

Abstract
Despite the importance of the mammalian neocortex for complex cognitive processes, we still lack a comprehensive description of its cellular components. To improve the classification of neuronal cell types and the functional characterization of single neurons, we present Patch-seq, a method that combines whole-cell electrophysiological patch-clamp recordings, single-cell RNA-sequencing and morphological characterization. Following electrophysiological characterization, cell contents are aspirated through the patch-clamp pipette and prepared for RNA-sequencing. Using this approach, we generate electrophysiological and molecular profiles of 58 neocortical cells and show that gene expression patterns can be used to infer the morphological and physiological properties such as axonal arborization and action potential amplitude of individual neurons. Our results shed light on the molecular underpinnings of neuronal diversity and suggest that Patch-seq can facilitate the classification of cell types in the nervous system.
Since Ramon y Cajal and others first systematically investigated the cellular structure of the brain more than a century ago1, it has become increasingly clear that different brain regions contain distinct neuronal cell types arranged in stereotypical circuits that underlie the functions that each brain area performs2. The gold standard for classification of neuronal cell types has been their complex and diverse morphology1–3. In particular, axonal geometry and projection patterns have been the most informative morphological features for predicting how a neuron is integrated into the local circuit (i.e., which other neurons it will connect to)3,4. In addition, different morphological cell types often display unique physiological properties such as distinctive firing patterns in response to sustained depolarizing current injection5. Cellular morphology and physiology can be directly correlated at the single-cell level using whole-cell patch-clamp recording6.
Recent advances in molecular biology, particularly high-throughput single-cell RNA-sequencing (RNA-seq)7,8 have begun to reveal the genetic programs that give rise to cellular diversity9 and have enabled de novo identification of cell types10, including neuronal subtypes in the neocortex and hippocampus11,12. However, as these approaches require dissociation of tissue to isolate single cells, it has been difficult to link molecularly defined neuronal subtypes to their corresponding electrophysiological and morphological counterparts. The integration of physiology with gene expression profiles has primarily relied on single-neuron reverse transcription PCR (RT-PCR) of neurons recorded in patch-clamp mode13, which is restricted to only a small number (up to ~50)14 of prespecified genes, or on spotted cDNA array15, which has a limited dynamic range, sensitivity and specificity compared to sequencing-based approaches and cannot detect novel transcripts or splice variants7. Previous attempts at unbiased, whole-transcriptome profiling using single-neuron RNA-seq after patch-clamp recording have so far been unsuccessful: one study sequenced in total three neurons from acute slices with a mean correlation of ~0.25 across samples16, reflecting difficulties in maintaining RNA integrity throughout electrophysiological recordings.
We thus set out to develop a protocol for combining whole-cell patch-clamp recordings with high-quality RNA-seq of single neurons, and focused on layer 1 (L1) of the mouse neocortex (Fig. 1a). L1 is known to contain only two main morphological classes of neurons, both of which are inhibitory interneurons, with their own distinct firing patterns and connectivity profiles: elongated neurogliaform cells (eNGCs) and single bouquet cells (SBCs)4. Using standard electrophysiology techniques and cortical slices, we first used a dataset of 72 L1 interneurons4, whose firing pattern we had recorded in response to sustained depolarizing current and for which we had also reconstructed their detailed morphology using avidin-biotin-peroxidase staining (Fig. 1b). Using this as training data, we built an automatic cell type classifier based on electrophysiological properties that could predict morphological cell class with ~98% accuracy (Fig. 1d,e). In a separate set of experiments, we carried out patch-clamping on an additional set of 67 L1 interneurons in acute cortical slices using the Patch-seq protocol. The protocol was developed to improve RNA yield by making use of an optimized mechanical recording approach (e.g., tip size, volume inside pipette) as well as a modified intracellular recording solution to extract and preserve as much full-length mRNA from each cell as possible (Supplementary Figs. 1 and 2). We recorded their firing patterns (Fig. 1c) and extracted their cell contents until the cell had visibly shrunken (Fig. 1g) for downstream RNA-seq analysis. Each neuron from this RNA-seq dataset was assigned to a neuronal class of either eNGC or SBC by blinded expert examination of the firing pattern and using the automated classifier described above. Both classifications were carried out independently and led to very similar cell type labels (Fig. 1f, r = 0.85, P < 10−12, n = 44). In addition, we recorded the electrophysiological properties of 32 L1 interneurons in vivo in anesthetized animals and extracted their cell contents for RNA-seq. Large fluctuations in the resting membrane potential, likely due to ongoing activity in the local circuit and/or fluctuations in cortical state17, made it difficult to classify neurons recorded in vivo based on their electrophysiological properties. Thus, these cells did not receive a cell type label. Although we aimed to target L1 interneurons, we occasionally recorded an excitatory neuron (n = 1 ex vivo and n = 7 in vivo) or astrocyte (n = 1 in vivo) near the L1/L2 border. Rather than discarding these samples, we proceeded with RNA-seq in the same manner as for the L1 interneurons and used them as additional controls to validate cell type–specific markers (see below). In addition, each experiment included at least one negative control, in which a recording pipette was inserted into the tissue but no cell was patched. The negative controls were processed in the same manner as the rest of the samples to determine the amount of contamination during sample collection and amplification (Supplementary Fig. 3).
Figure 1.
