed real-time PCR analysis (SSoFast EvaGreen Supermix, Bio-Rad) using Sybr Green and the CFX384 real time PCR detection system (Bio-Rad). We normalized to Gapdh and Tbp expression and analyzed the results by Bio-Rad CFX Manager software. We used the following primers (5′–3′): Dmd forward: CAACAACTCCTTCCCTAGTT, Dmd reverse: GCTCTGCCCAAATCATCT; Dag1 forward: CTCATTTCGAGTGAGCATTCC, Dag1 reverse: ACTGTGTGGGTCCCAGTGTAG; Sgca forward: ACACAGCGCAGTCCCTATAAC, Sgca reverse: CCAGGAACTCAGCTTGGTATG; Mark2 forward: GTCCGCAGGAACCTGAATGA, Mark2 reverse: CCCGAAACTCCTCCTTGTCC; Pard3 forward: CATGATCCAGCTCATTGTGG, Pard3 reverse: CGTTCTCGGTCATCCAGTTC; Pax7 forward: GACGACGAGGAAGGAGACAA, Pax7 reverse: ACATCTGAGCCCTCATCCAG; Myod1 forward: GGCTACGACACCGCCTACTA, Myod1 reverse: GTGGAGATGCGCTCCACTAT; Gapdh forward: CCCAGAAGACTGTGGATGG, Gapdh reverse: ACACATTGGGGGTAGGAACA; Tbp forward: AGAACAATCCAGACTAGCAGCA, Tbp reverse: GGGAACTTCACATCACAGCTC.
EDL fiber culture and siRNA transfection
We performed myofiber culture as described earlier55. Briefly, we carefully dissected EDL muscles and incubated the muscles in DMEM (Gibco) containing 0.2% collagenase I (Sigma) for 45 min. We detached the myofibers using gentle trituration with a glass pipet. We cultured myofibers for 0, 12, 24, 36, 42, or 72 h in DMEM containing 20% FBS (Wisent), 1% chick embryo extract (MP Biomedicals), and 2.5 ng/ml bFGF (Cedarlane). We transfected satellite cells on myofibers using lipofectamine RNAimax (Life technologies) with validated smartpool siRNAs for Mark2, Pard3, Dmd, or scramble (SCR) (Dharmacon). To ensure maximal efficiency, We performed two transfections at 4 h and 16 h after isolation of the myofibers as described earlier19.
Immunostaining and antibodies
For immunostaining, we first fixed EDL myofibers for 5 min in 2% PFA and permeabilized for 15 min in 0.1% Triton, 0.1 M Glycine in PBS. We then blocked in 5% horse serum in PBS for 1-2 h. We applied primary antibodies in blocking solution for 2 h at room temperature or at 4 °C over night. We subsequently washed the samples with PBS and stained with appropriate fluorescently labeled secondary antibodies (Alexa fluor 488, 546, or 647) for 1 h at room temperature. After washing with PBS, we stained the fibers with DAPI for 5 min and mounted the samples with Permafluor (Fisher). We used the following antibodies: mouse anti-Pax7 (DSHB), chicken anti-GFP (cat# ab13970, Abcam), mouse anti-Dmd C-terminal (MANDRA1, clone 7A10, DSHB), rabbit anti-Dmd C-terminal (cat# ab15277, Abcam), mouse anti-Dmd rod domain (clone Dy4/6D3, cat# VP-D508, vector labs), mouse anti-Dmd rod domain (clone Dy4/6D3, cat# DYS1-CE-S, Leica microsystems), mouse anti-Dmd N-terminal (MANEX1011B, clone 1C7, DSHB), mouse anti-Dag1 (cat# ab49515, Abcam), rabbit anti-Dag1 (cat# D1945, Sigma), rabbit anti-Mark2 (cat# 9118S, Cell signaling technology), rabbit anti-phospho-Mark2 AF647-conjugated (cat# bs-5742R, Bioss), rabbit anti-Pard3 (cat# 07-330, Millipore), rabbit anti-phospho-Aurk (cat# 2914S, Cell Signaling technology), rat anti-itga7 AF647-conjugated (clone R2F2, cat# 67-0010-10, AbLab), mouse anti-itga7 (cat# K0046-3, Cedarlane), rat anti-laminin (cat# L0663, Sigma), rabbit anti-Myod1 (C-20, cat# sc-304, Santa Cruz), rabbit anti-Myog (M225, cat# sc-576, Santa Cruz). We performed proximity ligation assays (PLA) using Duolink (Sigma) PLA probes mouse and rabbit according to the manufacturer instructions as described earlier19. We captured images of immunostainings on an Axio Observer.Z1 microscope equipped with a LSM510 META confocal laser scanner and a plan-Apochromat 63x/1.40 Oil DIC M27 objective or an Axioplan 2 microscope equipped with a plan-Neofluar 40x/1.30 Oil DIC and a plan-Neofluar 100x/1.30 Oil DIC objective. We analyzed images with Axiovision, Zen, and FIJI software and measured myofiber feret size as described previously56.
