

Abstract
Aim
Low immunogenicity remains a major obstacle in realizing the full potential of cancer vaccines. In this study, we evaluated CpG-coated tumor antigen (Tag)-encapsulating ‘bacteriomimetic’ nanoparticles (CpG-nanoparticle [NP]-Tag NPs) as an approach to enhance anti-tumor immunity.
Materials & methods
CpG-NP-Tag NPs were synthesized, characterized for their physicochemical properties and tested in vivo.
Results
We found CpG predosing followed by intraperitoneal (IP) immunization with CpG-NP-Tag NPs significantly attenuated tumor growth in female BALB/c mice compared with respective controls. Histopathological and Immunofluorescence data revealed CpG-NP-Tag tumors had lower proliferation, higher apoptotic activity, greater CD4+ and CD8+ T cell infiltration as well as higher IFN-γ levels as compared with control groups.
Conclusion
Our findings suggest CpG-NP-Tag NPs can enhance anti-tumor effect of nanoparticulate tumor vaccination system.
Keywords: antigen, breast cancer, immunotherapy, nanoparticle, NP, vaccines
Background
Nanoparticulate drug delivery systems for vaccines and immunotherapy are being widely studied [1]. Such systems have been tested in infectious disease models, viral infections as well as cancer [2]. Although cancer is a disease of self-cells, the immune system is capable of producing exquisite and specific response against it. However, such response is not enough for preventing the growth and spread of cancer [3]. Nanoparticle (NP)-mediated cancer immunotherapeutic approaches have shown promise in enhancement of host immune responses against cancer antigens and are being explored as a novel alternative approach for cancer treatment [4]. NPs made from a biocompatible polymer such as poly(lactic-co-glycolic acid) (PLGA) have been utilized to deliver antineoplastic drugs (e.g., chemotherapeutic agents), siRNA, peptide or DNA vaccines for treatment of cancer [5]. NP-based delivery systems in general offer several benefits such as improved pharmacokinetic profile, targeted delivery, protection of antigen from enzymatic degradation and improved stability of encapsulated cargo. Recent advances suggest that NPs can be efficiently used for vaccines and immunotherapeutic strategies [4].
Efficient delivery of Tag to antigen-presenting cells (APCs) though a major challenge; is a promising approach in the development of immune-targeted tumor vaccines. NPs potentiate the intracellular delivery of antigen which enhances the immune response significantly. Tumor lysates, peptides and antigens encapsulated within NPs have been shown to induce specific anti-tumor responses. For example, several studies have reported enhancement of immune responses against cancer by encapsulation of tumor-associated antigens (TAA), which are aberrantly expressed on cancer cells but not on normal cells [6–8]. DNA vector-encoding proteins overexpressed on tumor cells have also shown a similar effect [9]. In addition, studies have shown that nonspecific immune activation by repeated administration of bacterial antigen CpG-oligodeoxynucletide (ODN) controls tumor growth [10–12]. Administration of NPs encapsulating such antigens leads to activation of tumor-specific T-cell responses as well as enhanced secretion of cytokines responsible for efficient function of cytotoxic T cells [13]. Among APCs, dendritic cells play a major role in antigen processing and presentation. Therefore, some studies have utilized ligands specific to dendritic cells to target delivery of antigen encapsulating NP cargo to these cells. This allows further enhancement of immune responses against the antigen, resulting in anti-tumor immunity [14].
CpG-ODNs have been successfully employed as adjuvants to enhance anti-tumor immune defenses [15]. However, its use has also been shown to elicit adverse responses [16]. In particular, repeated administration of CpG has been shown to cause pain at site of injections as well as frequent headaches in healthy volunteers [17]. Consideration of the multiple exposures of CpG and other potent adjuvants is a critical factor in vaccine efficacy. Thus, the dose of CpG is a critical factor in vaccine efficacy. In this study, we engineered CpG-NP-Tag NPs using Bis (sulfosuccinimidyl) suberate (BS3) as a bridge to dock the CpG on the surface of the NPs encapsulated with membrane lysate of 4T1 murine mammary carcinoma cells as the Tag (Figure 1A) and tested its efficacy in generating anti-tumor immunity limiting CpG exposure. It is well known from literature that surface proteins of pathogens are primary antigens and are sensed as foreign by the host immunity system, which is very likely in tumor cells as well [18]. However, both tumor antigens and material used to make NPs is less immunogenic. Therefore, to enhance identification of NPs as foreign particles, we coated bacterial antigen CpG on surface to prepare ‘bacteriomimetic’ NPs. The principle of booster dose is also very well known in vaccination. Therefore, we administered a predose of CpG to the mice and CpG coated NP as a booster dose. The objective of this study was to synthesize ‘bacteriomimetic’ NPs by CpG surface functionalization of tumor antigen-encapsulating NPs and evaluate its efficacy in generating strong anti-tumor effect.
Figure 1. Formulation of ‘bacteriomimetic’ CpG-nanoparticle-tumor antigen (CpG-NP-Tag) nanoparticles.
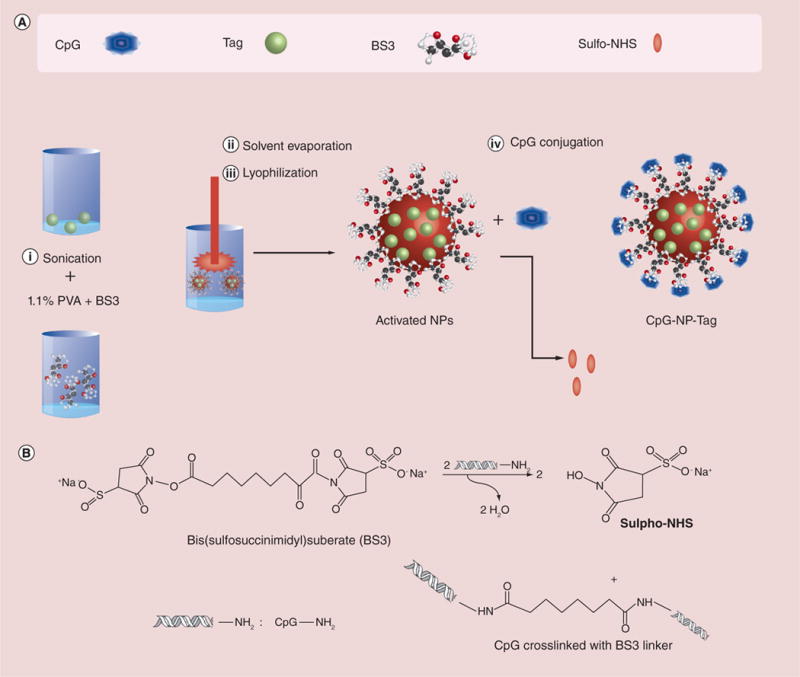
(A) Schematic of the steps involved in the formulation of CpG surface-functionalized Tag encapsulated [CpG-NP-Tag] encapsulating ‘bacteriomimetic’ nanoparticles) nanoparticle and components of the nanoparticle. (B) Schematic of the chemical reaction involved in the surface conjugation of CpG ligand.
CpG: Cytosine-phosphate-guanosine oligodeoxynucletide; NP: Nanoparticle; Tag: Tumor antigen.
Methods
Materials
Poly(d,l-lactide-co-glycolide) 50:50; inherent viscosity 0.7–0.9 dl/g; mw 50,000 was purchased from Lakeshore Biomaterials (AL, USA). Polyvinyl alcohol (PVA; mw 30,000–70,000; alcoholysis degree 88–99.9 (mol/mol%) was purchased from Sigma Aldrich (MO, USA). BS3 was purchased from Thermo Fisher Scientific (IL, USA). CpG-ODN 1826 (class B CpG Oligonucleotide-Murine TLR9 ligand) was obtained from InvivoGen (CA, USA). Roswell Park Memorial Institute (RPMI) 1640 media, Penicillin-Streptomycin (Pen-Strep) and fetal bovine serum (FBS) were obtained from Invitrogen (CA, USA). Na+/K+ ATPase and PGK antibodies were purchased from Cell Signaling, Inc (MA, USA). CD4+/CD8+ Alexa fluor 488 and IFN-γ Alexa fluor 488 conjugated anti-mouse antibodies as well as Ki67 eFluor® 615 conjugated anti-mouse antibody was purchased from ebioscience, Inc. (CA, USA). D-Luciferin K+ salt bioluminescent substrate for imaging was obtained from Perkin Elmer Inc. (MA, USA). In situ Cell Death Detection Kit, Fluorescein for terminal deoxynucleotidyltransferase (TdT)-mediated dUTP nick end labeling (TUNEL) assay kit was purchased from Roche Diagnostics (IN, USA).
