

Abstract
Pro-inflammatory cytokines tumor necrosis factor α (TNF-α) and Interleukin 6 (IL-6) are mediators in the development of many inflammatory diseases. To demonstrate that macrophages take up and respond to thermosensitive nanoparticle drug carriers we synthesized PEGylated poly(N-isopropylacrylamide–2-acrylamido-2-methyl-1-propanesulfonate) particles cross-linked with degradable disulfide (N,N′-bis(acryloyl)cystamine) (NGPEGSS). An anti-inflammatory peptide (KAFAK) was loaded and released from the thermosensitive nanoparticles and shown to suppress levels of TNF-α and IL-6 production in monocytes. Cellular uptake of fluorescent thermosensitive degradable nanoparticles and therapeutic efficacy of free KAFAK peptide compared to KAFAK loaded in PEGylated degradable thermosensitive nanoparticles was examined. The data suggests that the degradable, thermosensitive nanoparticles loaded with KAFAK may be an effective tool to treat inflammatory diseases.
Keywords: N-isopropylacrylamide, Thermosensitive polymer, Anti-inflammatory peptides, Inflammation
Graphical Abstract
INTRODUCTION
Immune cells constitute key players in the development of many inflammatory diseases such as rheumatoid arthritis1, ulcerative colitis2, atherosclerosis3, ischemia reperfusion injury4, transplantation rejection5, 6 and diabetes7. Immune cells primarily serve to protect against opportunistic infections, but when they become activated inappropriately, they can participate in the development of inflammatory diseases8, 9. During progression of these diseases, immune cells such as macrophages may release cytokines (interleukin-1 [IL-1], interleukin-6 [IL-6], tumor necrosis factor-alpha [TNF-α]), chemokines (e.g., monocyte chemotactic protein-1 [MCP-1]), digestive enzymes (e.g., collagenases), prostaglandins, and reactive oxygen species (ROS), which can aggravate or accelerate damage to the normal tissues10–12.
Treatment of inflammatory disorders using peptide drugs may be effective, by suppressing cell-mediated immunity and promoting anti-inflammatory effects13–15. However, gastric acid in the stomach and peptidases in the blood easily breakdown peptides into both fragments and single amino acids resulting in poor bioavailability of peptide16, 17. Other challenges, including potential peptide antigenicity may result in unwanted immune responses and limit the commercial applications of these drugs18, 19. Our laboratory previously developed the anti-inflammatory cell-penetrating peptide sequence KAFAKLAARLYRKALARQLGVAA (abbreviated KAFAK) that targets mitogen-activated protein kinase activate protein kinase 2 (MK2) to reduce expression of pro-inflammatory cytokines14. MK2, a member of the p38 mitogen-activated protein kinase pathway, controls activation of transcription factors that affect the stability of cytokine AU-rich mRNA and upregulates many pro-inflammatory cytokines such as, TNF-α and IL-620–22. However, in order to improve the efficacy and retention time of the peptide drug, alternative delivery approaches must be sought. For this purpose, we designed a biocompatible polymer as a drug carrier to protect the peptide from enzymatic degradation, thus increasing the half-life of the peptide therapeutic for diagnostics and treatment.
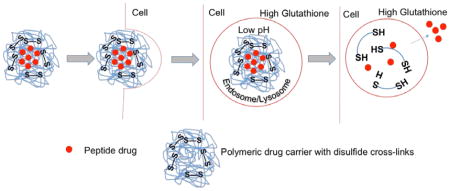
In recent years, there has been significant growth of basic and applied research in the area of polymers as drug carrier systems. There is increasing optimism that appropriate design of nanoparticle systems will bring advances in the diagnosis and treatment of disease23. Poly(N-isopropylacrylamide) (pNIPAm) is a thermosensitive polymer, which exhibits a lower critical solution temperature (LCST) at ~ 31–33°C24–26. Below the LCST, the pNIPAm backbone is hydrophilic, which results in nanoparticle swelling in aqueous environments. Above the LCST, the water is expelled as the pNIPAm collapses on itself. The LCST allows pNIPAm particles to be loaded with a drug by passive diffusion at temperatures below the LCST, to entrap the diffusible species above the LCST, and to slowly release the drug while in the collapsed state. Previous work from our lab demonstrated that incorporating sulfated monomer 2-acrylamido-2-methyl-1-propanesulfonic acid (AMPS) into the pNIPAm backbone increased loading efficiency of the positive-charged KAFAK peptide in poly(NIPAm-AMPS) nanoparticles27. To improve the stability of the nanoparticles, modification was used to render stealth properties by addition of poly(ethylene glycol)(PEG) in one pot polymerization. The stealth character of PEG is based on its excluded volume, or steric repulsion properties; the PEG segments serve to minimize the non-specific interactions with plasma protein28. An additional feature of the nanoparticle system is the incorporation of disulfide cross-links within the nanoparticle. Although stable in the oxidizing extracellular environment, once the nanoparticle is taken up by cells, the glutathione and low pH present in the intracellular compartment (endosome/lysosome) reduces the disulfide (SS) cross-links allowing nanoparticle degradation and subsequent peptide release29 (Graphical Abstract).
In this paper, we describe the synthesis and characterization of functionalized pPNIPAm-AMPS nanoparticles. Cellular uptake studies were carried out with fluorescent-labeled pNIPAm nanoparticles by the RAW 264.7 murine macrophage cell line. We examined whether using sulfated pNIPAm nanoparticles with disulfide cross-links (SS) and PEGylation (PEG) could improve the KAFAK peptide therapeutic efficacy as compared to free peptide.
EXPERIMENTAL SECTION
Materials
N-Isopropylacrylamide (≥ 98%, NIPAm) was acquired from Polysciences Inc. (Warrington, PA, USA). Dialysis membrane tubing was purchased from Spectrum Laboratories (Dominguez, CA). (1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (≥ 98%, HBTU), N-hydroxybenzotriazole (> 97%, HOBT), N,N′-methylenebisacrylamide (99%, MBA), sodium dodecyl sulfate (SDS; 10% w/v in water), 2-acrylamido-2-methyl-1-propanesulfonic acid (99%, AMPSA), dithiothreitol (98%, DTT), fluorescein o-acrylate (98%, F), N,N-Diisopropylethylamine (99%, DIPEA), potassium persulfate (99%, K2S2O8), N,N′-bis(acryloyl)cystamine (98%, BAC) and dimethyl sulfoxide (DMSO) were acquired from Sigma-Aldrich (St. Louis, MO, USA). N,O-dimethacryloyl hydroxylamine (98%, DMHA) were synthesized based on a previous protocol30. NIPAm, MBA, and AMPSA were stored under nitrogen at 4 °C. All water used in synthesis, dialysis, and testing was treated by a Milli-Q system (Millipore, Billerica, MA, USA; 18.2 MΩ·cm resistivity). Acrylate-PEG2000 was purchased from Laysan Bio (AL).