Characterization of satellite cell divisions
We fixed single myofibers after 36 h of in vitro culture, as described above. Satellite cells were identified by Pax7 expression. Pax7 staining becomes cytoplasmic in mitotic satellite cells after the dissociation of the nuclear envelope, but is still discernable. We identified mitotic satellite cells by positive p-Aurk staining, which labels cells from pro-metaphase to cytokinesis (Fig. 5a). We quantified as abnormal division any mitotic satellite cells with p-Aurk staining patterns that were not observed in WT cells, including monopolar, multipolar (>2), and abscission defects (Fig. 5b). We manually counted mitotic orientations according to the angle between the mitotic spindle and the tangential plane of the satellite cell’s attachment point to the myofiber (Supplementary Fig. 5a). Generally, apicobasal divisions were clearly oriented 60–90 degrees away from the myofiber whereas planar divisions were oriented 0–30 degrees along the myofiber (Supplementary Fig. 5b). Note, telophase cells that have undergone apicobasal divisions could become slanted or be pushed over during the staining process, therefore we assessed mitotic orientations of these telophase cells based on the position of the midbody found in the daughter cell that is attached to the myofiber57. We projected 3D z-stack images by maximum intensity using FIJI software.
Flow cytometry and Fluorescence Activated Cell Sorting (FACS)
We removed cardiotoxin-injured and contralateral hindlimb muscles and dissociated the muscles using the gentleMACS dissociator (Miltenyi Biotech). In short, we minced the muscles in 5 mL of collagenase/dispase solution (2.5 U/mL), placed them into gentleMACS C tubes (Miltenyi Biotech) where they were incubated with heating elements with a custom digestion program as follows: 3 min @ 60 rpm for further dissociation of chunks, 9 mins @ −30 rpm for digestion, 10 cycles of 5 sec @ ± 360rpm for trituration, and 12 min @ −30 rpm for secondary digestion. We filtered mononuclear cells through a 50 μm nylon filter, washed with FACS buffer (5% FBS with 1 mM EDTA in PBS), and stained with PE conjugated anti-Sca-1 (clone D7, cat# 553108, BD Biosciences), anti-CD45 (clone 30-F11, cat# 12-0451-83, BD Biosciences), anti-CD31 (clone 390, cat# 12-0311-81, BD Biosciences), anti-CD11b (clone M1/70, cat# 12-0112-81, BD Biosciences), APC conjugated anti-itga7 (clone R2F2, cat# 67-0010-10, AbLab), and Hoechst 33342. We gated satellite cells based on forward scatter and side scatter profiles, followed by negative lineage selection in PE for Sca-1, CD45, CD31, and CD11b and positive lineage selection in APC for itga7. We further purified this population by eliminating autofluorescent cells by gating APC against APC-Cy7 (Supplementary Fig. 6a)54. We established the gates based on the cytometric profiles of itga7+ YFP+ satellite cells from resting and injured muscles of WT and mdx Myf5-Cre:R26R-YFP mice. The forward and side scatter (FSC, SSC) gates resulting from this strategy also agree with our observation that activated satellite cells isolated from Pax7-zsGreen mice have higher SSC signal. We sorted satellite cells from Pax7-zsgreen mice based on zsgreen expression. We cytospun prospectively isolated cells onto slides immediately after isolation with the Cytospin 4 (Thermo Scientific) at 500 rpm for 10 min and analyzed for purity and expression of myogenic markers by immunofluorescence staining.
Statistical analysis
We performed experiments at least in biological triplicates unless otherwise stated. We replicated the data presented in the main figures at least twice in the laboratory. We did not exclude any animals from an experiment. We did not use any randomization process. The evaluator was blinded to the identity of the specific sample as far as the nature of the experiment allows it. We did not perform any power calculation analysis. Unless otherwise stated data displayed normal variance. We performed a 2-tailed Students t test to determine statistical significance. We used Wilcoxon Rank-sum test where the data were not normally distributed. Error bars denote SEM. The level of significance is indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.005.