Cell line
4T1 murine mammary carcinoma cell line was purchased from American Type Culture Collection (ATCC; VA, USA; see Supplementary Material Section 1.2 for ATCC cell-line characterization, available online at: www.futuremedicine.com/doi/full/10.2217/NNM.14.144) and was grown until 70% confluent in Roswell Park Memorial Institute (RPMI) media supplemented with 10% FBS and 1% Pen-Strep.
Membrane lysate preparation
4T1 cells were lysed using hypotonic buffer and dounce homogenizer followed by centrifugation at 5000 × g at 4°C for 15 min to pellet cell debris. Supernatant was collected and further centrifuged at 100,000 g for 1 h at 4°C using N55 rotor. After centrifugation, pellet (membrane fraction) was collected and washed with phosphate-buffered saline (PBS) followed by a second round of centrifugation. Final membrane fraction was resuspended in 100–150 μl of radioimmunoprecipitation assay (RIPA) buffer. Protein estimation was performed using Pierce™ BCA protein assay kit (Thermo Scientific, IL, USA).
Formulation of CpG-NP-Tag NPs
CpG-NP-Tag NPs were prepared using water-in-oil-in-water (w/o/w) double emulsion method employing solvent evaporation technique reported elsewhere [19–21] with slight modifications as described in section 1.1 of the Supplementary Material. CpG-NP-Tag NPs were further characterized and subsequently used for in vivo studies.
Characterization of NPs
Particle size, polydispersity index, zeta potential & encapsulation efficiency
Particle size was measured using Nanotrac ULTRA instrument by suspending NPs in PBS while polydispersity index (PDI; i.e., the width of the particle size distribution) was calculated using the formula: (σ/d)2 where σ represents the standard deviation and d indicates mean diameter. Zeta potential was measured using Zetasizer (Malvern Instruments Ltd, MA, USA). A known amount of NPs (0.25–0.5 mg) were resuspended in 1 ml distilled water and further diluted ten-times before measuring particle size and zeta potential. The quantity of Tag actually encapsulated was confirmed based on the amount of Tag (protein) extracted after degrading a fixed amount of NPs. 5 mg of NPs were degraded using 100 mM NaOH + 0.05% SDS (Sigma-Aldrich, MO, USA) by incubating at 37°C on a shaker. Samples were further centrifuged at 11,000 × g at 4°C for 10 min and the supernatants were tested for their protein content [22]. Protein estimations were done in triplicate using Bicinchonic acid (BCA) protein assay kit (Thermo Scientific, IL, USA) as per manufacturer’s instructions. Encapsulation efficiency was calculated as follows: amount of protein encapsulated / amount of protein used in encapsulation × 100%.
Transmission electron microscopy
Briefly, a known quantity of NPs (1 mg) were resuspended in distilled water and deposited on a transmission electron microscopy (TEM) grid employing uranyl acetate as negative stain. TEM images were taken using a Zeiss EM 910 transmission electron microscope at 80 keV.
Fluorescence correlation spectroscopy
For fluorescence correlation spectroscopy (FCS), 0.25 mg of NPs were resuspended in distilled water and incubated with 7 μg of CpG-fluorescein isothiocyanate (FITC) for 60–90 min followed centrifugation and washing at 11,000 × g for 15 min to remove excess CpG ligand. FCS measurements were done using Microtime 200 system from Picoquant GmbH (Berlin, Germany). NPs (approximately 0.25 mg) were diluted in distilled water and 30 μl of solution was dropped onto a 20 × 20 mm No. 1 coverslip (Menzel–Gläser, MA, USA). The focal height was adjusted to 20 μm above this coverslip using an Olympus i × 71 microscope and an Olympus 60 × 1.2 NA objective.
Syngenic breast cancer BALB/c mice model
BALB/c AnNHsd female mice were obtained from Harlan laboratories, Inc. (IN, USA) and housed at UNTHSC animal facility and allowed to acclimatize for a week prior to experimentation. All procedures were in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) at UNTHSC at Fort Worth. Mice were preimmunized intraperitoneally with CpG (600 μg/kg) followed by secondary immunization using respective NPs (Table 1) 7 days before tumor challenge. Dose of NPs (5 mg) was calculated based on the encapsulation efficiency to deliver Tag equivalent to the amount of CpG used during preimmunization. Mice immunized with the CpG-NP-Tag NPs (n = 5) comprised the test/treatment group while mice immunized with CpG-NP-Blank NPs (n = 5) and NP-Tag (n = 5) were considered as control groups. Subsequently, mice were challenged with 1 × 105 4T1-luciferase transfected cells (refer to Supplementary Material section 1.2 & Supple mentary Figure 1 for the transfection procedure). Primary tumor size was monitored over the course of 14 days following tumor challenge. Spleens and primary tumors were harvested for further studies (Figure 2).
Table 1.
Physicochemical characterization of nanoparticles.
| NP construct | Particle size (nm ± SD) | Zeta potential (mV ± SD) | PDI | Encapsulation efficiency (%) | PLC (μg/100 mg NP) |
|---|---|---|---|---|---|
| CpG-NP-Blank | 211.8 ± 79 | −4.22 ± 0.2 | 0.137 | – | – |
| NP-Tag | 221.3 ± 75 | −3.62 ± 0.4 | 0.115 | 32.2 ± 2.1 | 183 |
| CpG-NP-Tag | 227.4 ± 40 | −6.73 ± 0.3 | 0.031 | 32.2 ± 2.1 | 183 |
NP: Nanoparticle; PDI: Polydispersity Index; PLC: Protein (tumor antigen)-loading content; SD: Standard deviation.
Figure 2. Schematic representation of the study timeline followed for the in vivo studies.
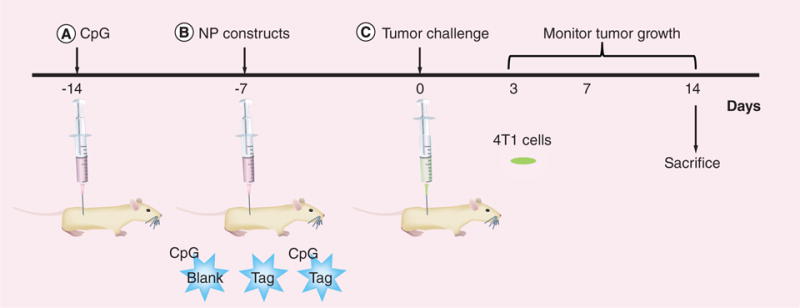
At around 5–6 weeks, female BALB/c mice (n = 5) were preimmunized intraperitoneally with CpG 14 days before tumor challenge followed by intraperitoneal immunization (7 days after CpG preimmunization) with the respective groups of nanoparticles. Mice were challenged subcutaneously with 105 4T1 luciferase transfected mammary carcinoma cells and the effect of nanoparticle immunization was evaluated on the rate of tumor growth and immune response for 14 days.
Rate of tumor growth & animal weight
Tumor size and animal weight were measured using vernier caliper and weighing balance respectively at different time intervals till day 14. Tumor size was measured in three different dimensions (mm): length, breadth and height. Tumor volume (mm3) was calculated using the following formula and the fold change in tumor volume was compared among the different groups of animals [23]:
Bioluminiscence imaging
Tumor growth was monitored using in vivo IVIS® Lumina bioluminescence imaging instrument (Caliper Life Sciences Inc., MA, USA) housed in UNTHSC animal facility. BALB/c mice were injected with D-luciferin (5 mg/kg) via IP route 10 min before taking images using IVIS Lumina and analyzed using Living Image software 3.0 (Perkin Elmer Inc. MA, USA).