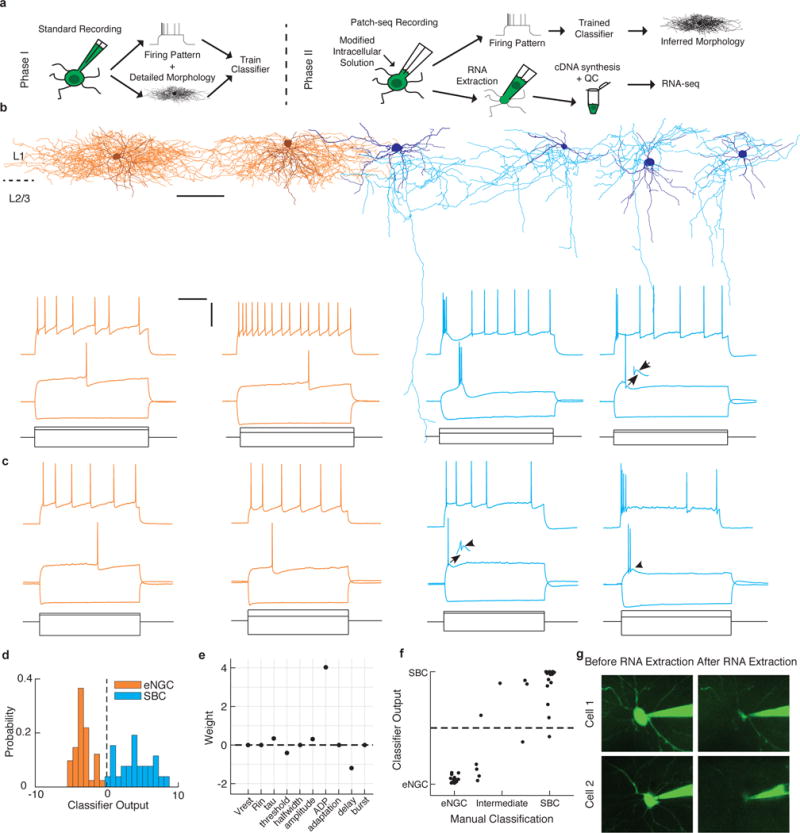
Two morphologically and electrophysiologically distinct neuronal classes in neocortical layer 1. (a) Schematic of experimental approach. (b) Representative examples of the morphology (top) and firing pattern (bottom) of the two main types of neurons found in L1: elongated neurogliaform cells (eNGCs, orange) and single bouquet cells (SBCs, cyan). For morphological reconstructions, the darker outline represents the somatodendritic region, and the lighter color is the axonal arbor; scale bar, 100 μm. For firing patterns, gray lines represent current steps used to elicit the firing patterns shown above; scale bars, 300 ms (horizontal bar), 40 mV and 500 pA (vertical bar); arrows denote prominent after-depolarization in SBCs. (c) Neurons recorded using Patch-seq protocol display similar firing responses as seen using standard electrophysiological techniques, as shown in b. (d) Output of automated cell type classifier robustly predicts morphological class based on electrophysiological features. (e) Weights of features used in the automated cell type classifier. (f) Results of the automated classifier highly correlate with an independent, blinded expert classification of the electrophysiological properties as “eNGC-like” or “SBC-like,” r = 0.91. (g) Example cells before and after RNA extraction.
After harvesting the cell contents, single-cell mRNA was converted to cDNA and used to generate sequencing libraries following a protocol similar to that of Smart-seq2 (ref. 18). Libraries with low cDNA yield (<200 pg/μl) or poor quality (<1,500 bp mean size) were excluded from further analysis (Supplementary Fig. 3; 50/108 cells and 32/32 negative controls). A higher fraction of in vivo samples was excluded (31/40) compared to ex vivo (19/68; Supplementary Fig. 3), likely owing to there being lower amounts of cDNA obtained as well as increased contamination during in vivo sample acquisition (i.e., the pipette must traverse more tissue and penetrate the dura to reach the target cell). We sequenced the 58 single-cell libraries that met our inclusion criteria; they corresponded to the following, all of which were recorded in patch-clamp mode, as indicated: 48 L1 interneurons in slices, 5 L1 interneurons in vivo, 1 pyramidal neuron in slices, 3 pyramidal neurons in vivo and 1 astrocyte in vivo. Analyses of the sequenced libraries revealed that, on average, 65% of reads mapped uniquely to the mouse genome, and 60% of those mapped within exons (Supplementary Fig. 4 and Supplementary Table 1). As expected, the pyramidal neuron and astrocyte samples showed clear differences in gene expression compared to the L1 interneurons (Fig. 2a and Supplementary Fig. 5), consistent with known cell type–specific markers19–22. We subsequently focused our analyses on the L1 interneurons, which expressed interneuron markers including Gad1, Reln and Cplx3 (refs. 23,24; Supplementary Figs. 5 and 6). We detected approximately 7,000 genes per interneuron (Fig. 2b), with an average Spearman correlation of 0.59 and 0.56 between ex vivo and in vivo cells, respectively (Fig. 2c), a higher number of genes detected per cell than a recent study using dissociated neurons12.
Figure 2.
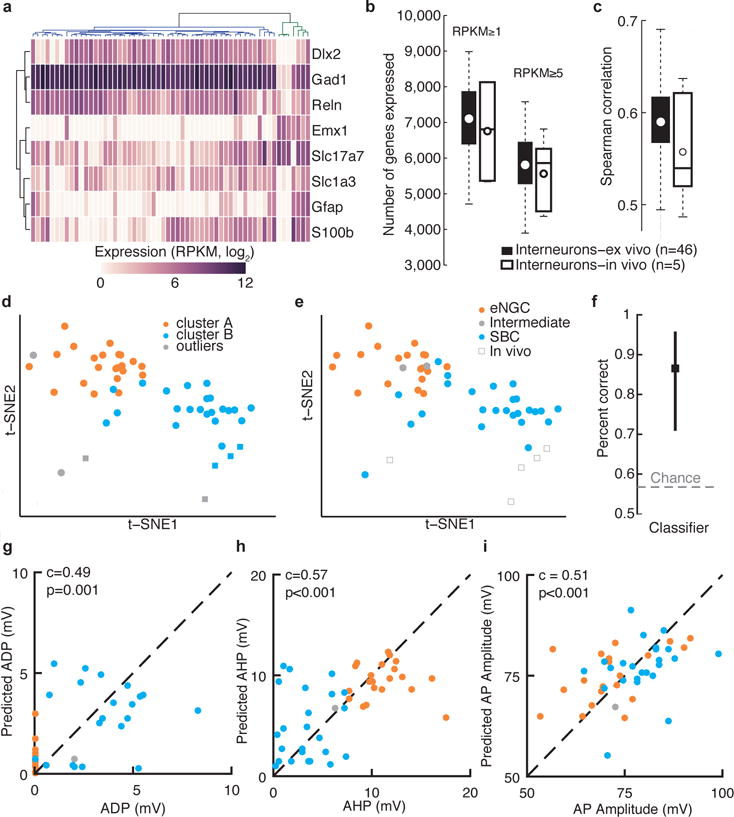
Single-neuron transcriptome profiles predict cell type and electrophysiological properties. (a) Clustering analysis separates interneurons (blue dendrogram subtree) from other neuronal classes (green dendrogram subtree; includes four pyramidal neurons, and one astrocyte) based on marker gene expression. Two L1 interneurons clustered with non-interneuron cell types, indicating possible contamination of these samples, and so these two cells were excluded from our analysis of interneuron subtypes. (b) Number of genes detected per neuron using two different expression thresholds for interneurons patch-clamp recorded ex vivo and in vivo. (c) Pairwise Spearman correlation across all detected genes for ex vivo and in vivo patch-clamp recorded interneurons. (d) Two-dimensional t-SNE representation of gene expression for all L1 interneurons. Cells are colored according to affinity propagation-based clustering in gene-space spanned by the 3,000 most variable genes, prior to dimensionality reduction. (e) The same two-dimensional map as in d, but with cells color-coded according to expert classification of cell type based on electrophysiological properties. Performance of GLMs using single-neuron gene expression to predict cell type (f), ADP (g), AHP (h), or action potential (AP) amplitude (i).