Cell Culture
RAW 264.7 cells (Murine macrophage cell line) were a gift from Dr. Philip Low’s lab - Chemistry, Purdue University. The cell lines were grown continuously as a monolayer in non-folate RPMI 1640 medium supplemented with 10% fetal bovine serum, penicillin (50 units/mL), and streptomycin (50 μg/mL) antibiotic at 37 °C maintained with 5% CO2.3
Peptide Synthesis and Purification
Therapeutic peptides (KAFAK) were synthesized on knorr resin by 9-fluorenylmethyloxycarbonyl (FMOC) solid phase methodology (see Supporting Scheme 1). Knorr amine resin was swollen with dichloromethane (DCM) followed by dimethylformamide (DMF). A solution of 25% piperidine in DMF was added to the resin, and nitrogen was bubbled for 15 min. The resin was washed with DMF (3x) and isopropyl alcohol (i-PrOH, 3x). For amino acid coupling, a solution of Fmoc-amino acid (3 equiv), (1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU, 3 equiv), N-hydroxybenzotriazole (HOBT, 3 equiv) and N,N-Diisopropylethylamine (DIPEA, 5 equiv) in DMF was added. Nitrogen was bubbled for 2 h, and resin was washed with DMF (3x), DCM (3x) and i-PrOH (3x). To deprotect Fmoc, a solution of 25% piperidine in DMF was added to the resin, and nitrogen was bubbled for 20 min, the solvent was removed and resin was washed with DMF (3x) and i-PrOH (3x). The above sequence was repeated for coupling of each amino acid. Final compound was cleaved from the resin with a cocktail of trifluoroacetic acid (Sigma-Aldrich, St. Louis, MO, U.S.A.), triisopropyl silane (TCI America, Boston, MA, U.S.A.), ethane dithiol (Alfa Asara, Ward Hill, Massachusetts, U.S.A.), and Milli-Q water. The cleaved mixture was concentrated under vacuum. The concentrated product was immediately precipitated in ether, recovered by centrifugation, solubilized in Milli-Q water, and lyophilized. Peptides were purified on an FPLC AKTA Explorer (GE Healthcare, Pittsburgh, PA, U.S.A.) with a 22/250 C18 prep-scale column (Grace Davidson, Deerfield, Illinois, U.S.A.) and an acetonitrile gradient with 0.1% trifluoroacetic acid. Purity of peptide were determined by HPLC to be > 98%. Peptide molecular weight was confirmed by matrix-assisted laser desorption ionization time-of-flight (MALDI TOF) mass spectrometry with a 4800 Plus MALDI TOF/TOF Analyzer (Applied Biosystems, Foster City, CA, U.S.A)
Preparation of nanoparticle
NIPAm-AMPS nanoparticles were synthesized as described elsewhere with slight modification27, 30, 31. Briefly, the nanoparticle compositions were formed by dissolving 192.1mg NIPAm, 2 mol% cross-linker, and 5 mol% AMPSA in 18ml degassed MilliQ water in a three-neck round bottom flask. 3 mol% PEG-acrylate and 2% cross-linker (BAC, MBA, DMHA) was pre-dissolved in 3% (v/v) of dimethyl sulfoxide (DMSO) in water for 10 mins before addition to polymer mixture. 41 μL of a 10% SDS in MilliQ water solution was added, and the mixture was heated to 65°C under nitrogen. Potassium persulfate (8.4mg) was dissolved in 2ml degassed MilliQ water and added after a 35 mins equilibration to initiate polymerization. After 5 h, the reaction was removed from heat and allowed to cool to room temperature. Particles were dialyzed against MilliQ water for 5 days using a 15,000 MWCO membrane.
For fluorescent nanoparticle synthesis, 1 mol% fluorescein o-acrylate (F) was pre-dissolved in 3% DMSO solution. The dye-DMSO solution was added to polymer mixtures and equilibrated for 30mins. Potassium persulfate dissolved in 2ml degassed MilliQ water was added to initiate polymerization. Particles were dialyzed against MilliQ water for 5 days using a 15,000 MWCO membrane. All sample were freeze-dried and kept in the dark for further experiments.
Nanoparticle Characterization
Size measurements were performed using dynamic light scattering (DLS) on a Nano-ZS90 Zetasizer (Malvern, Westborough, MA, USA). Zeta (ζ) potentials were measured at 25 °C using a Nano-ZS90 Zetasizer in capillary cells in PBS. To characterize their degradation, samples with an identical particle concentration were prepared in pH 7.4 PBS buffer. To one solution, an excess of DTT (1mM) was added and allowed to incubate for 24 h. Transmission electron microscopy (TEM) was conducted at the Purdue University Life Science Microscope facility on a FEI/Philips CM-100 transmission electron microscope at 100 kV using a uranyl acetate stain (UA) at pH 4.5. Discharged TEM sample grids were placed onto the top of a droplet of sample for 2 min. Then UA stain was added, and samples were briefly dried before imaging at room temperature.
Drug Loading and Release
Purified peptide was first dissolved in Milli-Q water to create a 1 mg/mL loading solution. Then this solution was added to 1 mg lyophilized nanoparticles. Next, the drug–nanoparticle loading solution complex was allowed to incubate for 24 h at 4 °C in the swollen state. After incubation particles underwent 1 h of centrifugation at 35 000 rpm (105 000 × g) in an Optima L-90k ultracentrifuge (Beckman Coulter, Indianapolis, IN, USA). The nanoparticle pellet was briefly resuspended in 1mL of Milli-Q water and was lyophilized. Measurement of free peptide released into the solution and the amount of peptide loaded were determined using fluorescence analysis with a fluoraldehyde o-phthalaldehyde (OPA) solution (Thermo Scientific, Waltham MA, USA). For KAFAK release studies, fluorescent measurements of drug release were taken every 15 min for the first hour, then at 1 h, 6 h, 12 h, and 24 h every day afterward for 4 days.