Histological analysis of tumors
For morphological examination, tissues were fixed in 4% paraformaldehyde, embedded in optimum cutting temperature (OCT) compound (Tissue-Tek, Sakura Fine-Tek, CA, USA) and sectioned using cryostat to obtain 5–8 μm thin sections. Hematoxylin and eosin (H & E) staining was performed using standard protocol. Images were captured using Olympus AX70 Provis microscope (NY, USA).
Proliferation rate
For proliferation study, 5–7 μm OCT embedded tumor sections were used. Sections were stained for nuclear proliferation marker, Ki 67 and were further quantified for tumor proliferation activity using NIH ImageJ software.
TUNEL assay
Apoptosis study was performed using fluorometric TUNEL (terminal deoxynucleotidyl transferase biotin dUTP nick end labeling) staining on 5–7 μm OCT embedded tumor sections. Data were further quantified for apoptotic activity using NIH Image J software.
Immunostimulatory efficacy of bacteriomimetic NPs
Immunostimulatory potential was evaluated by tumor quantification of CD4+ and CD8+ T-cell infiltration and IFN-γ cytokine production using Immunofluorescence staining technique.
Immunohistochemical staining of primary tumors
To check the CD4+ and CD8+ T-cell infiltration of primary tumor, 5–7 μm OCT embedded sections were stained with anti-CD4+ and anti-CD8+ alexa fluor 488-conjugated antibodies and immunofluorescence was observed using confocal microscopy. Confocal microscopy was conducted utilizing LSM 510 META (Carl Zeiss, NY, USA).
Intracellular IFN-γ staining of primary tumor
To evaluate the IFN-γ levels 5–7 μm tumor sections were permeabilized using 0.5% Triton-X and stained for IFN-γ using anti- IFN-γ antibody conjugated to Alexa fluor 488 and immunofluorescence was observed using confocal microscopy.
Statistical analysis
NP particle size characterization data was analyzed using Origin Pro 8.5 software (OriginLab, MA, USA). For evaluating biological assays GraphPad Prism 4.5 version (GraphPad Software Inc. CA, USA) was utilized. One way analysis of variance (ANOVA; p < 0.05) was used to analyze the in vivo tumor study data.
Results
Formulation of NPs
NPs were formulated employing a solvent evaporation method from a water/oil/water (w/o/w) emulsion as described in the ‘Materials & methods’ section (Figure 1A). Binding of CpG onto the surface of NPs was established using BS3 crosslinker. This approach takes advantage of the avidity of amine groups associated with the nucleobases cytosine/guanine of CpG DNA, which interact with the free carbonyl groups of BS3 to form an amide linkage (Figure 1B). We engineered three NP formulations: CpG-linked NP devoid of membrane antigen (CpG-NP-Blank), NPs encapsulating purified membrane fractions only (NP-Tag) and NPs encapsulating purified membrane fractions with CpG linked to the outer surface (CpG-NP-Tag; Table 1).
Assessment of 4T1 cell membrane fraction (Tag) purity
Membrane fractions of 4T1 cells (Tag) devoid of cytosolic protein were prepared to avoid nonspecific immune stimulation. Purity of membrane fraction was confirmed by probing for the membrane marker, Na+/K+ ATPase as well as a cytosolic marker PGK using SDS-PAGE (Figure 3A). Lane 1 shows unfractionated 4T1 cell lysate used as positive control for Na+/K+ ATPase and PGK. Lane 2 indicates purity of the membrane preparation demonstrated by the presence of Na+/K+ ATPase and the absence of PGK contamination within the membrane preparation.
Figure 3. Physicochemical characterization of nanoparticles.
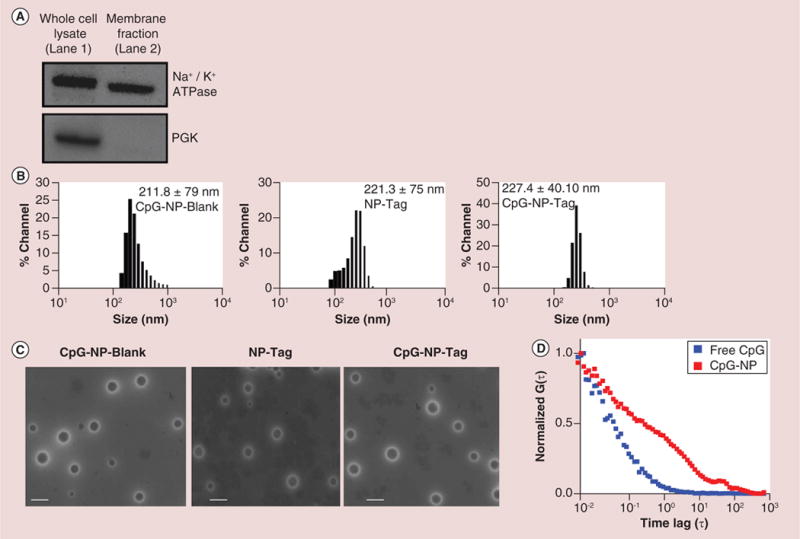
(A) Evaluation of purity of membrane preparation as a tumor antigen (Tag) determined by immunoblotting for Na+/K+ ATPase and phosphoglycerate kinase. (B) Particle size of CpG-Blank, nanoparticle (NP)-Tag and CpG-NP-Tag NPs as measured by dynamic light scattering. (C) Transmission Electron Micrographs of CpG-Blank, NP-Tag and CpG-NP-Tag NPs (Scale bar: 200 nm). (D) Fluorescence correlation spectroscopy curves of bound and free CpG.
NP: Nanoparticle; PGK: Phosphoglycerate kinase; Tag: Tumor antigen.
Surface functionalization & characterization of NPs
NP formulations were characterized for particle size, PDI, zeta potential, encapsulation efficiency, surface functionalization and surface morphology. The average particle size of the NPs was found to be in the range of 200–220 nm with PDI of 0.1 (Figure 3B & Table 1) [24]. The zeta potential was found to be slightly positive for the uncoated NP-Tag NPs as compared with CpG-Blank or CpG-NP-Tag NPs as indicated in Table 1. Encapsulation efficiency of Tag was on average 32 ± 2% with protein (Tag) loading content (PLC) of 183 μg/100 mg of NPs. Results from the TEM indicated that the particles were nonagglomerated, spherical and had a uniform size distribution (Figure 3C). Surface functionalization of NP was confirmed via Fluorescence Correlation Spectroscopy (FCS; Figure 3D). FITC-labeled CpG was used in these experiments. Figure 3D shows the FCS curves of free CpG (diffusion coefficient: 250 μM2/s) and NP bound CpG (diffusion coefficient: 3 μM2/s) clearly indicating slower diffusion of bound CpG proving successful conjugation of CpG to NPs.
Effect of NP immunization on tumor growth & morphology
Rate of tumor growth
During and at the end of the study, tumor size was measured using digital vernier caliper and bioluminiscence imaging. Fold change in tumor size was found to be significantly smaller in mice immunized with ‘bacteriomimetic’ CpG-NP-Tag NPs (test sample) as compared with mice immunized with CpG-NP-Blank NPs (control sample) (p = 0.0292) which correlated with bioluminescence intensity of imaged primary tumors (Figure 4A & Supplementary Figure 2). The bioluminescence was higher for CpG-NP-Blank (8.77 × 106 photons/s/cm2/steradian [p/s/cm2/sr]) and NP-Tag (3.809 × 106 p/s/cm2/sr) tumors as compared with CpG-NP-Tag tumors (2.058 × 106 p/s/cm2/sr). Ex vivo examination of primary tumors 14 days post-tumor challenge also demonstrated significantly smaller primary tumors in size and weight for CpG-NP-Tag immunized mice (p = 0.0133) as compared with NP-Tag and CpG-NP-Blank (Figure 4B & C).
Figure 4. Effect of nanoparticle immunization on tumor growth and morphology.