Supplementary Material
Acknowledgments
We thank J. Dilworth and L. Megeney for careful reading of the manuscript. We thank Jennifer Ritchie for animal husbandry. N.A.D. is supported by a Postdoctoral Fellowship from the Canadian Institutes of Health Research. Y.X.W. is supported by fellowships from QEII-GSST and the Canadian Institutes of Health Research. J.v.M. was supported by a grant from the Deutsche Forschungsgemeinschaft, C.F.B. was supported by a grant from the Swiss National Science Foundation. C.E.B. is supported by a Postdoctoral Fellowship from the Ontario Institute for Regenerative Medicine. M.A.R. holds the Canada Research Chair in Molecular Genetics. These studies were carried out with support of grants to M.A.R. from the US National Institutes for Health [R01AR044031], the Canadian Institutes for Health Research [MOP-12080, MOP-81288], E-Rare-2: Canadian Institutes of Health Research/Muscular Dystrophy Canada [ERA-132935], the Muscular Dystrophy Association, the Stem Cell Network, and the Government of Ontario Ministry of Research and Innovation (MRI) [ORF-RE05-084].
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
N.A.D., and Y.X.W. designed and carried out experiments, analyzed results and wrote the manuscript. J.v.M. designed and conducted experiments and analyzed results. A.P., C.F.B., and C.E.B. conducted experiments. M.A.R. designed experiments, analyzed results, wrote the manuscript and provided financial support.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Anderson MS, Kunkel LM. The molecular and biochemical basis of Duchenne muscular dystrophy. Trends Biochem Sci. 1992;17:289–292. doi: 10.1016/0968-0004(92)90437-e. [DOI] [PubMed] [Google Scholar]
- 2.Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve. 2000;23:1456–1471. doi: 10.1002/1097-4598(200010)23:10<1456::aid-mus2>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 3.Koenig M, et al. Complete cloning of the duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- 4.Serrano AL, et al. Cellular and molecular mechanisms regulating fibrosis in skeletal muscle repair and disease. Curr Top Dev Biol. 2011;96:167–201. doi: 10.1016/B978-0-12-385940-2.00007-3. [DOI] [PubMed] [Google Scholar]
- 5.Cohn RD, et al. Disruption of Dag1 in Differentiated Skeletal Muscle Reveals a Role for Dystroglycan in Muscle Regeneration. Cell. 2002;110:639–648. doi: 10.1016/s0092-8674(02)00907-8. [DOI] [PubMed] [Google Scholar]
- 6.Sacco A, et al. Short Telomeres and Stem Cell Exhaustion Model Duchenne Muscular Dystrophy in mdx/mTR Mice. Cell. 2010;143:1059–1071. doi: 10.1016/j.cell.2010.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Webster C, Blau HM. Accelerated age-related decline in replicative life-span of Duchenne muscular dystrophy myoblasts: implications for cell and gene therapy. Somat Cell Mol Genet. 1990;16:557–565. doi: 10.1007/BF01233096. [DOI] [PubMed] [Google Scholar]
- 8.Kottlors M, Kirschner J. Elevated satellite cell number in Duchenne muscular dystrophy. Cell Tissue Res. 2010;340:541–548. doi: 10.1007/s00441-010-0976-6. [DOI] [PubMed] [Google Scholar]
- 9.Reimann J, Irintchev A, Wernig A. Regenerative capacity and the number of satellite cells in soleus muscles of normal and mdx mice. Neuromuscul Disord. 2000;10:276–282. doi: 10.1016/s0960-8966(99)00118-2. [DOI] [PubMed] [Google Scholar]
- 10.Chakkalakal JV, et al. Early forming label-retaining muscle stem cells require p27kip1 for maintenance of the primitive state. Development. 2014;141:1649–1659. doi: 10.1242/dev.100842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamashita K, et al. The 8th and 9th tandem spectrin-like repeats of utrophin cooperatively form a functional unit to interact with polarity-regulating kinase PAR-1b. Biochem Biophys Res Commun. 2010;391:812–817. doi: 10.1016/j.bbrc.2009.11.144. [DOI] [PubMed] [Google Scholar]
- 12.Masuda-Hirata M, et al. Intracellular polarity protein PAR-1 regulates extracellular laminin assembly by regulating the dystroglycan complex. Genes Cells. 2009;14:835–850. doi: 10.1111/j.1365-2443.2009.01315.x. [DOI] [PubMed] [Google Scholar]