In Vitro Cell Imaging
For confocal microscopy studies, RAW 264.7 cells were seeded in 8 wells culture plates and maintained at 37 °C. Samples were performed in triplicate. RAW 264.7 cells incubated with fluorescein labeled nanoparticles were incubated for 4 h or 24 h at 37 °C in culture medium. Culture media used in nanoparticle experiments contained FBS (as indicated in the cell culture section) unless otherwise noted. After washing with medium to remove any unbound nanoparticles, the cells were imaged using confocal microscopy (Olympus BH-2). Cell samples were subjected to the trypan blue dye exclusion method to check cell viability. Lysotracker DND-99 (Life Technologies, Grand Island, NY, USA) staining was performed according to the manufacturer’s protocol to confirm that nanoparticles were taken up into endosomal/lysosomal intracellular compartments. Semi-quantitative fluorescence measurements were used to compare uptake between PEGylated and non-PEGylated nanoparticles. Image analysis by ImageJ determined the number of fluorescent pixels (area) and the integrated fluorescence intensity for the green channel normalized to the red channel within each field of view (n = 3).
Cytokine Analysis of In Vitro Inflammatory Model
RAW 264.7 cells were seeded at a density of 1× 104 cells/mL into 96-well plate for 4 hours. Cells were then incubated with various concentrations of KAFAK drug or KAFAK loaded nanoparticle in media with or without 10% fetal bovine serum (FBS). The amount of nanoparticles used was adjusted based on the concentration of drugs loaded, thus keeping the concentration of drugs consistent for both free drug and KAFAK loaded nanoparticles. Cells were activated with 2 ug/mL lipopolysaccharide (LPS) 12 h after cells were seeded. Control samples received PBS in place of LPS. After 24 h incubation in the cell culture incubator, the supernatant was collected and stored at −80 °C until cytokine analysis could be performed. CellTiter assays were performed according to manufacturer’s protocol to quantitatively check NP cytotoxicity (supplementary figure S3).
Pro-inflammatory cytokine TNF-α and IL-6 production was determined by sandwich immunoassay methods using commercially available electrochemiluminescent detection system, plates, and reagents (Meso-Scale Discovery (MSD), Gaithersburg, Maryland). For each assay, samples were analyzed and compared with known concentrations of protein standard based on manufacturer protocol. Plates were analyzed using the SECTOR Imager 2400.
Statistical Analysis
Student’s t-tests were used to determine statistical significance between treatment groups using a significance level of α = 0.05. Data is expressed as mean values ± standard deviation unless otherwise noted.
RESULTS
Nanoparticle Characterization and Drug Loading
Previous work from our laboratory showed that utilizing 5% AMPS co-monomer content in pNIPAm nanoparticles resulted in the maximum KAFAK loading capacity27. Incorporating 5% AMPS into our disulfide PEGylated (NGPEGSS) and non-PEGylated (NGSS) nanoparticles yielded loading efficiencies of ~31.1% and ~29.7% respectively (Table 1). The KAFAK loading efficiency of nanoparticles using 2% of MBA and DMHA cross-linker ranged from 33 – 37% (all loading capacities are statistically equivalent).
Table 1.
ζ Potential and Drug Loading
| Nanoparticle | Size (nm) ± stdev | Drug loading (%) ± stdev | Zeta potential (mV) ± stdev |
|---|---|---|---|
| NGSS | 178 ± 10.1 | 24.1 ± 4.1 | −4.56 ± 2.35 |
| NGPEGSS | 223 ± 9.7 | 34.6 ± 3.7 | −3.81 ± 2.01 |
| NGMBA | 226 ± 9.5 | 34.1 ± 4.6 | −5.57 ± 1.13 |
| NGPEGMBA | 246 ± 8.9 | 36.7 ± 4.3 | −4.28 ± 1.56 |
| NGDMHA | 217 ± 7.8 | 31.4 ± 5.3 | −7.49 ± 2.11 |
| NGPEGDMHA | 237 ± 8.2 | 34.7 ± 7.2 | −6.05 ± 1.91 |
As shown in the TEM micrographs collected from the disulfide PEGylated (NGPEGSS) and non-PEGylated (NGSS) nanoparticles (Figure 1), the nanoparticles were spherical, which is consistent with previous work with both non-degradable MBA and degradable DMHA cross-link pNIPAm nanoparticles. 27, 30 In addition, The TEM images in Figure 1 illustrate gel integrity in the absence (A and C) or degradation in the presence (B and D) of DTT for disulfide cross-linked nanoparticles. DLS measurements of the nanoparticles size confirmed that nanoparticles maintained integrity even after lyophilization from deionized Milli-Q water (Figure 2B).
Figure 1.
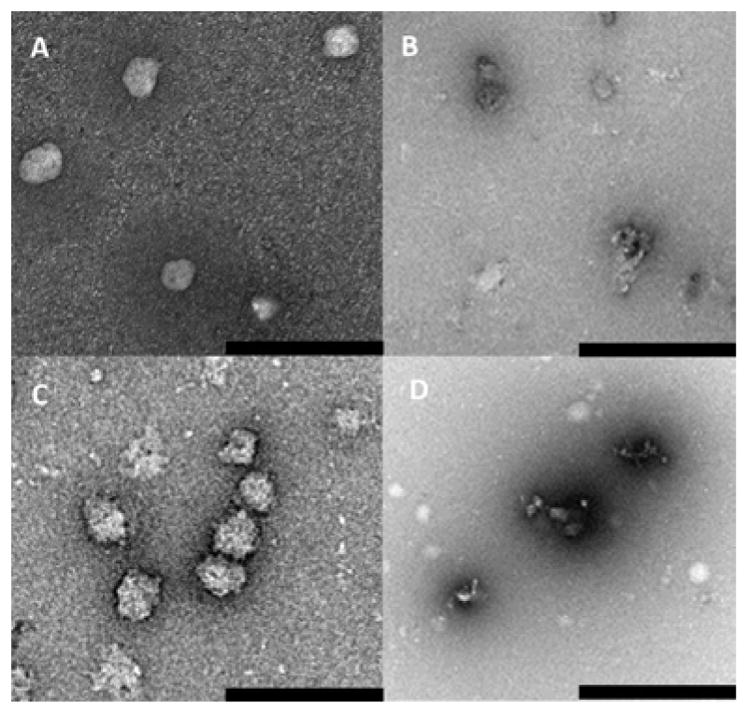
TEM micrographs of disulfide cross-linker nanoparticles containing 2 mol% BAC (SS) and 5 mol% AMPS, pH 7.4 at 25°C, after 24 h (A) NGSS; (B) NGSS, presence of DTT; (C) NGPEGSS; (D) NGPEGSS, presence of DTT. Scale bar is 1 μm.