To explore the interneuron transcriptomes and to resolve the molecular cell classes in an unbiased manner, we performed clustering and dimensionality-reduction analysis using the 3,000 most variable genes (Supplementary Table 2). Affinity propagation was employed to cluster cells in this high-dimensional gene space (without prespecifying the number of clusters). We reduced the dimensionality of the data to visualize the resulting clusters using t-distributed stochastic neighbor-hood embedding (t-SNE). We identified two molecular interneuron clusters (Fig. 2d) with high correspondence to the eNGC and SBC classification (41/47 cells, 87%; Fig. 2d,e). Random subsampling of the data demonstrated that the two cell classes could be robustly distinguished using as few as 31 cells (Supplementary Fig. 7). In addition, we asked whether we could predict cell class based on single-cell gene expression using a regularized generalized linear model (GLM; Supplementary Table 3). The classifier performed at approximately 86% accuracy for predicting cell type (Fig. 2f). Although our sample size was clearly sufficient to identify the two major physiological cell classes in L1, it is still possible that additional molecular subclasses exist within each of these broad groups. Together, these results demonstrate a strong agreement between cell type assignments based on morphological, electrophysiological and transcriptional profiles.
Next we asked whether specific physiological properties could also be predicted using single-neuron gene expression data. We trained a sparse, regularized GLM for each of seven quantitative electrophysiological measurements using the single-cell transcriptome data as input (using the most variable 50–250 genes across cells as input). Three of these (after-hyperpolarization amplitude (AHP), after-depolarization amplitude (ADP) and action potential amplitude (Amp)) could be predicted based on differential gene expression, as shown by the correlation between cross-validated predictions and the ground truth for individual neurons (Fig. 2g–i and Supplementary Table 3). The remaining variables (membrane time constant, adaptation index, action potential width and resting membrane potential) could not be modeled using gene expression data, suggesting that variability along these features may reflect factors other than differential gene expression, or that a larger dataset is needed to infer these properties from single-cell gene expression.
Transcriptome analyses of cells collected in vivo assigned many of them to a specific cell class (Fig. 2e) and suggested a shift in gene expression compared to cells collected ex vivo (Fig. 2e, t-SNE, second component) that may reflect an increased stress response in the acute slice preparation (e.g., increased Fos expression ex vivo compared to in vivo; Supplementary Fig. 8). Notably, these results demonstrate that high-quality whole-transcriptome data can be obtained even from single neurons in intact animals, and that the gene expression profile within a cell class is largely preserved across in vivo and ex vivo preparations. Extension of cell type classification to include dynamic functional properties such as receptive fields and tuning properties, which can only be measured in vivo, may ultimately lead to better understanding of cell types in terms of their role in information processing in the cortex.
Previous studies suggest that late-spiking eNGCs express Reelin whereas burst-spiking SBCs express vasoactive intestinal peptide (VIP)25. However, other studies show that Reelin is found in similar proportions in both cell types, and only about 20% of burst-spiking cells express VIP26. We found that neither of these markers was very useful for distinguishing eNGCs from SBCs at the mRNA level (Fig. 3a and Supplementary Fig. 6), calling into question whether single-neuron RT-PCR and protein-level studies are well suited for predicting which transcripts show the greatest differences in expression between cell types. Single-cell differential expression analysis (SCDE)27 identified several genes that are differentially expressed between the two cell types (Fig. 3b, Supplementary Fig. 9 and Supplementary Table 4). In addition, gene set enrichment analysis (GSEA) revealed that genes involved in cell-cell signaling (transmembrane and extracellular proteins, receptors, ion channels and intracellular signaling molecules) were particularly upregulated in SBCs, whereas genes involved in RNA processing and mitochondrial function were upregulated in eNGCs (Fig. 3c and Supplementary Table 5). These findings are consistent with previous reports that eNGCs communicate nonspecifically with all cell types using volume transmission, whereas SBCs form highly selective synapses onto particular neuronal types4,28,29. In particular, our results predict that increased expression of cell adhesion molecules including Cdh18, Cdh4 and ALCAM, and synaptic regulatory proteins such as Syndig1 (ref. 30) may play an important role in the shaping of the synaptic specificity of SBCs4,28. Taken together, these results demonstrate that whole-transcriptome profiling of patch-clamp-recorded neurons is a useful approach to identify novel molecular markers for well-defined physiological and morphological cell classes, and to generate hypotheses regarding the molecular mechanisms of cell type diversity.
Figure 3.
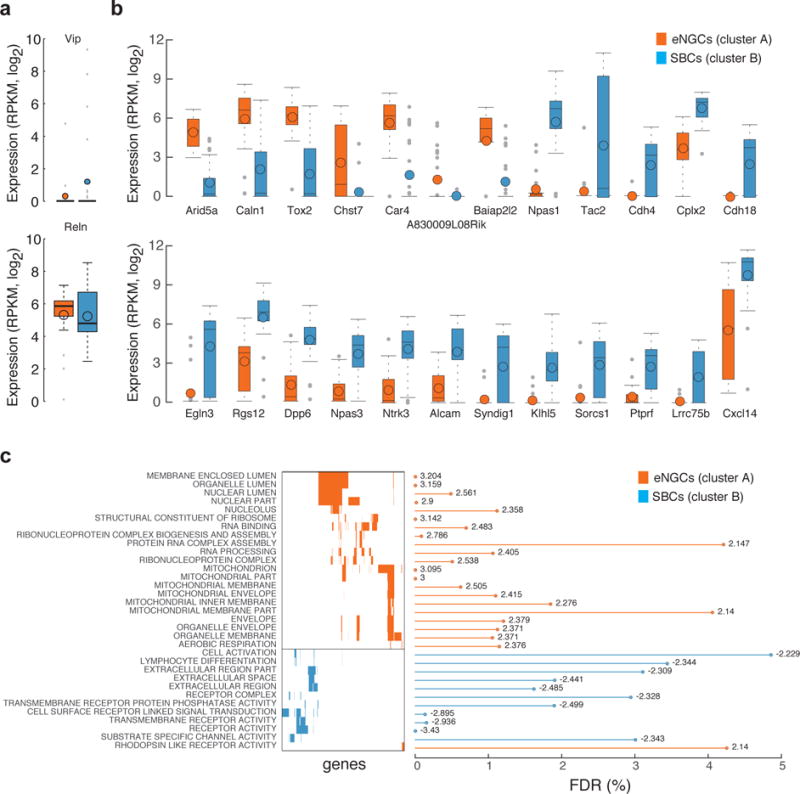
Differential gene expression analysis reveals novel markers for L1 interneuron classes. (a) Boxplots summarize the cell-type expression level of previous marker genes (Vip and Reelin). (b) Boxplots with expression levels across cell types for differentially expressed genes identified between the two affinity propagation clusters. (c) Gene categories that were significantly enriched in SBCs or eNGCs based on gene set enrichment analysis. The gene matrix illustrates gene overlap among categories; the bar plot shows the false discovery rates and the numbers indicate normalized enrichment scores per category from GSEA.