- 13.Neumüller RA, Knoblich JA. Dividing cellular asymmetry: asymmetric cell division and its implications for stem cells and cancer. Genes Dev. 2009;23:2675–2699. doi: 10.1101/gad.1850809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knoblich JA. Asymmetric cell division: recent developments and their implications for tumour biology. Nat Rev Mol Cell Biol. 2010;11:849–860. doi: 10.1038/nrm3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goulas S, Conder R, Knoblich JA. The Par complex and integrins direct asymmetric cell division in adult intestinal stem cells. Cell Stem Cell. 2012;11:529–540. doi: 10.1016/j.stem.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Troy A, et al. Coordination of Satellite Cell Activation and Self-Renewal by Par-Complex-Dependent Asymmetric Activation of p38α/β MAPK. Cell Stem Cell. 2012;11:541–553. doi: 10.1016/j.stem.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miranda AF, et al. Immunocytochemical study of dystrophin in muscle cultures from patients with Duchenne muscular dystrophy and unaffected control patients. Am J Pathol. 1988;132:410–416. [PMC free article] [PubMed] [Google Scholar]
- 18.Huard J, Labrecque C, Dansereau G, Robitaille L, Tremblay JP. Dystrophin expression in myotubes formed by the fusion of normal and dystrophic myoblasts. Muscle Nerve. 1991;14:178–182. doi: 10.1002/mus.880140213. [DOI] [PubMed] [Google Scholar]
- 19.Bentzinger CF, et al. Fibronectin regulates Wnt7a signaling and satellite cell expansion. Cell Stem Cell. 2013;12:75–87. doi: 10.1016/j.stem.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Asymmetric Self-Renewal and Commitment of Satellite Stem Cells in Muscle. Cell. 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rocheteau P, Gayraud-Morel B, Siegl-Cachedenier I, Blasco M, Tajbakhsh S. A Subpopulation of Adult Skeletal Muscle Stem Cells Retains All Template DNA Strands after Cell Division. Cell. 2012;148:112–125. doi: 10.1016/j.cell.2011.11.049. [DOI] [PubMed] [Google Scholar]
- 22.Ono Y, et al. Slow-dividing satellite cells retain long-term self-renewal ability in adult muscle. J Cell Sci. 2012;125:1309–1317. doi: 10.1242/jcs.096198. [DOI] [PubMed] [Google Scholar]
- 23.Nishijo K, et al. Biomarker system for studying muscle, stem cells, and cancer in vivo. FASEB J Off Publ Fed Am Soc Exp Biol. 2009;23:2681–2690. doi: 10.1096/fj.08-128116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fredriksson S, et al. Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol. 2002;20:473–477. doi: 10.1038/nbt0502-473. [DOI] [PubMed] [Google Scholar]
- 25.Hurov JB, et al. Immune system dysfunction and autoimmune disease in mice lacking Emk (Par-1) protein kinase. Mol Cell Biol. 2001;21:3206–3219. doi: 10.1128/MCB.21.9.3206-3219.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu MS, Johnston CA. Molecular pathways regulating mitotic spindle orientation in animal cells. Dev Camb Engl. 2013;140:1843–1856. doi: 10.1242/dev.087627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang G, Jiang Q, Zhang C. The role of mitotic kinases in coupling the centrosome cycle with the assembly of the mitotic spindle. J Cell Sci. 2014;127:4111–4122. doi: 10.1242/jcs.151753. [DOI] [PubMed] [Google Scholar]
- 28.Carmena M, Earnshaw WC. The cellular geography of Aurora kinases. Nat Rev Mol Cell Biol. 2003;4:842–854. doi: 10.1038/nrm1245. [DOI] [PubMed] [Google Scholar]
- 29.Fukada S, et al. Molecular signature of quiescent satellite cells in adult skeletal muscle. Stem Cells Dayt Ohio. 2007;25:2448–2459. doi: 10.1634/stemcells.2007-0019. [DOI] [PubMed] [Google Scholar]
- 30.Liu L, et al. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013;4:189–204. doi: 10.1016/j.celrep.2013.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tennyson CN, Klamut HJ, Worton RG. The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat Genet. 1995;9:184–190. doi: 10.1038/ng0295-184. [DOI] [PubMed] [Google Scholar]
- 32.Lewandowski KT, Piwnica-Worms H. Phosphorylation of the E3 ubiquitin ligase RNF41 by the kinase Par-1b is required for epithelial cell polarity. J Cell Sci. 2014;127:315–327. doi: 10.1242/jcs.129148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knoblich JA. Mechanisms of Asymmetric Stem Cell Division. Cell. 2008;132:583–597. doi: 10.1016/j.cell.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 34.Kwon M, et al. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008;22:2189–2203. doi: 10.1101/gad.1700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yennek S, Burute M, Théry M, Tajbakhsh S. Cell Adhesion Geometry Regulates Non-Random DNA Segregation and Asymmetric Cell Fates in Mouse Skeletal Muscle Stem Cells. Cell Rep. 2014;7:961–970. doi: 10.1016/j.celrep.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 36.Marumoto T, Zhang D, Saya H. Aurora-A — A guardian of poles. Nat Rev Cancer. 2005;5:42–50. doi: 10.1038/nrc1526. [DOI] [PubMed] [Google Scholar]