Figure 2.

NGPEGSS degradation after 24h A) Reduction of disulfide cross-linker in the presence of DTT; B) DLS Size distribution of NGPEGSS in Milli-Q water (Red) and NGPEGSS in the presence of DTT (Green)
Following 24 h incubation in Milli-Q water, disulfide nanoparticles did not show significant signs of degradation; however, when the disulfide cross-linked nanoparticles were exposed to the reductant DTT, DLS measurements confirmed that the nanoparticles fragmented (Figure 2B), which is consistent with the TEM data shown in Figure 1B & D. As evidence of longer term stability in the absence of a reducing agent, the hydrodynamic diameter data depicted in Figure 3A showed that the PEGylated and non-PEGylated nanoparticles did not degrade or aggregate when stored at 25°C for over 2 days in Milli-Q water. The data also demonstrated a general trend of increased size with incorporation of PEG. In summary, all nanoparticles synthesized with various cross-linkers (2 mol%) maintained size stability up to 48 hours at pH 7.4 @ 25 °C and during lyophilization; only the disulfide cross-linked nanoparticles were susceptible to degradation in reducing environments.
Figure 3.


Degradation over time in different environments of PEG- and non-PEG-poly(NIPAM–AMPS) nanoparticles with various cross-linker over time measurement: (A) Nanoparticle in pH 7.4; (B) Non-PEG nanoparticle in presence of DTT; (C)PEGylated nanoparticle in presence of DTT. Bars represent average ± SEM (n = 3).
As determined using DLS, degradation of the nanoparticles occurred over time when exposed to reducing conditions (Figure 3B and 3C). The data in Figure 3B and 3C demonstrated that over a two day period, NGSS lost ~31% of their size and NGPEGSS lost ~ 32% of their size. Nanoparticles formed with 2% monomer feed of other cross-linkers, non-degradable MBA and high-pH sensitive (pH > 5.0) DMHA, were found to maintain their size in the presence of DTT with very little degradation (< 5%). Both non-degradable (MBA) and disulfide (SS) particle size remained intact when particles were exposed to conditions at pH 9.0 suggesting that little degradation occurred at basic pH, whereas high-pH sensitive degradable DMHA cross-linked nanoparticles degraded over time under basic conditions, which is consistent with previous studies (Supplement figure 1)30.
With respect to nanoparticle stability, negative surface charge (ζ potential) can prevent agglomeration. Changing cross-link type does not have a profound impact on ζ potential for particles when using 2 mol% cross-linker content as shown in Table 1. All nanoparticles possess a negative ζ potential as expected due to the incorporation of the sulfated AMPS, although the range of ζ potentials did not vary much among the different nanoparticle formulations.
Figure 4 shows DLS size measurements of disulfide nanoparticles as a function of temperature. The ability of pNIPAm-AMPS nanoparticles to respond to temperature was maintained with the incorporation of PEG and disulfide cross-links. Barlett et al.27 used NIPAm as the primary constituent of a bulk polymer of nanoparticles and found the phase transition temperature, or LCST, shifted to higher temperatures as the AMPS content of the polymer increased. However, we found that addition of PEG to the bulk nanoparticle formulation does not affect the LCST. Nanoparticles synthesized using non-degradable (MBA) cross-linker were used as a control and were shown to have a LCST consistent with literature around 31–33°C34 (data not shown). The LCSTs of the synthesized disulfide nanoparticles were in the range of 35–43°C as shown in Figure 4 demonstrating that the addition of the disulfide crosslinks shifts the LCST to slightly higher temperatures.
Figure 4.

Dynamic light scattering hydrodynamic diameter temperature sweep from 20°C to 50°C A) NGSS; B) NGPEGSS. Bars represent average ± SEM (n = 3).
Drug Release Studies
Due to the strong affinity of KAFAK for sulfated moieties as reported previously, KAFAK loading capacity and release are controlled in part by incorporation of 5% AMPS30. NIPAm was used as the polymer backbone to ensure the high molecular weight drug KAFAK was able to efficiently diffuse into the nanoparticle cores while particles were in the swollen state below the LCST, while above the LCST, at physiological conditions, the peptide remained encapsulated and protected from proteases. To test the drug delivery efficacy, release studies of KAFAK from the nanoparticles were conducted in different environments at 37°C. As shown in Figure 5, the amount of KAFAK released in a pH 7.4 environment was significantly lower than the amount of KAFAK released in a reducing environment using (Figure 5A) DTT or (Figure 5B) low pH 4.0 environment where disulfide bonds were expected to reduce and release loaded drug. After 24 hours, disulfide particles released nearly ~24% of their loaded drug in a reducing environment compared to ~7% at pH 7.4. Taken together these observations demonstrate that the loading and release mechanism is driven by both cross-link density and the electrostatic interactions between KAFAK and the copolymer. The integrity of the KAFAK following release from disulfide-cross-linked nanoparticles was confirmed by MALDI TOF MS (data not shown).
Figure 5.


Drug release profiles of KAFAK containing particles from NGSS and NGPEGSS in pH 4.0 and pH 7.4 over a 96 h period from at 37 °C. Bars represent average ± SEM (n = 3)
Cellular Uptake of Fluorescent Nanoparticles
To investigate cellular uptake and toxicity of disulfide nanoparticles, fluorescein-labeled disulfide nanoparticles (NGSSF and NGPEGSSF) were synthesized. An intracellular uptake study was performed with the fluorescently labeled disulfide nanoparticles (Figure 6). The uptake and localization of the particles in RAW 264.7 cells were visualized by confocal laser scanning microscope after 24 h of incubation with particles (Figure 7III). The internalization of the particles is illustrated in Figure. 7 AIII and BIII. The optical sections (z-plane) depicted in Figure 7 represent a plane of focus interior to the cell membrane demonstrating that the nanoparticles have been internalized rather than accumulated in or at the cell membrane. The fluorescein-labeled nanoparticles were taken up and were distributed throughout the cell within 24 hours of exposure, but no uptake was observable in the first 4 hours. However, similar to previous reports35 cellular uptake of small molecule fluorescein was observed within 2 hours of incubation (Supplement Figure 2), which suggests that the fluorescence observed in our uptake study is due to labeled particles and not from potentially unreacted fluorophore since unreacted fluorophore should be observable at earlier time points. No evidence of localization within the cell nuclei was observed. The punctate staining seen in Figure 7 AIII and BIII, suggests that particles accumulated into the endosomes/lysosomes, but it is also possible that further agglomeration occurred intracellularly. This was verified by using a red lysotracker probe to confirm co-localization of labeled nanoparticles in endosomes/lysosomes (Figure 7C,D). Semi-quantitative measurements of fluorescence intensity comparing green fluorescent pixel area normalized to red fluorescent pixel area showed that there was a statistically significant difference (p < 0.01) in the amount of co-localization for PEGylated compared to non-PEGylated nanoparticles with PEGylated nanoparticles exhibiting significantly less co-localization (Supplemental Figure S4); perfect co-localization would result in a red to green fluorescence area ratio of 1. Similarly, green fluorescence intensity integration measurements normalized to red fluorescence intensity showed significantly less PEGylated compared to non-PEGylated nanoparticle uptake (p < 0.01) (Supplemental Figure S4).