A number of the identified differentially expressed genes are also associated with human disease. For example, the transcription factors Npas1 and Npas3 are highly expressed in SBCs but not in eNGCs (Fig. 3b). Notably, these proteins have been implicated in autism spectrum disorders and schizophrenia and were previously shown to regulate the generation of specific neocortical interneurons31,32. SBCs also preferentially express Dpp6 and Cplx2 (Fig. 3b). Dpp6 (Dipeptidyl-peptidase 6) is an auxiliary subunit of the Kv4 family of voltage-gated K+ channels implicated in autism spectrum disorders that regulates channel function and dendrite morphogenesis33, whereas Cplx2 (Complexin-2) is a presynaptic protein linked to schizophrenia that controls neurotransmitter release and presynaptic differentiation34. Our observation that four disease genes implicated in neuropsychiatric illness are significantly (P-adjusted < 0.05) upregulated in SBCs, in combination with previous studies suggesting that SBCs may play an important role in the detection of salient sensory information and the mediation of top-down influences28, raises the question of whether dysfunction of SBCs may contribute to the pathophysiology of these disorders. The ability to map disease-associated genes onto specific neuronal cell types will open entirely new lines of inquiry and pave the way for a more precise, circuit-level understanding of neuropsychiatric disorders.
In summary, Patch-seq is a technique that enables whole-cell patch-clamp recordings and high-quality RNA-seq of individual neurons. Using this approach, we demonstrate that cellular morphology, physiology and gene expression can be integrated at the single-cell level to generate a comprehensive profile of neuronal cell types, using neocortical L1 interneurons as a test case. In addition, we identify several molecular markers that can be used to target these cell types for further study, generate new hypotheses regarding their differentiation and link specific cell types to neuropsychiatric illness. Our approach can be used broadly to characterize neuronal cell types in any brain region, in different mouse models of disease and even in nongenetically tractable organisms such as primates. The ability to perform unbiased, whole-genome transcriptome analysis and physiological characterization of individual neurons might help to resolve long-standing questions in the field of neuroscience and will enable new directions of investigation.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Animals
All experiments were carried out in accordance with, and with approval from, the Animal Care and Use Committee (IACUC) at Baylor College of Medicine (BCM). The dataset for electrophysiological and morphological characterization of L1 interneurons came from a much larger study of neuronal cell types across all cortical layers4.
For the RNA-seq experiments, we aimed to collect approximately 20–30 samples of each cell type, because previous studies have demonstrated that this number is typically sufficient to separate different cell classes within high-dimensional gene space12. A total of ten mice (seven males and three females) aged 15–70 d were used for these experiments (five for ex vivo and five for in vivo experiments). The majority were wild-type C57Bl/6 mice obtained from the BCM Center for Comparative Medicine (CCM, n = 7/10 mice). A few animals (n = 3/10) were Viaat-Cre; ROSA26-LSL-tdTomato (Ai9) double heterozygotes, in which all interneurons express the fluorescent reporter tdTomato35, in order to facilitate targeted patch-clamp recording of L1 interneurons. The Viaat-Cre line was obtained from H. Zoghbi (BCM) and maintained on an Fvb background. The Ai9 line was obtained from the Jackson Laboratory (JAX stock #007909). Thus, the Viaat-Cre/Ai9 offspring used for experiments were on a mixed C57Bl6/Fvb background. The differences in gene expression we observed between cell types were unrelated to the genetic background, sex or age of the animals (data not shown).
Slice electrophysiology and RNA extraction
Brain slices were prepared as previously described4 with modifications to improve recovery from adult (>P20) tissue36. Animals were deeply anesthetized with 3% isoflurane. Following decapitation the brain was quickly removed and placed into cold (0–4 °C) oxygenated physiological solution. For juvenile tissue, a standard physiological solution was used containing (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 1 MgCl2, 25 dextrose and 2 CaCl2, pH 7.4. For adult tissue, a modified NMDG solution was used containing (in mM): 93 NMDG, 93 HCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 10 MgSO4 and 0.5 CaCl2, pH 7.4. Parasagittal slices 300 μm thick were cut from the tissue blocks using a microslicer (Leica VT 1200). For adult tissue, slices were kept in oxygenated NMDG solution for 10–15 min, and then transferred to oxygenated standard physiological solution. For juvenile tissue, slices were immediately stored in oxygenated standard physiological solution. All slices were kept at 37.0 ± 0.5 °C in oxygenated physiological solution for ~0.5–1 h before recordings. During the recording sessions the slices were submerged in a custom chamber. The slices were stabilized with a fine nylon net that was attached to a platinum ring. The recording chamber was perfused with oxygenated physiological solution throughout the experiments. The half-time for the bath solution exchange was ~6 s, and the temperature of the bath solution was maintained at 34.0 ± 0.5 °C.
To collect an initial dataset of only morphology and electrophysiology for a large number of neurons, we carried out whole-cell recordings according to previously described techniques4. Patch recording pipettes (5–7 MΩ) were filled with intracellular solution containing 120 mM potassium gluconate, 10 mM HEPES, 4 mM KCl, 4 mM MgATP, 0.3 mM Na3GTP, 10 mM sodium phosphocreatine and 0.5% biocytin (pH 7.25). Whole-cell recordings were made using a Quadro EPC 10 amplifier (HEKA Electronic, Lambrecht, Germany). A built-in LIH 8+8 interface board (HEKA) was used to achieve simultaneous A/D and D/A conversion of current, voltage, command and triggering signal. PatchMaster software (HEKA) and custom-written MATLAB-based programs (Mathworks) were used to operate the recording system and perform online and offline analysis of the electrophysiology data. For each cell, we also recorded its spiking response to a sustained depolarizing current.