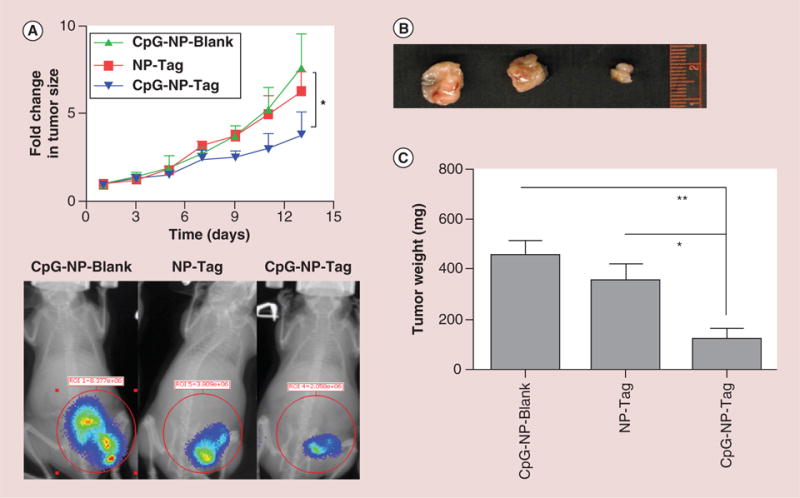
(A) Fold change in tumor volume (mm3) as compared with day 1 post-tumor challenge, in mice treated with different NP formulation and its correlation with bioluminiscence imaging tracking tumor growth at day 12 after tumor challenge (*p < 0.05). BALB/c mice were injected with d-luciferin (5 mg/kg) via intraperitoneal route 10 min before taking images using IVIS Lumina XR (Caliper Life Sciences Inc., MA, USA). (B) Tumor tissue of animals immunized with respective groups of NPs harvested on day 14 after tumor challenge. (C) Weight (mg) of isolated tumors of mice immunized with the respective groups of NPs harvested at day 14 after tumor challenge (*p < 0.05; **p < 0.01).
NP: Nanoparticle; Tag: Tumor antigen.
Gross histopathological changes in breast tumor tissue
Tumor sections were stained with H & E dyes to study the histopathology features of tumor tissues. Tumors from mice administered with CpG-NP-Blank and NP-Tag NPs (i.e., control groups) showed polymorphic nuclei, decreased cellular organization and poor differentiation as compared with tumors from CpG-NP-Tag-administered mice. Additionally, tumors from CpG-NP-Blank and NP-Tag were intensely stained for the nuclear stain, hematoxylin relative to eosin in comparison to tumors from CpG-NP-Tag-treated mice indicative of higher proliferative activity and aggressive phenotype in control groups (Figure 5 & Supplementary Figure 3) [25].
Figure 5. Hematoxylin and eosin staining of tumor tissues isolated from different groups.
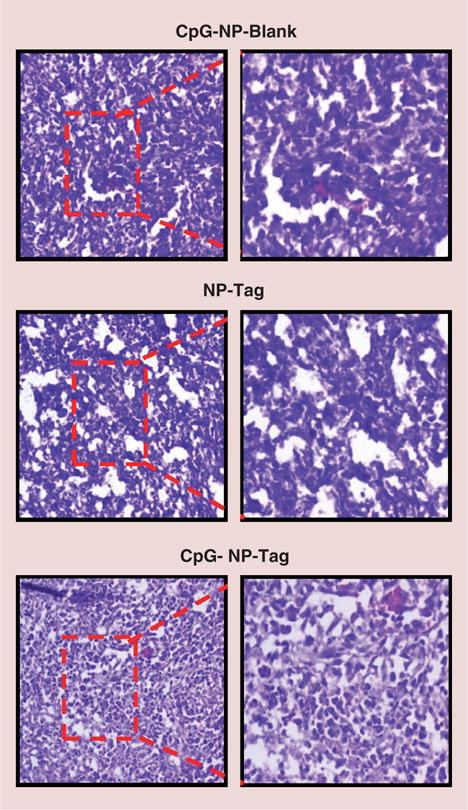
Frozen tumor tissue sections (5–8 μm) were stained with hematoxylin and eosin and images (10× and 20×) were taken using Olympus AX70 Provis microscope (NY, USA).
NP: Nanoparticle; Tag: Tumor antigen.
Proliferative activity
To assess the proliferative status of tumors, tissue sections were stained for the proliferation marker, Ki67. Quantitative evaluation of immunofluorescence indicated a higher tumor proliferation rate of tumor cells in mice immunized with CpG-NP-Blank NPs and NP-Tag as compared with mice immunized with our test sample ‘bacteriomimetic’ CpG-NP-Tag NPs (Figure 6A).
Figure 6. Effect of nanoparticle immunization on tumor cell proliferation and survival.
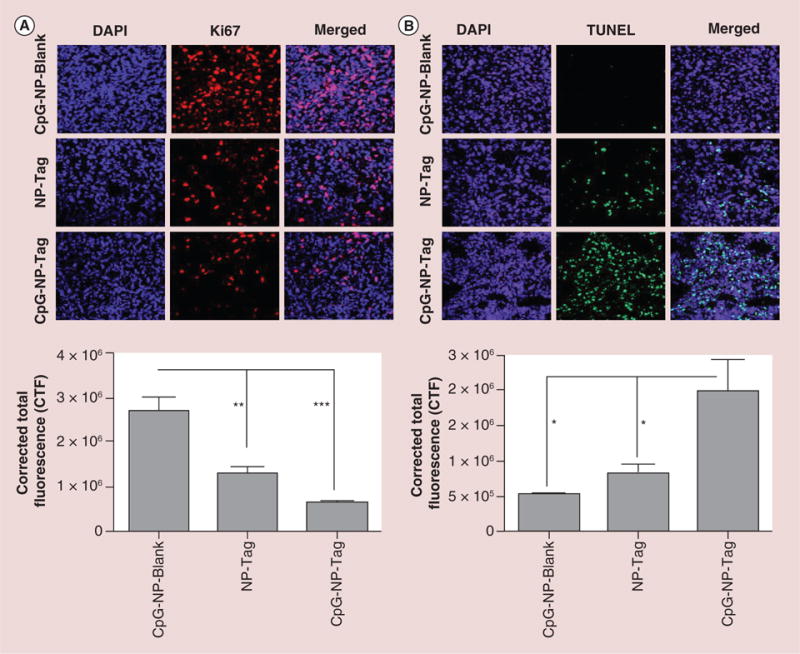
(A) Representative images (40×) of Ki67 stained tumor tissues sections showing rate of tumor proliferation and Quantitative analysis indicating proliferation rate of the tumor tissue harvested from the different groups analyzed using NIH ImageJ software (**p < 0.01; ***p < 0.001). (B) Representative images of TUNEL staining of the tumor sections showing the induction of apoptosis and Quantitative analysis of the apoptotic activity using TUNEL assay analyzed using NIH ImageJ software (*p < 0.05).
CTF: Corrected total fluorescence; DAPI: (4′,6-diamidino-2-phenylindole) fluorescent nuclear DNA stain; NP: Nanoparticle; Tag: Tumor antigen; TUNEL: terminal deoxynucleotidyltransferase (TdT)-mediated dUTP nick end labeling (TUNEL) assay.
Apoptosis assay
Cell death presumably by immune-mediated host responses due to immunization should involve tumor killing. To verify if the decrease in proliferation rate observed in CpG-NP-Tag tumors correlated with cell death, we performed TUNEL assay. The number of apoptotic nuclei was significantly higher in our test sample CpG-NP-Tag as compared with CpG-NP-Blank and NP-Tag (p < 0.05; Figure 6B). These findings demonstrate that conjugation of CpG on Tag encapsulating NPs functions in additive manner where the apoptotic effect cannot be achieved by using them individually.
Immunostimulatory efficacy of ‘bacteriomimetic’ CpG-NP-Tag NPs
CD4+ and CD8+ T cell infiltration of primary tumor
To evaluate if our test formulation CpG-NP-Tag NPs enhanced T lymphocyte infiltration, we assessed the presence of CD4+ and CD8+ T-cell infiltrates within subcutaneous tumors isolated on day 14. CpG-NP-Tag tumors demonstrated a significant infiltration of CD4+ (Figure 7A) as well as CD8+ T cells (Figure 7B) relative to tumor of controls NP-Tag and CpG-NP-Blank-treated mice (p < 0.05).