Figure 6.

Fluorescence Imaging of disulfide nanoparticles: A) White light B) Fluorescence image; I) NGSS II) fluorescence NGSSF III) NGPEGSS IV) fluorescence NGPEGSSF.
Figure 7.


Confocal fluorescence microscopy of RAW 264.7 cells incubated @ 37°C with AI) Control, with non-fluorescent NGSS, AII) fluorescent NGSSF for 4 h C) AIII) fluorescent NGSSF for 24 h, BI) Control, incubated with non-fluorescent NGPEGSS, BII) fluorescent NGPEGSSF for 4 h C) AIII) fluorescent NGPEGSSF for 24 h. Cells were visualized with confocal fluorescence microscope (λex = 490nm, λem= 520nm).
Lysotracker DND-99 fluorescence indicating co-localization of fluorescent NGSSF (C III and IV) and NGPEGSSF (D III and IV) in endolysosomal compartments. (C I) NGSSF and (D I) NGPEGSSF uptake in RAW 264.7 cells. (C II) and (D II) are lysotracker labeled endolysosomes.
The cytotoxicity of the fluorescently labeled and unlabeled disulfide nanoparticles was studied in the RAW 264.7 cell line. The dye exclusion assay36 was carried out by analyzing the uptake of trypan blue, virtually all cells remain viable after incubating with NGSS and NGPEGSS for 2 days as determined by lack of trypan blue uptake by the cells. The study demonstrated that addition of PEG or disulfide into the particles does not impact macrophage viability in vitro. CellTiter assays also showed that there were no significant differences in cell number after exposure to nanoparticles (Supplemental Figure S3).
Cytokine assay
It has previously been reported that the intracellular inhibition of MK2 by a single dose of 40μM KAFAK results in suppression of pro-inflammatory cytokines TNF-α and IL-6 secretion14, 37. As shown in Figure 8, studies of RAW 264.7 murine macrophage cell line confirmed that treatment with KAFAK free peptide is serum dependent and suggests that the peptide is susceptible to enzymatic degradation. This reduces the therapeutic effectiveness in the presence of serum in vitro. KAFAK peptides released from the disulfide nanoparticles were protected from proteases and remained active in vitro. The data in supplemental Figure S3 and Figure 5 demonstrate that more KAFAK was released from degradable disulfide nanoparticle than non-degradable particles in a reducing environment. This is consistent with a low therapeutic availability seen with release from non-degradable (NGMBA and NGPEGMBA) nanoparticles as reported previously30, 37. The data in Figure 8 demonstrate that sufficient peptide could be released from the degradable particles to reduce IL-6 and TNF-α expression in vitro. In addition, the data also indicate that PEGylated nanoparticles yielded the greatest inhibition of IL-6 and TNF-α production after 24h. Levels of IL-6 were reduced by ~67% and TNF-α by ~64% for KAFAK loaded NGPEGSS, whereas ~40% and ~44% reduction respectively was observed for the non-degradable (MBA) PEG-nanoparticle counterpart.
Figure 8.

TNF-α (Top) and IL-6 (Bottom) production (pg/ml) in RAW 264.7 murine macrophage model treated with free KAFAK peptide (40 μM), KAFAK (40 μM) containing degradable nanoparticle (NGSS + KAFAK, NGPEGSS+ KAFAK) and non-degradable (NGMBA+KAFAK, NGPEGMBA + KAFAK) nanoparticle. Blank control was incubated in FBS containing media only. All other samples were treated with LPS, and FBS containing media. Bars represent average ± SD (n = 3). * denotes statistical significance (ρ < 0.05) between nanoparticle groups and free peptide treatment, between nanoparticle groups and LPS only treatment, + between disulfide crosslinker nanoparticles and non-degradable MBA crosslinker nanoparticles, and ≠ between PEGylated nanoparticles and their non-PEGylated counterparts.
DISCUSSION
KAFAK-loaded pNIPAm nanoparticles have been reported to protect KAFAK from enzymatic degradation, thus increasing drug half-life in the presence of serum14, 27. We have reported27, 30 the ability to produce nanoparticles with sizes ranging between 100 and 400 nm in solution. Nanoparticle size can be essential for cellular permeability during drug delivery. As reported previously,38, 39 particles in the size range of 160–400 nm show promise for intra-cellular delivery in inflamed tissue. With the ability to tune nanoparticles to a size range of 180–260 nm, we anticipate efficient cellular permeability of our nanoparticles. Observation by confocal microscopy showed that cellular uptake of fluorescent nanoparticles by RAW 264.7 cells occurred within 24 hours of particle exposure. Semi-quantitative fluorescence measurements of FITC nanoparticles and lysotracker red indicated that there is statistically less co-localization of PEGylated nanoparticles with endolysosomes compared to non-PEGylated particles. This suggests that the PEGylated nanoparticles may either be due to delayed endosomal trafficking, a separate mode of uptake, or increased endolysosomal escape. Further studies are needed to elucidate the mechanisms causing differences in cellular uptake of PEGylated and non-PEGylated nanoparticles.
Incorporating PEG into a nanoparticle can endow the particle with several advantages. PEG is known to be biocompatible can protect materials from unwanted adsorption and cell adhesion. Gulati et al28 reported that a longer blood circulation lifetime of PEGylated nanoparticles with higher molecular weight PEG chains (Mw >1000) as compared to those with lower molecular weight PEG. It was reported that by incorporation of 2 – 5 mol% PEG into pNIPAm nanoparticles, the non-specific plasma protein interactions were minimized28, 40. Thus, PEGylated pNIPAm nanoparticles represent a promising system that can be employed as a drug delivery carrier. To further support PEGylated pNIPAm nanoparticles for use in drug delivery, the data in Figure 2 demonstrated that NGSS and NGPEGSS nanoparticles were stable at physiological pH 7.4 for 48 hours in non-reducing conditions; however in a reductive environment, like the intracellular space, the nanoparticles were degraded.