To obtain electrophysiology and transcriptome data from single neurons, we made additional modifications to improve RNA recovery37. A series of pilot experiments were carried out to test various protocol modifications, including (i) the inclusion of RNase inhibitor in the intracellular patch-clamp solution, (ii) silanization of the glass capillaries37 used for patch-clamping, (iii) dNTP concentration of the lysis buffer, (iv) inclusion of the nucleus during the extraction process, (v) pipette tip size and (vi) volume of intracellular solution (Supplementary Fig. 1). Here we describe the final protocol used for sample acquisition from L1 interneurons in this study. Glass capillaries (2.0 mm OD, 1.16 mm ID, Sutter Instruments) were autoclaved prior to pulling patch-clamp pipettes, all work surfaces including micromanipulator pieces were thoroughly cleaned with DNA-OFF (Takara Cat. #9036) and RNase Zap (Life Technologies Cat. #AM9780) and great care was taken to maintain an RNase-free environment during sample collection. Recording pipettes of 2–4 MΩ resistance were filled with RNase-free intracellular solution containing: 123 mM potassium gluconate, 12 mM KCl, 10 mM HEPES, 0.2 mM EGTA, 4 mM MgATP, 0.3 mM NaGTP, 10 mM sodium phosphocreatine, 20 μg/ml glycogen, and 1 U/μl recombinant RNase inhibitor (Takara Cat.no. 2313A), pH ~7.25. To maximize RNA recovery, it was critical to use a small volume of intracellular solution in the patch-clamp pipette (ideally less than 0.3 μl, but certainly less than 1 μl, Supplementary Fig. 1f). Loading the pipette with such a small volume could be done reliably by hand using a standard backfilling approach with some practice, but raises additional challenges such as ensuring the patch-clamp electrode reaches the internal solution. Addition of EGTA to the intracellular solution scavenges free calcium and thus reduces the activity of any RNases present in the solution, and glycogen serves as an RNA carrier13,37. Addition of RNase Inhibitor directly to the internal solution significantly improved cDNA yield (Supplementary Fig. 1a). Importantly, recordings could be carried out using this modified internal solution for up to 60 min without affecting the health of the cells as indicated by their resting membrane potential (Supplementary Fig. 2). RNA was collected at the end of the recording (typically 2–3 min from break-in to RNA extraction) by applying light suction until the cell had visibly shrunken (Fig. 1g). Often, the entire nucleus and most of the cytoplasm could be seen entering the pipette (Fig. 1g) and this was associated with a high yield of cDNA (Supplementary Fig. 1d). We achieved an optimal balance between maintaining stable recordings (easier with small pipette tips) and obtaining high quality RNA samples (easier with large pipette tips, see Supplementary Fig. 1e) when the tip of pipette was approximately one-quarter to one-third the size of the cell body. For experiments where it is necessary to hold the cell for longer than ~20 min, smaller pipette tips may be required. If any extracellular contents were observed to enter the pipette, the pipette and its contents were discarded. Otherwise, the contents of the pipette were ejected using positive pressure into an RNase-free PCR tube containing 4 μl of RNase-free lysis buffer consisting of: 0.1% Triton X-100, 5 mM (each) dNTPs, 2.5 μM Oligo-dT30VN (5′-AAGCAGTGGTATCAACGCAGAGTACT30VN-3′, where ‘N’ is any base and ‘V’ is either A, C or G), 1 U/μl RNase inhibitor, and 1 × 10−5 dilution of ERCC RNA Spike-In Mix (Life Technologies Cat. #4456740). We aimed to collect approximately equal numbers of each cell type from individual animals and/or experiments, so that any differences between the cell types could not be attributed to interanimal or interexperimental differences.
Morphological reconstruction
Following slice recordings, morphological examination was carried out using light microscopy according to previously described protocols4. In brief, the slices were fixed by immersion in freshly prepared 2.5% glutaraldehyde/4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS) at 4 °C for at least 48 h, and subsequently processed with the avidin-biotin-peroxidase method in order to reveal cell morphology4. The morphology of the cells was reconstructed and analyzed using a 100× oil-immersion objective lens and camera lucida system (Neurolucida, MicroBrightField).
Surgical procedure for in vivo experiments
Anesthesia was induced with 3% isoflurane and maintained with 1.5–2% isoflurane during the surgical procedure. Anesthetized mice were placed in a stereotaxic head holder (Kopf Instruments) and body temperature was maintained at 37 °C throughout the surgery using a homeothermic blanket system (Harvard Instruments). After shaving the scalp, bupivicane (0.05 cc, 0.5%, Marcaine) was applied subcutaneously, and after 10–20 min an approximately 1 cm2 area of skin was removed above the skull and the underlying fascia was scraped and removed. The wound margins were sealed with a thin layer of surgical glue (VetBond, 3M), and a headbar was attached to the skull with dental cement (Dentsply Grip Cement). At this point, the mouse was removed from the stereotax and the skull was held stationary on a small platform by means of the newly attached headbar. Using a surgical drill and HP 1/2 burr, a ~3 mm craniotomy was made over V1 (2.7 mm lateral of the midline, contacting the lambdoid suture), and the exposed cortex was washed with artificial cerebrospinal fluid (ACSF) containing (in mM): 125 NaCl, 5 KCl, 10 Glucose, 10 HEPES, 2 CaCl2 and 2 MgSO4, pH ~ 7.4. The craniotomy was sealed with a coverslip containing a 500 μm-diameter hole that had been previously drilled with a diamond-tipped burr (Coltene/Whaledent) to allow access with patch-clamp pipette(s).