Figure 7. Immunostimulatory efficacy of ‘bacteriomimetic’ CpG-nanoparticle-tumor antigen (CpG-NP-Tag) nanoparticles.
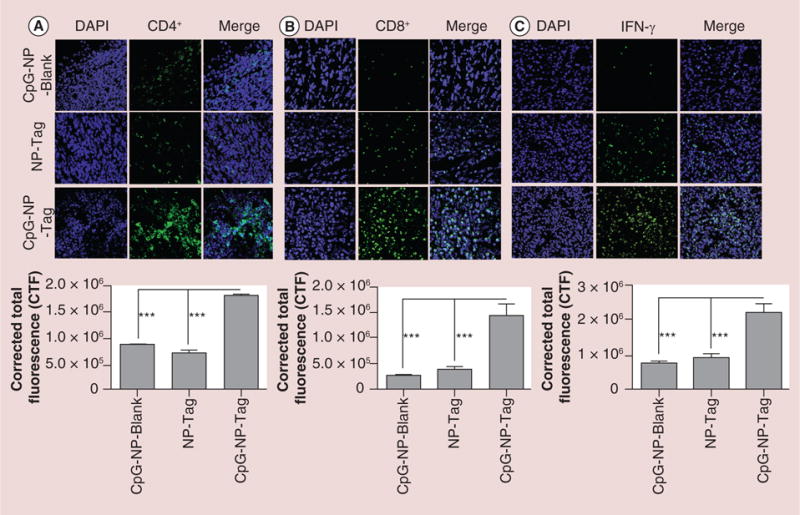
(A) Representative images (40×) of CD4+ T-cell infiltration at tumor site indicating immunostimulatory potential of CpG-NP-Tag NPs and Quantification of tumor CD4+ T-cell infiltration analyzed using NIH ImageJ software (***p < 0.001). (B) Representative images of CD8+ T-cell infiltration at tumor site indicating immunostimulatory potential of CpG-NP-Tag NPs and quantification of tumor CD8+ T-cell infiltration analyzed using NIH ImageJ software (***p < 0.001). (C) Representative images of IFN-γ intracellular staining at tumor site and quantification of tumor IFN-γ levels analyzed using NIH ImageJ software (***p < 0.001; ****p < 0.0001).
CpG: Cytosine-phosphate-guanosine oligodeoxynucletide; CTF: Corrected total fluorescence; DAPI: (4′,6-diamidino-2-phenylindole) fluorescent nuclear DNA stain; IFN: Interferon; NP: Nanoparticle; Tag: Tumor antigen.
IFN-γ in primary tumor tissue
IFN-γ is an essential cytokine mediator of immune cell-mediated tumor cell cytotoxicity [26–28]. We investigated the presence of IFN-γ by intracellular staining of tissue sections. CpG-NP-Tag tumors demonstrated higher levels of IFN-γ as compared with NP-Tag and CpG-NP-Blank tumors (p < 0.05; Figure 7C).
Discussion
Deficits in immune surveillance against cancer caused by inherent tumor escape mechanisms or by toxicities due to cancer treatment (e.g., chemoradiotherapy) have raised interests in developing novel immune-based therapies. Most notable are tumor antigen-based cancer vaccines employed to trigger tumor-specific immune responses [29,30]. Such vaccines have taken advantage of nanotechnology to produce nanoparticulate delivery systems capable of anchoring vaccine components including TAAs, tumor lysates, plasmid DNA and immune stimulants such as Toll-like receptor agonists to strengthen immune responses against various tumor malignances [31].
NP technology has advanced drug delivery by providing enhanced stability and protection of target antigens from proteolytic degradation. In addition, NPs offers the advantage of co-delivery of antigen along with immune adjuvants. This benefits optimal induction of immune responses by simultaneous targeted delivery to APCs, which is paramount for effective induction of tumor-specific immunity [32–34]. Studies conducted by Blander and Medzhitov, and Schlosser et al. emphasize the importance of codelivery in improving vaccine efficacy [34,35]. The submicron size of NPs offers many benefits over microparticles including higher uptake, increased surface area-to-volume ratio and decreased clearance of particulate vaccines by the reticular system that would negatively impact immune recognition [31]. Additionally, it has been reported that co-delivery of adjuvant moieties such as CpG and antigen (on separate NPs) enhances dendritic cell (DC) cross-presentation compared with free adjuvant [36]. In contrast, we have utilized PLGA-based NP system for codelivery of CpG and tumor antigen (on the same NP) to test the immunostimulatory potential of Tag-encapsulated CpG surface-functionalized (CpG-NP-Tag) NPs in a prophylactic murine breast cancer model.
‘Bacteriomimetic’ (CpG-NP-Tag) NPs were engineered by decorating the surface of NPs with a bacterial ligand CpG, a commonly used adjuvant in cancer vaccines (Figure 1A). CpG surface functionalization was confirmed using FCS (Figure 3D). FCS autocorrelation function analyzes translational diffusion of a fluorescent species through a confocal volume of very dilute sample (nanomolar concentration). It is understood that small molecular size biomolecules will diffuse faster than high molecular weight biomolecules. Hence we hypothesized that free CpG will diffuse faster and will have higher diffusion coefficient than when it is bound to a NP. We observed that free CpG has diffusion coefficient of about 250 μM2/s and bound CpG has 3 μM2/s, showing successful conjugation to NP surface.
Electric charge properties influence cellular interactions as well as NP stability. We observed that CpG-NP-Tag and CpG-NP-Blank particles were slightly negatively charged as compared with uncoated NP-Tag particles (Table 1). The shift toward negative charge is due to surface coating with negatively charged CpG DNA. The negative shift is beneficial for the internalization of the NPs by APCs while neutral or positive charge is not favorable for phagocytic or endocytic activity [19]. Studies conducted by Kasturi et al. suggest that NPs up to 300 nm are capable of inducing a potent T-cell-mediated (CD4+ and CD8+) immune response and protect mice against influenza infection [37]. It is also known that particles less than 500 nm are efficiently endocytosed by DCs [31] either actively (>200 nm) or passively (≤200 nm), whereas the larger particles (≥500 nm) are preferentially cleared by macrophages of reticuloendothelial system (RES) [24]. Particle size of CpG-NP-Tag NPs was in the range of 200–220 nm (Figure 3B) and PDI of 0.03 indicating monodispersity of the NP formulation. Encapsulation efficiency was found to be approximately 32%. For hydrophilic drugs, peptide or protein encapsulation efficiency is relatively lower as compared with hydrophobic substances in PLGA matrix. Specifically, encapsulation efficiency of hydrophilic species lies in the range of 30–60% compared with a range of 80–90% encapsulation efficiency of hydrophobic species [38]. Taking into consideration that co-encapsulation of Tag and CpG might compromise the encapsulation efficiency of both components; we prepared surface functionalized activated NPs using BS3 cross-linker for CpG ligand attachment.
CpG-ODNs are known to generate antigen-specific memory immune responses [10]. In our studies we found that administration of CpG alone prior to a single CpG-NP-Tag immunization was conducive for promoting an effective immune response leading to slower tumor progression. In contrast, multiple CpG-NP-Tag administration typical of traditional immunization protocols demonstrated significantly faster tumor growth compared with our method of immunization (data not shown). This finding suggests that exposure of CpG combined with NP-Tag could promote immune-tolerogenic responses and that priming with CpG alone followed by administration of a booster dose of the ‘bacteriomimetic’ NPs construct is optimal for inducing anti-tumor immune responses. Previous reports show that CpG-ODN as a single agent is also efficient in reducing progression of tumors [10,39]. However, in those studies the CpG-ODN was given repeatedly over the duration of tumor growth monitoring. Our study shows that a single dose of CpG-ODN followed by the administration of a booster dose of CpG-NP-Tag NPs before tumor challenge can significantly reduce tumor progression. Whereas administration of a CpG predose followed by booster dose of CpG-NP-Blank, does not provide the same response (Figure 4A). This suggests that the effect seen in CpG-NP-Tag mice is not only due to CpG, but also the Tag encapsulated in NPs leading to tumor-specific immune response. We also monitored the weight of the animals after CpG preimmunization, NP immunization, post tumor challenge until day 14. We found that the weight of the animals was not affected significantly over the entire duration of the study indicating that the CpG preimmunization as well as NP immunization was safe and did not lead to any adverse effects or toxicities (Supplementary Figure 4). Based on our findings, it will be prudent to understand how various vaccination models using various NPs vaccine constructs provide optimal tumor defenses.