Previous work by our group has found that the ‚ potential, measured in MilliQ water, of similar pNIPAm nanoparticles with varying AMPS% correlated with drug loading efficiency due to the contribution of electrostatic interactions with the peptide27, 30; specifically, the more negative-charged particles increased loading of the positively charged KAFAK peptide. Changes in the AMPS% variable in these previous studies resulted in a greater range of zeta potentials. In our study, we investigate different formulations that keep the AMPS% constant at 5%. As a result, the range of zeta potentials for our formulations was not as large and did not produce a statistically significant difference in drug loading efficiency. Previous studies27, 30 also show that incorporating AMPS monomer into the particles shifted the LCST to higher temperatures as the AMPS content of the polymer increased. The higher LCST values of the formulations make them suitable for targeting of drugs to inflammation in combination with hyperthermia. As reported by Meyer et al.,41 pNIPAm nanoparticles could be used for both passive and active targeting to cancer cells, where passive accumulation was achieved through enhanced permeability and retention (EPR) effect, and active accumulation was influenced via external temperature changes. When tumors were heated locally to 42°C, the accumulation of thermosensitive nanoparticle with a LCST of 40°C was approximately two fold greater than the concentration of the same polymer in tumors that were not heated42, 43. While active targeting was not a goal of the current work, the work by Meyer et al.41 suggests that thermal targeting could be used with the thermosensitive particles described here.
KAFAK peptide treatment suppressed pro-inflammatory TNF-α and IL-6 production. However, in the presence of fetal bovine serum, the KAFAK peptide was less effective at inhibiting production of pro-inflammatory cytokines (Figure 8). The pNIPAm-AMPS carrier was shown previously to increase half-life of KAFAK in serum27, 37. It was also shown that the addition of negatively charged sulfate groups through incorporation of AMPS and the addition of cross-linking increased KAFAK loading efficiency and nanoparticle stability27, 30. However, in earlier studies27, 30 the high affinity of KAFAK to nanoparticles and lack of core degradation resulted in successful release of less than half of the loaded peptides. To attempt to overcome this limitation reducible disulfide cross-links were introduced into the nanoparticles. In the presence of intracellular glutathione and low pH in the late endosome/lysosome compartments, the disulfide bonds are reduced leading to nanoparticle degradation, thus enabling release of a higher percentage of KAFAK over time following cellular uptake. Ru Cheng et al.,44 reported highly efficient intracellular protein release in cancer cells when using pNIPAm nanoparticles with disulfide cross-links as opposed to a non-degradable cross-links. In order to capitalize on the physiological stability and release efficiency of KAFAK during cellular uptake, we explored the use of reducing disulfide cross-links in pNIPAm particles (Graphical abstract). As anticipated, the data in Figure 5A and 5B indicated that indeed a higher percentage of KAFAK released from disulfide nanoparticles than from the non-degradable (MBA) counterpart at low pH (Figure 5C). The release of KAFAK from the degradable nanoparticles reaches a maximum of ~24% after 24 hours. Despite releasing a significantly greater proportion of drug compared to nanoparticles without degradable crosslinks, most of the loaded drug is likely tightly associated with the core of the nanoparticle, which does not degrade as rapidly as the rest of the particle. Lack of core degradation is perhaps due to a higher reactivity ratio of the crosslinker as compared to the other monomers which results in a densely crosslinked core. As shown in Figure 8, KAFAK delivered from disulfide degradable nanoparticles effectively reduced pro-inflammatory cytokine production during the 24-hour incubation, and was more effective at reducing cytokine secretion than when KAFAK was delivered from non-degradable particles, or as free peptide. It is likely that the increased efficacy seen with the encapsulated peptide is due to protection of the peptide from enzymatic degradation41, 45; however, it is also possible that the peptide is simply protected from becoming inactivated by binding to serum proteins27, 28. This protection from non-specific interactions with serum proteins may also contribute to the increased anti-inflammatory efficacy of PEGylated nanoparticles compared to their non-PEGylated counterparts, but future studies are needed to quantify these effects.
Finally, the advantage of retaining the cell-penetrating peptide region of the peptide drug is two fold. First, the cell-penetrating peptide sequence, KAFAKLAARLYR, contains two lysines and two arginines that facilitate binding to the sulfated nanoparticles. Second, some peptide releases from the particles prior to uptake by the macrophages, and while not completely protected from the extracellular environment, a portion of the released peptide is still able to enter cells and function as an anti-inflammatory peptide27, 30.
CONCLUSION
The data described herein demonstrated that pNIPAm-AMPS nanoparticles are an effective tool for delivery of anti-inflammatory cell-penetrating peptide in vitro. By functionalizing particles with both PEG and disulfide cross-links (SS), the nanoparticles were effective both peptide loading and subsequent release of KAFAK in physiological intracellular environments after being taken up into the endosomal/lysosomal compartment. Importantly, the particles improved the anti-inflammatory efficacy of the peptide presumably by protecting the peptide from enzymatic degradation in the presence of serum. Furthermore, pNIPAm nanoparticles did not elicit a significant cytotoxic response in RAW 264.7 macrophage cells, indicating that these particles were biocompatible in nature.
This system has the potential to be an effective tool as a delivery vehicle for cationic peptides by taking full advantage of the increased loading efficiency, stealth characteristics and controlled release that is possible in this modified thermosensitive system. Future work will further optimize the system with respect to PEG and disulfide concentration to fine-tune the release of KAFAK and other enzymatically susceptible therapeutics.
Supplementary Material
Acknowledgments
We wish to thank Dr. Rush Barlett II and Jim McMasters for their valuable assistance. We also thank Dr. Philip Low (Chemistry-Purdue University) for providing cell samples. This work was supported by a grant from NIH.
ABBREVIATIONS
- NG
pNIPAm-AMPS
- SS
disulfide cross-linker
- NGSS
pNIPAm-AMPS-disulfide
- NGPEGSS
PEGylated-pNIPAm-AMPS-disulfide
- NGPEGSS
PEGylated-pNIPAm-AMPS-disulfide
- NGSSF
pNIPAm-AMPS-disulfide-fluorescein
- NGPEGSSF
PEGylated-pNIPAm-AMPS-disulfide-fluorescein
- NGMBA
nondegradable pNIPAm-AMPS-MBA
- NGPEGMBA
nondegradable PEGylated-pNIPAm-AMPS-MBA
Footnotes
Notes
Moerae Matrix, Inc. has a worldwide exclusive license to the MK2 inhibitor peptide. A. Panitch owns greater than 5% of Moerae Matrix, Inc.