In vivo electrophysiology and RNA extraction
Patch-clamp pipettes were pulled from borosilicate glass (1.5 mm OD × 0.86 mm ID, Sutter Instruments) to an impedance of 4–7 MΩ. Pipettes were filled with the same RNase-free modified internal solution used for slice electrophysiology (see above) with the addition of Alexa 488 or 598 (10–50 μM) to allow visualization of the pipette and extracellular space. A manometer (Fisher Scientific 06-664-19) and custom-built pressure manifold allowed fast switching between high pressures while entering the bath and penetrating the dura (~150 mbar), and low pressures (~20–50 mbar) while advancing the pipette through the cortex under two-photon guidance, which helped to reduce the overall volume of intracellular solution ejected from the pipette. Bias currents were zeroed once the pipette was placed in the bath. Gigaseals were allowed to stabilize for 3–5 min before break-in. Compensating for tissue distortion by retracting the pipette ~10 μm during this time resulted in improved access and more stable recordings. Membrane potential (Vm) was not adjusted for the liquid junction potential. The procedure for RNA extraction was essentially identical to that described for slice recordings, relying on two-photon guidance in this case to visualize when the cell contents had entered the pipette (Fig. 1g).
Library construction and sequencing
We converted the RNA collected from patch-clamped neurons into complementary DNA (cDNA) using the Smart-seq2 protocol38. Briefly, poly(A)+ RNA was reverse transcribed using a tailed oligo(dT) primer (5′-AAGCAGTGGTATCAACGCAGAGTACT(30)VN-3′, where V represents A, C or G) and Moloney murine leukemia virus reverse transcriptase (MMLV RT). When the reverse transcriptase reaches the 5′ end of an RNA molecule, the terminal transferase activity of MMLV adds several nontemplated C nucleotides to the 3′ end of the cDNA molecule. These additional C nucleotides base-pair with the template switching oligo (TSO, 5′-AAGCAGTGGTATCAACGCAGAGTACATrGrG+G-3′, where rG indicates riboguanosines and +G indicates a locked nucleic acid (LNA)-modified guanosine), allowing the reverse transcriptase to switch templates and continue transcribing to the end of the TSO. The resulting first-strand cDNA molecule thus contains the full-length mRNA as well as universal priming sites. After 18 cycles of amplification, 1 ng of purified cDNA was used to construct sequencing libraries using our in-house Tn5-mediated tagmentation39. Briefly, cDNA was fragmented by Tn5 transposase at 55 °C for 8 min, followed by incubation with 5 μl 0.2% SDS for 5 min at room temperature. Whole volume (25 μl) was then used for enrichment PCR with ten cycles of amplification. The PCR master mix included 10 μl of Fidelity Buffer (5×), 1.5 μl dNTPs (10 mM), 1 μl KAPA HiFi DNA polymerase (1 unit/μl, all three reagents from KAPA Biosystems) and 1 μl each of Index i7 (0.1 μM) and Index i5 (0.1 μM). Quality control was performed on both the amplified cDNA and the final library using a Bioanalyzer (Agilent). cDNA samples containing less than 200 pg/μl cDNA from 300–9,000 bp (15 μl total volume), or with an average size (considering the band from 300–9,000 bp) less than 1,500 bp were not sequenced (n = 50/108 samples). The DNA were sequenced from single end (43 bp) together with both i5 and i7 indices (6 bp each) using an Illumina HiSeq 2000. Investigators were blinded to cell type during library construction and sequencing.
Read processing and quantification of gene expression
Reads were aligned to the mouse genome (mm10 assembly) using STAR (v2.4.1c)40, with default settings except for the use of the automatic two-pass alignment strategy (–twopassMode Basic) to increase the sensitivity of splice junction identification and quantification. We discarded cells (n = 2) with less than 50,000 sequenced reads. We next verified that uniquely aligned reads in the Patch-seq libraries came from mRNAs by overlaying aligned read coordinates with annotated NCBI RefSeq genes. All libraries had >68% of reads mapping to known exons and introns, in line with standard single-cell RNA-seq libraries generated from lysed whole cells. Uniquely aligned reads were used to quantify gene expression levels using rpkmforgenes41. Expression levels were normalized to the number of reads per kilobase of transcript per million total reads (RPKM value), using NCBI RefSeq (downloaded on 24th of June 2014) gene and transcript models. We detected on average ~7,000 genes per L1 interneuron above 1 RPKM. To filter out noise from the expression data, we omitted genes that did not have an expression value of at least 1 RPKM in more than one sample. After the noise extraction, the dimensionality of the complete dataset was reduced to ~16,000 genes, and when considering only L1 interneurons it was reduced to ~15,000 genes. We next log2 transformed the RPKM values, and performed all computational analyses in log2-space. Investigators were blinded to cell type during read processing and quantification of gene expression.
Identification of highly variable genes
We ranked genes according to biological variation across the L1 interneurons, after controlling for the relationship between mean expression and technical variability42. We made an in-house R implementation of the method previously described42, with the addition of first replacing the largest positive outlier with the second highest value (performed to reduce the occurrence of genes with expression in a single cell from being selected in the most variable genes). The ranked list of genes is available in Supplementary Table 2 and, as expected, contains many genes with important functions in interneurons or genes that have been linked to psychiatric disorders.
Clustering analysis
The hierarchical clustering based on marker gene expression (Fig. 2a) was performed using the clustermap implementation in the python package seaborn, with Pearson correlation as metric and average linkage for calculating cluster distances. The package internally uses the scientific python module cluster (scipy.cluster) for clustering and the matplotlib packages for visualization. We employed the affinity propagation algorithm to cluster L1 interneurons based on their gene expression similarities using the negative Euclidean distance as metric (MATLAB implementation, available at the authors homepage: http://www.psi.toronto.edu/index.php?q=affinity%20propagation). The clustering was performed in the high-dimensional space spanned by the 3,000 most variable genes (described in the above section). In order to treat every cell as a potential cluster center, the preference parameter was set to the median similarity. The algorithm partitioned the interneurons into two main groups and additionally four clusters containing only one cell each (those four cells were labeled as outliers). The resulting clustering (Fig. 2d) is in good agreement with both the output of the automatic classifier and the blinded expert classification of the L1 interneurons to eNGCs and SBCs (Fig. 2e,f). In order to test if the number of samples analyzed is sufficient to distinguish the two cell types and provide a robust classification, we repeated the clustering analysis using subsets from the 46 ex vivo L1 interneurons (Supplementary Fig. 7). The number of sample sizes tested ranged from 41 to 11 cells, with a decrement step of 5 and 250 iterations for each different sample size. In each subsampling, cells were selected at random and used as input to the affinity propagation algorithm. The resulting clustering was compared with the clusters obtained initially from the whole dataset to score the cells that were classified correctly. The same procedure was repeated considering all the L1 interneurons, yielding similar results (data not shown).