To avoid nonspecific immune stimulation, we used 4T1 mouse carcinoma cells for preparation of Tag instead of human cancer cell line depicting a syngeneic BALB/c murine model as summarized in Figure 2. The growth of subcutaneous 4T1 tumors was monitored over time. In vivo tumor growth rate was significantly (p < 0.05) decreased in CpG-NP-Tag immunized mice than CpG-NP-Blank mice (Figure 4A). Post tumor harvest, it was evident that CpG-NP-Tag tumors were significantly smaller in size as well as weight than respective control groups (Figure 4B & C). Further validation by H & E staining showed CpG-NP-Tag treated mice tumors were well differentiated and less aggressive (less nuclear staining; Figure 5). To confirm this, tumor sections were stained for the proliferation marker, Ki67. As expected, tumors from CpG-NP-Tag group showed significantly less proliferative activity (Figure 6A). Furthermore, the decreased proliferation of CpG-NP-Tag tumors may be a consequence of cell death via apoptosis. We performed TUNEL assay on tumor tissue sections and found that CpG-NP-Tag NPs induce apoptotic tumor cell death (Figure 6B). Thus, CpG-NP-Tag NPs were found to be effective in attenuating tumor growth by inhibiting the proliferation and inducing apoptotic death of tumor cells.
Considering the importance of cell-mediated immunity in anti-tumor defense mechanisms, vaccine strategies aim toward activating tumor-specific CD8+ T cells (CTLs). A number of studies also substantiate the central role of CD4+ T cells in mounting an effective immune response [40,41]. As helper T cells, CD4+ T cells contribute to anti-tumor activity by releasing a range of cytokines such as IL-2 and IFN-γ and hence are critical in optimal activation and priming of CTLs. Furthermore, CD4+ T cells are known to help in clonal expansion of CTLs and thus help in maintenance of immune memory leading to prolonged tumor protection [40,41]. Not only overall production of CD4+ cells, but also its infiltration at the site of disease is of importance. The cytokines produced by CD4+ cells help in potentiation of cytotoxic effect and increases survival of CD8+ cells [41–44]. Uptake of NPs by receptor mediated endocytosis as well as by other mechanisms by APCs such as dendritic cells leads to both cytosolic and endosomal delivery of antigen leading to cross-presentation, capable of production of both CD4+ and CD8+ T cells [31,45]. We found that CpG-NP-Tag tumors showed higher infiltration of CD4+ (Figure 7A) and CD8+ T cells (Figure 7B) as compared with NP-Tag and CpG-NP-Blank tumors. Moreover, spleen flow cytometric analysis (see Supplementary Material section 1.3) also showed a higher CD4+ and CD8+ T-cell response in CpG-NP-Tag immunized mice (Supplementary Figure 5). We observed higher IFN-γ levels within the CpG-NP-Tag tumor tissue as compared with NP-Tag and CpG-NP-Blank tumors (Figure 7C). IFN-γ is known to induce MHC class I expression of cancer cells and thereby potentiates tumor specific immune responses [28]. Thus, CpG-NP-Tag NPs induce a potent CD4+ T cell response as well as higher levels of IFN-γ, which potentially aids in triggering a stronger CD8+ T-cell response leading to apoptotic death of tumor cells.
Ligation of the intracellular TLR-9 receptor by CpG results in the up-regulation of MHC molecules (MHC I and II), costimulatory surface molecules (CD80 and CD86) and increased cytokine production by APCs required for optimal induction of antigen-specific CD4+ and CD8+ T-cell responses [15] resulting in tumorlytic responses by CD8+ and CD4+ T cells [44]. The current study demonstrated, significantly higher infiltrating CD4+ and CD8+ T cells, as well as increased IFN-γ levels within the tumor micro-environment. These findings were associated with increased tumor cell death by apoptosis.
T-cell-mediated mechanisms are critical in vaccine-based therapies. In addition, humoral antibody responses mainly through the induction of antibody-dependent cell cytotoxicity (ADCC) are also known to mediate anti-tumor immune responses [46]. Thus, we also measured serum IgG antibody titers (see Supplementary Material section 1.4) of NP-treated tumor bearing mice. CpG-NP-Tag as well as NP-Tag mice showed relatively higher total serum IgG levels as compared with CpG-NP-Blank mice supporting the efficacy of CpG-NP-Tag NPs to trigger humoral immune responses along with cell-mediated immunity (Supplementary Figure 6). Collectively, these studies indicate that CpG-NP-Tag (bacteriomimetic) NPs have an inhibitory effect on tumor proliferation and possess immunostimulatory potential indicated by their ability to stimulate CD4+ and CD8+ T-cell-mediated response which might aid the apoptotic killing of tumor cells as well as promote a higher antibody response.
Conclusion
In this study, our goal was to engineer CpG surface functionalized Tag-containing ‘bacteriomimetic’ NPs (CpG-NP-Tag) and to investigate their immune potentiating ability against breast tumor. We formulated and characterized CpG-NP-Tag NPs and tested their immunostimulatory efficacy in vivo in a murine breast cancer model. Our results indicate that a combined approach of Tag encapsulation and CpG surface functionalization of PLGA NPs, with a predose of CpG enhances anti-tumor immunity of NPs. We believe that NP-mediated vaccination strategies could be efficiently used as adjuvant therapy along with surgery, radiation and chemotherapy [47,48]. This study also suggests that vaccination strategies whereby coating of well-known immunogenic antigens such as bacterial antigen CpG on NPs could be used to enhance anti-tumor responses. In our future studies, we plan to delineate the mechanistic details involved in enhancement of anti-tumor immunity imparted by the ‘bacteriomimetic’ NPs. Further validation of this system in a therapeutic model and delineation of anti-tumor mechanisms associated with NP-based delivery approaches will be of translational significance for cancer immunotherapy.
Future perspective
Targeted immunotherapeutic approaches to combat cancer have gained considerable importance in the past decade. Particulate cancer vaccines based on tumor antigens and adjuvants have shown promising results by triggering robust specific anti-tumor immune responses in animal models. Such systems can be exploited easily for co-delivery of antigen as well as adjuvant to facilitate activation and maturation of APCs. Additionally, the surface of such vaccine carriers can be modified with immune adjuvants or APC-targeting ligands for enhanced immune response. In the present study we have shown that surface functionalization of tumor antigen encapsulating PLGA NPs with bacterial immune stimulant CpG enhances the immunostimulatory efficacy of these particles. Proof-of-concept studies from this paper will aid to development of nanoparticulate immunotherapy agents for prophylactic as well as therapeutic applications. The use of CpG coated tumor antigen-containing ‘bacteriomimetic’ NPs will serve as a novel technique to evoke a dual immune response – nonspecific immune stimulation by the use of common bacterial/viral antigens such as CpG/hemagglutinin (HA) peptide and Tag-specific T-cell-mediated response eventually providing a robust and a long-lasting immune response. Thus, ‘bacteriomimetic’ NPs will serve as a platform for the development of immune-based therapeutic vaccines in future which could be efficiently used as an adjuvant therapy along with surgery, radiation and chemotherapy. Validation of this system in mice model will be of translational importance for breast cancer therapy as well as other cancer models. Recent advances in molecular characterization of tumor for personalized therapy can help to identify a particular TAA in patients. Such information about antigens and proposed model of bacteriomimetic drug delivery system can be implemented for personalized vaccination/immune therapy. Such technology can be readily translated into humans by using a commonly used vaccine component, such as flu vaccine, on the surface of NPs encapsulating established TAAs to produce strong immune response against tumor. In the future this strategy could be extended to other models such as infectious/viral diseases including HIV. Understanding the mechanism through which these NPs work could provide more insight in the area of such nanocarrier vaccines. Our laboratory is currently conducting immunology-based functional assays to further investigate the mechanism behind enhanced anti-tumor efficacy of the ‘bacteriomimetic’ NPs.