References
- 1.Avdeev AS, Novikov AA, Aleksandrova EN, Panasiuk E, Nasonov EL. The importance of cytokine profile characteristics for evaluating the therapeutic effectiveness of monoclonal antibodies against IL-6 receptors in patients with rheumatoid arthritis. Klin Med (Mosk) 2014;92(1):28–34. [PubMed] [Google Scholar]
- 2.Takac B, Mihaljevic S, Stefanic M, Glavas-Obrovac L, Kibel A, Samardzija M. Importance of interleukin 6 in pathogenesis of inflammatory bowel disease. Coll Antropol. 2014;38(2):659–64. [PubMed] [Google Scholar]
- 3.Ayala-Lopez W, Xia W, Varghese B, Low PS. Imaging of atherosclerosis in apoliprotein e knockout mice: targeting of a folate-conjugated radiopharmaceutical to activated macrophages. J Nucl Med. 2010;51(5):768–74. doi: 10.2967/jnumed.109.071324. [DOI] [PubMed] [Google Scholar]
- 4.Gueler F, Park JK, Rong S, Kirsch T, Lindschau C, Zheng W, Elger M, Fiebeler A, Fliser D, Luft FC, Haller H. Statins attenuate ischemia-reperfusion injury by inducing heme oxygenase-1 in infiltrating macrophages. Am J Pathol. 2007;170(4):1192–9. doi: 10.2353/ajpath.2007.060782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patella M, Anile M, Del Porto P, Diso D, Pecoraro Y, Onorati I, Mantovani S, De Giacomo T, Ascenzioni F, Rendina EA, Venuta F. Role of cytokine profile in the differential diagnosis between acute lung rejection and pulmonary infections after lung transplantationdagger. Eur J Cardiothorac Surg. 2014 doi: 10.1093/ejcts/ezu395. [DOI] [PubMed] [Google Scholar]
- 6.Xiao L, He X, Zhou W, Fan W, Wei Y, Gao Y, Ma X, Kong X, Han Y, Xu X, Huang H, Wang X, Shi B. Expression of T lymphocyte cytokines in peripheral blood of renal transplant recipients. Zhonghua Yi Xue Za Zhi. 2014;94(32):2506–9. [PubMed] [Google Scholar]
- 7.Gomez-Banoy N, Mockus I, Aranzalez LH, Zambrano JM. Changes to circulating inflammatory cytokines in response to moderate exercise. J Sports Med Phys Fitness. 2014 [PubMed] [Google Scholar]
- 8.Jang JC, Nair MG. Alternatively Activated Macrophages Revisited: New Insights into the Regulation of Immunity, Inflammation and Metabolic Function following Parasite Infection. Curr Immunol Rev. 2013;9(3):147–156. doi: 10.2174/1573395509666131210232548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vural A, Kehrl JH. Autophagy in macrophages: impacting inflammation and bacterial infection. Scientifica (Cairo) 2014;2014:825463. doi: 10.1155/2014/825463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 11.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 12.Varin A, Gordon S. Alternative activation of macrophages: immune function and cellular biology. Immunobiology. 2009;214(7):630–41. doi: 10.1016/j.imbio.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 13.Quiniou C, Dominguez-Punaro M, Cloutier F, Erfani A, Ennaciri J, Sivanesan D, Sanchez M, Chognard G, Hou X, Rivera JC, Beauchamp C, Charron G, Vilquin M, Kuchroo V, Michnick S, Rioux JD, Lesage S, Chemtob S. Specific targeting of the IL-23 receptor, using a novel small peptide noncompetitive antagonist, decreases the inflammatory response. Am J Physiol Regul Integr Comp Physiol. 2014;307(10):R1216–30. doi: 10.1152/ajpregu.00540.2013. [DOI] [PubMed] [Google Scholar]
- 14.Brugnano JL, Chan BK, Seal BL, Panitch A. Cell-penetrating peptides can confer biological function: regulation of inflammatory cytokines in human monocytes by MK2 inhibitor peptides. J Control Release. 2011;155(2):128–33. doi: 10.1016/j.jconrel.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 15.Lee E, Shin A, Kim Y. ANTI-INFLAMMATORY ACTIVITIES OF CECROPIN A AND ITS MECHANISM OF ACTION. Arch Insect Biochem Physiol. 2014 doi: 10.1002/arch.21193. [DOI] [PubMed] [Google Scholar]
- 16.Bundgaard H. (C) Means to enhance penetration: (1) Prodrugs as a means to improve the delivery of peptide drugs. Adv Drug Delivery Rev. 1992;8(1):1–38. [Google Scholar]
- 17.Scott Swenson E, Curatolo WJ. (C) Means to enhance penetration: (2) Intestinal permeability enhancement for proteins, peptides and other polar drugs: mechanisms and potential toxicity. Adv Drug Delivery Rev. 1992;8(1):39–92. [Google Scholar]
- 18.Bhaskar Kompella U, Lee VHL. (C) Means to Enhance Penetration: (4) Delivery systems for penetration enhancement of peptide and protein drugs: design considerations. Adv Drug Delivery Rev. 1992;8(1):115–162. doi: 10.1016/s0169-409x(00)00137-x. [DOI] [PubMed] [Google Scholar]
- 19.Shen WC, Wan J, Ekrami H. (C) Means to enhance penetration: (3) Enhancement of polypeptide and protein absorption by macromolecular carriers via endocytosis and transcytosis. Adv Drug Delivery Rev. 1992;8(1):93–113. [Google Scholar]
- 20.Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68(2):320–44. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engel K, Ahlers A, Brach MA, Herrmann F, Gaestel M. MAPKAP kinase 2 is activated by heat shock and TNF-alpha: in vivo phosphorylation of small heat shock protein results from stimulation of the MAP kinase cascade. J Cell Biochem. 1995;57(2):321–30. doi: 10.1002/jcb.240570216. [DOI] [PubMed] [Google Scholar]
- 22.Brugnano J, McMasters J, Panitch A. Characterization of endocytic uptake of MK2-inhibitor peptides. J Pept Sci. 2013;19(10):629–38. doi: 10.1002/psc.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.C & EN Volume. 2014;92(45):10–13. [Google Scholar]