Dimensionality reduction techniques
To reduce the dimensionality of the gene expression data in an unbiased manner, we employed both Principal Component Analysis (using the pca function in MATLAB) and t-Distributed Stochastic Neighbor Embedding (t-SNE) (using the MATLAB implementation of t-SNE) to project the gene space onto two dimensions. For t-SNE analyses we adjusted the default parameters according to the dimensionality of our dataset and provided the principal components as input for the initialization of cells. The number of principal components was selected so that the 60% of total variability in the data will be retained during the preprocessing step, corresponding to the first 28 principal components when analyzing all 58 cells and to 23 principal components when considering only L1 interneurons. The parameter for the perplexity of the Gaussian distributions (number of neighboring cells considered effective in the algorithm) was set to 20 when analyzing all 58 cells and to 10 when comparing L1 interneurons. Very similar two-dimensional maps were generated with different numbers of genes or parameters indicating that the separation is robust. The dimensionality reduction of all 58 cells (Supplementary Fig. 5a,b) used as input the normalized expression of all 16,000 genes, whereas the analyses of the L1 interneurons (Fig. 2d,e) used as input the 3,000 most variable genes.
Cell type classifier based on electrophysiological properties
To build an automatic cell type classifier based on electrophysiological properties, we trained an L1-regularized logistic regression43 with 20-fold cross-validation on a set of 72 cells, for which cell-type labels were inferred from reconstructed morphologies (Fig. 1b). As input, we used resting membrane potential, input resistance, action potential (AP) decay constant, AP threshold, AP amplitude, after-depolarization, adaptation index, presence of delayed spiking and presence of bursting. All variables were z-scored before training the classifier. We used the implementation provided by lassoGlm in MATLAB with a binomial output distribution. We applied the “1 SE” rule43 and chose the most strongly regularized decoder within 1 SE of the decoder with the best decoding performance. Most weight was placed on after depolarization amplitude and delayed spiking (Fig. 1e). We verified the performance of the classifier by training the classifier only on a randomly selected stratified subset of the data (46 cells) and evaluated it on a held-out validation set (26 cells). On the training set the classifier performs at nearly 100% correct, on the validation set it achieves a performance of ~92%.
We then applied the decoder to the electrophysiological data collected during the RNA extraction process and classified the cells. The correlation between a manual expert classification and our automatic classification was very high (correlation between nonthresholded classifier output and manual score: 0.91). Both the manual expert classification and the automatic cell type classification were carried out blinded to the gene expression profiles of the neurons.
Predicting cell type and physiological properties from gene expression
We used regularized generalized linear models with a binomial output distribution (i.e., logistic link function) to predict cell type, or with a normal output distribution (i.e., linear link function) to predict continuous physiological properties (after-depolarization amplitude, after-hyperpolarization amplitude, membrane time constant, AP width, AP amplitude, natural log of the adaptation index (second/first inter-spike interval), and resting membrane potential). As input we used between 50 and 250 genes with highest relative variability (see section “Identification of highly variable genes,” above). We used the elastic-net algorithm with alpha = 0.95 (i.e., a high degree of sparsity in the weights), regularizing the weights with a mix of L2 and L1 regularization. The elastic-net penalty is particularly well suited for coping with highly correlated predictors43. The genes used for predicting each physiological property are reported in Supplementary Table 3.
To obtain a realistic estimate of how well we can predict cell type or physiological properties, we used nested cross-validation. In the outer cross-validation loop, we iterated over each individual cell, evaluating the prediction for that cell with a model trained on all but that cell (‘leave-one-out cross-validation’). Cross-validation in the inner loop was used to select the optimal amount of regularization. The evaluated model is selected based on the 1-SE rule to prevent overfitting43. As sparse models can have a substantial bias in their outcome, we refit the model using only the selected genes. Performance was measured using percent correct for binary features (Fig. 2f) and Spearman rank correlation for continuous features (Fig. 2g–i). 95%-confidence intervals on percent correct scores (Fig. 2f) are Clopper-Pearson intervals44.
Differential gene expression analysis
To identify genes that drive the separation of L1 interneurons into two distinct molecular classes (Fig. 3), we performed differential expression analysis between the two main clusters identified by the affinity propagation clustering. We further extended the analysis to test for differential expression between the ex vivo and in vivo interneurons, plus between the different cell groups based on the electrophysiological properties. In particular, we tested for genes that differ between the two cell types as classified by blinded expert examination of the firing pattern, delayed-spiking and nondelayed-spiking interneurons, as well as the burst-spiking and non-burst-spiking interneurons. The results obtained from all these comparisons are summarized in Supplementary Table 4. For the differential analysis, we used the R/Bioconductor package SCDE27 with the default settings, except for increasing the transcription magnitude to 500 for increased sensitivity. The raw read counts were provided as input, as the SCDE algorithm requires integer values that should not be normalized. Genes with zero reads across the samples being compared were discarded. We fit the error models using either a common set of genes (“common fit”) or two different sets of genes, one for each group (“independent fit”). Both approaches generated very similar results and we report the results from the independent fit. We used the MATLAB toolbox aboxplot for drawing boxplots.
Gene set enrichment analysis
We investigated whether the differentially expressed genes within the two neuronal clusters (clusters A and B) were enriched for those that coded for proteins annotated with particular biological functions. We used three different types of tests and all results are summarized in Supplementary Table 5. First, we performed gene set enrichment analysis (GSEA), which is a threshold-independent (and therefore list-independent) method for finding gene set enrichments. The method was used with default settings except for providing a pre-ranked list of genes, as the differential expression test in GSEA was not designed for single-cell RNA-seq data. Therefore, we used the z-score from the SCDE analysis between the two cell types as gene ranking metric. Leading Edge Analysis (LEA) was performed afterwards using the output (FDR < 5%) from the GSEA, to identify the genes that were members of more than one significantly enriched set: the leading-edge subsets. The significant categories (FDR < 5%) are reported in Figure 3c and in sheet 1 of Supplementary Table 5. Second, we tried two variants of list-based gene ontology enrichment analyses. To this end, we selected the top 100 or 200 differentially expressed genes from each cell-type and computed the overlaps within the Gene Ontology annotations (Biological Process, Cellular Component and Molecular Function). We performed these analyses either using interneuron-expressed genes (sheets 2–3, Supplementary Table 5) or all genes as background (sheets 4–5, Supplementary Table 5) using DAVID. Using all genes as background is the standard procedure, but has the drawback that significant gene categories might resemble more interneuron-expressed genes than the differential expression between the two cell types. To alternatively use interneuron-expressed genes as background should preclude such confounding effects, but can be less powerful and instead depends on how the background set is chosen. Therefore, we generated the background set of interneuron-expressed genes to include those with expression above 5 RPKM in at least 20 cells (resulting in 6,300 genes) and uploaded these to DAVID to perform the gene set enrichment tests.