Supplementary Material
Executive summary.
Biodegradable NPs as vaccine delivery systems
US FDA-approved biodegradable polymer-based nanoparticles (NPs) have been effectively used as tumor antigen (Tag) delivery vehicles to enhance anti-tumor immune response.
Surface modifications of such NPs with immunostimulatory adjuvants/ligands such as CpG-ODN (bacterial ligand) or viral hemagglutinin (HA) peptide can enhance the immune-potentiating ability of such NPs.
Formulation of ‘bacteriomimetic’ NPs
Poly(D,L-lactide-co-glycolide) (PLGA)-based CpG surface-functionalized Tag-encapsulated (CpG-NP-Tag) ‘bacteriomimetic’ NPs can be formulated successfully using w/o/w double emulsion technique employing BS3 as chemical crosslinker to dock CpG ligand on the surface of the NP.
Effect of CpG-NP-Tag immunization on breast tumor growth
CpG-NP-Tag NP immunization following CpG preimmunization (priming) significantly decreases tumor growth rate in a prophylactic syngenic BALB/c mice model of breast cancer by inhibiting the proliferation and inducing apoptotic death of cancer cells.
Validation of this system in prophylactic setting can serve as a platform for future NP-based cancer vaccines.
Immune potentiating ability of CpG-NP-Tag NPs
CpG-NP-Tag immunization increases tumor infiltration of both CD4+ as well as CD8+ T cell.
CpG-NP-Tag immunization increases the levels of pro-inflammatory cytokine, IFN-γ in the tumor microenvironment.
CpG-NP-Tag NPs potentiate anti-tumor immunity by improving cell mediated (CD4+ and CD8+) T-cell response in IFN-γ dependent manner.
Conclusion
Surface functionalization of Tag containing NPs with CpG-ODN enhances the immunostimulatory efficacy and anti-tumor immunity.
CpG-NP-Tag increases production and tumor infiltration of CD4+ and CD8+ T cell.
CpG-NP-Tag treatment controls tumor growth and induces apoptosis in mice model of breast cancer.
Acknowledgments
The authors would like to thank Xiangle Sun from core facility, and Alakananda Basu, Dong-Ming Su and Ryan Rich for their technical help.
Financial & competing interests disclosure
This research was supported in part by National Institute on Minority Health and Health Disparities Grant 1P20 MD006882 (to JK Vishwanatha), Department of Molecular and Medical Genetics at UNTHSC and Predoctoral Fellowship (BC093521) from DOD Breast Cancer Research Program (SI Thamake).
Footnotes
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
References
Papers of special note have been highlighted as:
• of interest
- 1.Gregory AE, Titball R, Williamson D. Vaccine delivery using nanoparticles. Front Cell Infect Microbiol. 2013;3:13. doi: 10.3389/fcimb.2013.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li AV, Moon JJ, Abraham W, et al. Generation of effector memory T cell-based mucosal and systemic immunity with pulmonary nanoparticle vaccination. Sci Transl Med. 2013;5(204):204ra130. doi: 10.1126/scitranslmed.3006516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blattman JN, Greenberg PD. Cancer immunotherapy: a treatment for the masses. Science. 2004;305(5681):200–205. doi: 10.1126/science.1100369. [DOI] [PubMed] [Google Scholar]
- 4.Krishnamachari Y, Geary SM, Lemke CD, et al. Nanoparticle delivery systems in cancer vaccines. Pharm Res. 2011;28(2):215–236. doi: 10.1007/s11095-010-0241-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sahoo SK, Labhasetwar V. Biodegradable plga/pla nanoparticles for anti-cancer therapy (chapter 14) In: Amiji MM, editor. Nanotechnology For Cancer Therapy. CRC Press Taylor and Francis Group; FL, USA: 2006. pp. 243–249. [Google Scholar]
- 6•.Schnurr M, Galambos P, Scholz C, et al. Tumor cell lysate-pulsed human dendritic cells induce a T-cell response against pancreatic carcinoma cells: an in vitro model for the assessment of tumor vaccines. Cancer Res. 2001;61(17):6445–6450. Valuable information on designing experiments for establishing an in vitro model for cancer vaccination studies provided in this particular research article. [PubMed] [Google Scholar]
- 7•.Jaini R, Kesaraju P, Johnson JM, Altuntas CZ, Jane-Wit D, Tuohy VK. An autoimmune-mediated strategy for prophylactic breast cancer vaccination. Nat Med. 2010;16(7):799–803. doi: 10.1038/nm.2161. Emphasizes the importance of prophylactic breast cancer vaccination, the model employed in the current study and how unique tumor antigens could be utilized for designing such vaccines. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Machluf N, Arnon R. Therapeutic MUC1-based cancer vaccine expressed in flagella – efficacy in an aggressive model of breast cancer. World J Vaccines. 2012;2(3):109–120. [Google Scholar]
- 9.Fioretti D, Iurescia S, Fazio VM, Rinaldi M. DNA vaccines: developing new strategies against cancer. J Biomed Biotechnol. 2010;2010:174378. doi: 10.1155/2010/174378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10•.Baines J, Celis E. Immune-mediated tumor regression induced by CpG-containing oligodeoxynucleotides. Clin Cancer Res. 2003;9(7):2693–2700. Focuses on anti-tumor effects of CpG, a TLR-9 agonist, which is widely used as an immune stimulant in cancer vaccines and is also used in the current study as an immune adjuvant to decorate the surface of NPs, thereby bestowing bacteriomimetic nature to the CpG-NP-Tag NPs. [PubMed] [Google Scholar]
- 11.Carpentier A, Laigle-Donadey F, Zohar S, et al. Phase 1 trial of a CpG oligodeoxynucleotide for patients with recurrent glioblastoma. Neuro Oncol. 2006;8(1):60–66. doi: 10.1215/S1522851705000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mangsbo SM, Ninalga C, Essand M, Loskog A, Tötterman TH. CpG therapy is superior to BCG in an orthotopic bladder cancer model and generates CD4 T-cell immunity. J Immunother. 2008;31(1):34–42. doi: 10.1097/CJI.0b013e3181587d29. [DOI] [PubMed] [Google Scholar]
- 13.Nikitczuk KP, Schloss RS, Yarmush ML, Lattime EC. PLGA-polymer encapsulating tumor antigen and CpG DNA administered into the tumor microenvironment elicits a systemic antigen-specific IFN-γ response and enhances survival. J Cancer Ther. 2013;4(1):280–290. doi: 10.4236/jct.2013.41035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klippstein R, Pozo D. Nanotechnology-based manipulation of dendritic cells for enhanced immunotherapy strategies. Nanomedicine. 2010;6(4):523–529. doi: 10.1016/j.nano.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 15.Bode C, Zhao G, Steinhagen F, Kinjo T, Klinman DM. CpG DNA as a vaccine adjuvant. Expert Rev Vaccines. 2011;10(4):499–511. doi: 10.1586/erv.10.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mueller M, Reichardt W, Koerner J, Groettrup M. Coencapsulation of tumor lysate and CpG-ODN in PLGA-microspheres enables successful immunotherapy of prostate carcinoma in TRAMP mice. J Control Release. 2012;162(1):159–166. doi: 10.1016/j.jconrel.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 17.Wang W, Singh M. Selection of adjuvants for enhanced vaccine potency. World J Vaccines. 2011;1(2):33–78. [Google Scholar]
- 18.Abbas AK, Lichtman AH, Pillai S. Immunity to tumors (Chapter 17) In: Schmitt B, editor. Cellular and Molecular Immunology. 7th. Elsevier/Saunders Co; MO, USA: 2011. pp. 391–395. [Google Scholar]
- 19•.Thamake SI, Raut SL, Ranjan AP, Gryczynski Z, Vishwanatha JK. Surface functionalization of PLGA nanoparticles by non-covalent insertion of a homobifunctional spacer for active targeting in cancer therapy. Nanotechnology. 2011;22(3):035101. doi: 10.1088/0957-4484/22/3/035101. Provides in-depth information and explains the chemistry of ligand binding on surface of PLGA nanoparticles using BS3 cross-linker, employed to formulate bacteriomimetic CpG-NP-Tag NPs. [DOI] [PubMed] [Google Scholar]