- 24.Constantin M, Bucatariu S, Harabagiu V, Popescu I, Ascenzi P, Fundueanu G. Poly(N-isopropylacrylamide-co-methacrylic acid) pH/thermo-responsive porous hydrogels as self-regulated drug delivery system. Eur J Pharm Sci. 2014;62:86–95. doi: 10.1016/j.ejps.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 25.Mishra S, De A, Mozumdar S. Synthesis of thermoresponsive polymers for drug delivery. Methods Mol Biol. 2014;1141:77–101. doi: 10.1007/978-1-4939-0363-4_4. [DOI] [PubMed] [Google Scholar]
- 26.Yang J, van Lith R, Baler K, Hoshi RA, Ameer GA. A thermoresponsive biodegradable polymer with intrinsic antioxidant properties. Biomacromolecules. 2014;15(11):3942–52. doi: 10.1021/bm5010004. [DOI] [PubMed] [Google Scholar]
- 27.Bartlett RL, 2nd, Medow MR, Panitch A, Seal B. Hemocompatible poly(NIPAm-MBA-AMPS) colloidal nanoparticles as carriers of anti-inflammatory cell penetrating peptides. Biomacromolecules. 2012;13(4):1204–11. doi: 10.1021/bm300173x. [DOI] [PubMed] [Google Scholar]
- 28.Gulati N, Rastogi R, Dinda AK, Saxena R, Koul V. Characterization and cell material interactions of PEGylated PNIPAAM nanoparticles. Colloids Surf B Biointerfaces. 2010;79(1):164–73. doi: 10.1016/j.colsurfb.2010.03.049. [DOI] [PubMed] [Google Scholar]
- 29.Jain AK, Gund MG, Desai DC, Borhade N, Senthilkumar SP, Dhiman M, Mangu NK, Mali SV, Dubash NP, Halder S, Satyam A. Mutual prodrugs containing bio-cleavable and drug releasable disulfide linkers. Bioorg Chem. 2013;49:40–8. doi: 10.1016/j.bioorg.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Bartlett RL, 2nd, Panitch A. Thermosensitive nanoparticles with pH-triggered degradation and release of anti-inflammatory cell-penetrating peptides. Biomacromolecules. 2012;13(8):2578–84. doi: 10.1021/bm300826v. [DOI] [PubMed] [Google Scholar]
- 31.Gaulding JC, Smith MH, Hyatt JS, Fernandez-Nieves A, Lyon LA. Reversible Inter- and Intra-Microgel Cross-Linking using Disulfides. Macromolecules. 2012;45(1):39–45. doi: 10.1021/ma202282p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ding H, Wu F, Huang Y, Zhang Z-r, Nie Y. Synthesis and characterization of temperature-responsive copolymer of PELGA modified poly(N-isopropylacrylamide) Polymer. 2006;47(5):1575–1583. [Google Scholar]
- 33.Tauer K, Gau D, Schulze S, Völkel A, Dimova R. Thermal property changes of poly(N-isopropylacrylamide) microgel particles and block copolymers. Colloid and Polymer Science. 2009;287(3):299–312. [Google Scholar]
- 34.Gan D, Lyon LA. Tunable swelling kinetics in core--shell hydrogel nanoparticles. J Am Chem Soc. 2001;123(31):7511–7. doi: 10.1021/ja010609f. [DOI] [PubMed] [Google Scholar]
- 35.Naha PC, Bhattacharya K, Tenuta T, Dawson KA, Lynch I, Gracia A, Lyng FM, Byrne HJ. Intracellular localisation, geno- and cytotoxic response of polyN-isopropylacrylamide (PNIPAM) nanoparticles to human keratinocyte (HaCaT) and colon cells (SW 480) Toxicol Lett. 2010;198(2):134–43. doi: 10.1016/j.toxlet.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 36.Strober W. Current Protocols in Immunology. John Wiley & Sons, Inc; 2001. Trypan Blue Exclusion Test of Cell Viability. [DOI] [PubMed] [Google Scholar]
- 37.Bartlett RL, 2nd, Sharma S, Panitch A. Cell-penetrating peptides released from thermosensitive nanoparticles suppress pro-inflammatory cytokine response by specifically targeting inflamed cartilage explants. Nanomedicine. 2013;9(3):419–27. doi: 10.1016/j.nano.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Neal DP, Hirsch LR, Halas NJ, Payne JD, West JL. Photo-thermal tumor ablation in mice using near infrared-absorbing nanoparticles. Cancer Lett. 2004;209(2):171–6. doi: 10.1016/j.canlet.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 39.Sandanaraj BS, Gremlich HU, Kneuer R, Dawson J, Wacha S. Fluorescent nanoprobes as a biomarker for increased vascular permeability: implications in diagnosis and treatment of cancer and inflammation. Bioconjug Chem. 2010;21(1):93–101. doi: 10.1021/bc900311h. [DOI] [PubMed] [Google Scholar]
- 40.Tziampazis E, Kohn J, Moghe PV. PEG-variant biomaterials as selectively adhesive protein templates: model surfaces for controlled cell adhesion and migration. Biomaterials. 2000;21(5):511–20. doi: 10.1016/s0142-9612(99)00212-4. [DOI] [PubMed] [Google Scholar]
- 41.Meyer DE, Shin BC, Kong GA, Dewhirst MW, Chilkoti A. Drug targeting using thermally responsive polymers and local hyperthermia. Journal of Controlled Release. 2001;74(1–3):213–224. doi: 10.1016/s0168-3659(01)00319-4. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J, Chen H, Xu L, Gu Y. The targeted behavior of thermally responsive nanohydrogel evaluated by NIR system in mouse model. J Control Release. 2008;131(1):34–40. doi: 10.1016/j.jconrel.2008.07.019. [DOI] [PubMed] [Google Scholar]
- 43.Chilkoti A, Dreher MR, Meyer DE, Raucher D. Targeted drug delivery by thermally responsive polymers. Adv Drug Deliv Rev. 2002;54(5):613–30. doi: 10.1016/s0169-409x(02)00041-8. [DOI] [PubMed] [Google Scholar]
- 44.Cheng R, Meng F, Ma S, Xu H, Liu H, Jing X, Zhong Z. Reduction and temperature dual-responsive crosslinked polymersomes for targeted intracellular protein delivery. Journal of Materials Chemistry. 2011;21(47):19013–19020. [Google Scholar]
- 45.Bromberg LE, Ron ES. Temperature-responsive gels and thermogelling polymer matrices for protein and peptide delivery. Adv Drug Delivery Rev. 1998;31(3):197–221. doi: 10.1016/s0169-409x(97)00121-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.