- 37.Kollu S, Abou-Khalil R, Shen C, Brack AS. The Spindle Assembly Checkpoint Safeguards Genomic Integrity of Skeletal Muscle Satellite Cells. Stem Cell Rep. doi: 10.1016/j.stemcr.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galluzzi L, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steigemann P, et al. Aurora B-Mediated Abscission Checkpoint Protects against Tetraploidization. Cell. 2009;136:473–484. doi: 10.1016/j.cell.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 40.Ross J, et al. Defects in Glycosylation Impair Satellite Stem Cell Function and Niche Composition in the Muscles of the Dystrophic Largemyd Mouse. STEM CELLS. 2012;30:2330–2341. doi: 10.1002/stem.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Irintchev A, Zweyer M, Wernig A. Impaired functional and structural recovery after muscle injury in dystrophic mdx mice. Neuromuscul Disord NMD. 1997;7:117–125. doi: 10.1016/s0960-8966(96)00422-1. [DOI] [PubMed] [Google Scholar]
- 42.Hayashiji N, et al. G-CSF supports long-term muscle regeneration in mouse models of muscular dystrophy. Nat Commun. 2015;6 doi: 10.1038/ncomms7745. [DOI] [PubMed] [Google Scholar]
- 43.Florencia G, Verónica F, Viviana D, Irene S. Dystrophin deletions and cognitive impairment in Duchenne/Becker muscular dystrophy. Neurol Res. 2004;26:83–87. doi: 10.1179/016164104773026589. [DOI] [PubMed] [Google Scholar]
- 44.De Stefano ME, Leone L, Lombardi L, Paggi P. Lack of dystrophin leads to the selective loss of superior cervical ganglion neurons projecting to muscular targets in genetically dystrophic mdx mice. Neurobiol Dis. 2005;20:929–942. doi: 10.1016/j.nbd.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 45.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441:1068–1074. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, et al. Dystrophin is a tumor suppressor in human cancers with myogenic programs. Nat Genet. 2014;46:601–606. doi: 10.1038/ng.2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Long C, et al. Prevention of muscular dystrophy in mice by CRISPR/Cas9–mediated editing of germline DNA. Science. 2014;345:1184–1188. doi: 10.1126/science.1254445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang B, Li J, Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci. 2000;97:13714–13719. doi: 10.1073/pnas.240335297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J, et al. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat Genet. 1999;21:133–137. doi: 10.1038/5089. [DOI] [PubMed] [Google Scholar]
- 50.Tajbakhsh S, et al. Gene targeting the myf-5 locus with nlacZ reveals expression of this myogenic factor in mature skeletal muscle fibres as well as early embryonic muscle. Dev Dyn Off Publ Am Assoc Anat. 1996;206:291–300. doi: 10.1002/(SICI)1097-0177(199607)206:3<291::AID-AJA6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 51.Tallquist MD, Weismann KE, Hellström M, Soriano P. Early myotome specification regulates PDGFA expression and axial skeleton development. Dev Camb Engl. 2000;127:5059–5070. doi: 10.1242/dev.127.23.5059. [DOI] [PubMed] [Google Scholar]
- 52.Srinivas S, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Von Maltzahn J, Jones AE, Parks RJ, Rudnicki MA. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc Natl Acad Sci U S A. 2013;110:16474–16479. doi: 10.1073/pnas.1307680110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pasut A, Oleynik P, Rudnicki MA. Isolation of muscle stem cells by fluorescence activated cell sorting cytometry. Methods Mol Biol Clifton NJ. 2012;798:53–64. doi: 10.1007/978-1-61779-343-1_3. [DOI] [PubMed] [Google Scholar]
- 55.Pasut A, Jones AE, Rudnicki MA. Isolation and culture of individual myofibers and their satellite cells from adult skeletal muscle. J Vis Exp JoVE. 2013:e50074. doi: 10.3791/50074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Briguet A, Courdier-Fruh I, Foster M, Meier T, Magyar JP. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul Disord NMD. 2004;14:675–682. doi: 10.1016/j.nmd.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 57.Lee CY, et al. Drosophila Aurora-A kinase inhibits neuroblast self-renewal by regulating aPKC/Numb cortical polarity and spindle orientation. Genes Dev. 2006;20:3464–3474. doi: 10.1101/gad.1489406. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.