- 20.Ranjan AP, Zeglam K, Mukerjee A, Thamake S, Vishwanatha JK. A sustained release formulation of chitosan modified PLCL: poloxamer blend nanoparticles loaded with optical agent for animal imaging. Nanotechnology. 2011;22(29):295104. doi: 10.1088/0957-4484/22/29/295104. [DOI] [PubMed] [Google Scholar]
- 21.Thamake SI, Raut SL, Gryczynski Z, Ranjan AP, Vishwanatha JK. Alendronate coated poly-lactic-co-glycolic acid (PLGA) nanoparticles for active targeting of metastatic breast cancer. Biomaterials. 2012;33(29):7164–7173. doi: 10.1016/j.biomaterials.2012.06.026. [DOI] [PubMed] [Google Scholar]
- 22•.Prasad S, Cody V, Saucier-Sawyer JK, et al. Optimization of stability, encapsulation, release, and cross-priming of tumor antigen-containing PLGA nanoparticles. Pharm Res. 2012;29(9):2565–2577. doi: 10.1007/s11095-012-0787-4. Elaborately explains the methods used for the physicochemical characterization of Tag encapsulating NPs which were extensively referred to characterize the bacteriomimetic NPs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feldman JP. Quantitative methods inquires: a mathematical model for tumor volume evaluation using two-dimensions. J Appl Quant Methods. 2009;4(4):455–462. [Google Scholar]
- 24.Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Nanoparticles target distinct dendritic cell populations according to their size. Eur J Immunol. 2008;38(5):1404–1413. doi: 10.1002/eji.200737984. [DOI] [PubMed] [Google Scholar]
- 25.Basavanhally A, Ganesan S, Feldman M, et al. Multi-field-of-view framework for distinguishing tumor grade in ER breast cancer from entire histopathology slides. IEEE Trans Biomed Eng. 2013;60(8):2089–2099. doi: 10.1109/TBME.2013.2245129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belardelli F, Ferrantini M, Proietti E, Kirkwood JM. Interferon-alpha in tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002;13(2):119–134. doi: 10.1016/s1359-6101(01)00022-3. [DOI] [PubMed] [Google Scholar]
- 27.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6(11):836–848. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 28.Martini M, Testi MG, Pasetto M, et al. IFN-γ-mediated upmodulation of MHC class I expression activates tumor-specific immune response in a mouse model of prostate cancer. Vaccine. 2010;28(20):3548–3557. doi: 10.1016/j.vaccine.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 29.Erica Jackson, Hatem Soliman. Realizing the promise of breast cancer vaccines. Vaccine: Dev Ther. 2012;2:35–41. [Google Scholar]
- 30.Buonaguro L, Petrizzo A, Tornesello ML, Buonaguro FM. Translating tumor antigens into cancer vaccines. Clin Vaccine Immunol. 2011;18(1):23–34. doi: 10.1128/CVI.00286-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31•.Akagi T, Baba M, Akashi M. Biodegradable nanoparticles as vaccine adjuvants and delivery systems: regulation of immune responses by nanoparticle-based vaccine. In: Kunugi S, Yamaoka T, editors. Advanced Polymer Science (247) Polymers in Nanomedicine. Springer-Verlag Berlin Heidelberg; London, New York, USA: 2012. pp. 31–64. Excellent book chapter about NP-based cancer vaccines providing immense information about how the biodegardable NPs could be exploited as a delivery route in immunotherapeutic intervention. [Google Scholar]
- 32•.Prasad S, Cody V, Saucier-Sawyer JK, et al. Polymer nanoparticles containing tumor lysates as antigen delivery vehicles for dendritic cell-based anti-tumor immunotherapy. Nanomedicine. 2011;7(1):1–10. doi: 10.1016/j.nano.2010.07.002. Illustrates how polymer nanoparticles containing crude tumor antigens such as tumor cell lysates could be used for cancer immunotherapy thus providing an insight that tumor cell membrane fractions (used in the current study) could also be employed in similar fashion with slight modification for an enhanced immune response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamdy S, Haddadi A, Hung RW, Lavasanifar A. Targeting dendritic cells with nano-particulate PLGA cancer vaccine formulations. Adv Drug Deliv Rev. 2011;63(10):943–955. doi: 10.1016/j.addr.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 34.Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440(7085):808–812. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 35.Fischer S, Schlosser E, Mueller M, et al. Concomitant delivery of a CTL-restricted peptide antigen and CpG ODN by PLGA microparticles induces cellular immune response. J Drug Target. 2009;17(8):652–661. doi: 10.1080/10611860903119656. [DOI] [PubMed] [Google Scholar]
- 36.de Titta A, Ballester M, Julier Z, et al. Nanoparticle conjugation of CpG enhances adjuvancy for cellular immunity and memory recall at low dose. Proc Natl Acad Sci USA. 2013;110(49):19902–19907. doi: 10.1073/pnas.1313152110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kasturi SP, Skountzou I, Albrecht RA, et al. Programming the magnitude and persistence of antibody responses with innate immunity. Nature. 2011;470(7335):543–547. doi: 10.1038/nature09737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pistel K, Kissel T. Effects of salt addition on the microencapsulation of proteins using W/O/W double emulsion technique. J Microencapsul. 2000;17(4):467–483. doi: 10.1080/026520400405723. [DOI] [PubMed] [Google Scholar]
- 39.Gnjatic S, Sawhney NB, Bhardwaj N. TLR agonists: are they good adjuvants? Cancer J. 2009;16(4):382–391. doi: 10.1097/PPO.0b013e3181eaca65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ding Z, Zhou G. Cytotoxic chemotherapy and CD4. Clin Dev Immunol. 2012;2012 doi: 10.1155/2012/890178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ostrand-Rosenberg S. CD4 T lymphocytes: a critical component of anti-tumor immunity. Cancer Invest. 2005;23(5):413–419. [PubMed] [Google Scholar]
- 42•.Melief CJ. ‘License to kill’ reflects joint action of CD4 and CD8 T cells. Clin Cancer Res. 2013;19(16):4295–4296. doi: 10.1158/1078-0432.CCR-13-1432. This translational communication in Clinical Cancer Research emphasizes the fact that epitopes generating both ‘helper’ CD4 T-cell response as well as CTL response simultaneously are more beneficial for producing a robust cell-mediated anti-tumor response rather than single CTL response. [DOI] [PubMed] [Google Scholar]
- 43.Yo-Ping L, Chung-Jiuan J, Shu-Ching C. The roles of CD4 T cells in tumor immunity. ISRN Immunology. 2011;2011:1–6. [Google Scholar]
- 44.Zhang S, Zhang H, Zhao J. The role of CD4 T cell help for CD8 CTL activation. Biochem Biophys Res Commun. 2009;384(4):405–408. doi: 10.1016/j.bbrc.2009.04.134. [DOI] [PubMed] [Google Scholar]
- 45.Yu MK, Park J, Jon S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics. 2012;2(1):3. doi: 10.7150/thno.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dermime S, Armstrong A, Hawkins RE, Stern PL. Cancer vaccines and immunotherapy. Br Med Bull. 2002;62(1):149–162. doi: 10.1093/bmb/62.1.149. [DOI] [PubMed] [Google Scholar]
- 47.Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. 2013;105(4):256–265. doi: 10.1093/jnci/djs629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamby LS, McGrath PC. Improved survival with adjuvant immunotherapy after surgical resection in a murine model. Ann Surg Oncol. 1994;1(4):307–313. doi: 10.1007/BF02303569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.