Supplementary Material
Acknowledgments
We thank A. Morgan for technical assistance. This study was supported by grants DP1EY023176, P30EY002520, T32EY07001, and DP1OD008301 to A.S.T.; grants from the Swedish Research Council and the Swedish Foundation for Strategic Research (FFL4) to R.S.; grant R01MH103108 to A.S.T. and K.F.T.; grant R01NS062829 to K.F.T.; the McKnight Scholar Award to A.S.T.; and the Arnold and Mabel Beckman Foundation Young Investigator Award to A.S.T. C.R.C. was supported by grants F30MH095440, T32GM007330 and T32EB006350. M.B. and P.B. were supported by the Deutsche Forschungsgemeinschaft (DFG, EXC 307) and the German Federal Ministry of Education and Research (BMBF; BCCN Tübingen, FKZ 01GQ1002).
Footnotes
Accession codes. ArrayExpress: E-MTAB-4092.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
C.R.C. collected RNA samples, generated cDNA libraries, assisted with analysis and drafted the manuscript. A.P. performed the computational analyses of RNA-seq data. X.J. performed the ex vivo patch-clamp experiments, reconstructed neuronal morphologies and analyzed electrophysiological properties of the neurons. P.B. built the automated cell type classifier and generalized linear models. Q.D. and M.Y. generated cDNA and sequencing libraries. J.R. and S.S. performed the in vivo patch-clamp experiments. M.B. supervised the machine learning analysis. A.S.T., R.S. and K.F.T. supervised all experiments and analyses. All authors contributed to writing the paper.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Cajal SR, Pasik P, Pasik T. Texture of the Nervous System of Man and the Vertebrates. Springer; 2002. [Google Scholar]
- 2.Ascoli GA, et al. Petilla Interneuron Nomenclature Group Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat Rev Neurosci. 2008;9:557–568. doi: 10.1038/nrn2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burkhalter A. Many specialists for suppressing cortical excitation. Front Neurosci. 2008;2:155–167. doi: 10.3389/neuro.01.026.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang X, et al. Principles of connectivity among morphologically defined cell types in adult neocortex. Science. 2015;350:aac9462. doi: 10.1126/science.aac9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Connors BW, Gutnick MJ. Intrinsic firing patterns of diverse neocortical neurons. Trends Neurosci. 1990;13:99–104. doi: 10.1016/0166-2236(90)90185-d. [DOI] [PubMed] [Google Scholar]
- 6.Neher E, Sakmann B. Single-channel currents recorded from membrane of denervated frog muscle fibres. Nature. 1976;260:799–802. doi: 10.1038/260799a0. [DOI] [PubMed] [Google Scholar]
- 7.Tang F, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6:377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 8.Sandberg R. Entering the era of single-cell transcriptomics in biology and medicine. Nat Methods. 2014;11:22–24. doi: 10.1038/nmeth.2764. [DOI] [PubMed] [Google Scholar]
- 9.Fishell G, Heintz N. The neuron identity problem: form meets function. Neuron. 2013;80:602–612. doi: 10.1016/j.neuron.2013.10.035. [DOI] [PubMed] [Google Scholar]
- 10.Jaitin DA, et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343:776–779. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Darmanis S, et al. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci USA. 2015;112:7285–7290. doi: 10.1073/pnas.1507125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeisel A, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- 13.Sucher NJ, Deitcher DL, Baro DJ, Warrick RM, Guenther E. Genes and channels: patch/voltage-clamp analysis and single-cell RT-PCR. Cell Tissue Res. 2000;302:295–307. doi: 10.1007/s004410000289. [DOI] [PubMed] [Google Scholar]
- 14.Toledo-Rodriguez M, Markram H. Single-cell RT-PCR, a technique to decipher the electrical, anatomical, and genetic determinants of neuronal diversity. Methods Mol Biol. 2014;1183:143–158. doi: 10.1007/978-1-4939-1096-0_8. [DOI] [PubMed] [Google Scholar]
- 15.Subkhankulova T, Yano K, Robinson HP, Livesey FJ. Grouping and classifying electrophysiologically-defined classes of neocortical neurons by single cell, whole-genome expression profiling. Front Mol Neurosci. 2010;3:10. doi: 10.3389/fnmol.2010.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiu S, et al. Single-neuron RNA-Seq: technical feasibility and reproducibility. Front Genet. 2012;3:124. doi: 10.3389/fgene.2012.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGinley MJ, et al. Waking state: rapid variations modulate neural and behavioral responses. Neuron. 2015;87:1143–1161. doi: 10.1016/j.neuron.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Picelli S, et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- 19.Chan CH, et al. Emx1 is a marker for pyramidal neurons of the cerebral cortex. Cereb Cortex. 2001;11:1191–1198. doi: 10.1093/cercor/11.12.1191. [DOI] [PubMed] [Google Scholar]
- 20.Fremeau RT, Jr, et al. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–260. doi: 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- 21.Marshak DR. S100 beta as a neurotrophic factor. Prog Brain Res. 1990;86:169–181. [PubMed] [Google Scholar]
- 22.Bignami A, Eng LF, Dahl D, Uyeda CT. Localization of the glial fibrillary acidic protein in astrocytes by immunofluorescence. Brain Res. 1972;43:429–435. doi: 10.1016/0006-8993(72)90398-8. [DOI] [PubMed] [Google Scholar]
- 23.Stühmer T, Anderson SA, Ekker M, Rubenstein JL. Ectopic expression of the Dlx genes induces glutamic acid decarboxylase and Dlx expression. Development. 2002;129:245–252. doi: 10.1242/dev.129.1.245. [DOI] [PubMed] [Google Scholar]
- 24.Alcántara S, et al. Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J Neurosci. 1998;18:7779–7799. doi: 10.1523/JNEUROSCI.18-19-07779.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyoshi G, et al. Genetic fate mapping reveals that the caudal ganglionic eminence produces a large and diverse population of superficial cortical interneurons. J Neurosci. 2010;30:1582–1594. doi: 10.1523/JNEUROSCI.4515-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma J, Yao XH, Fu Y, Yu YC. Development of layer 1 neurons in the mouse neocortex. Cereb Cortex. 2014;24:2604–2618. doi: 10.1093/cercor/bht114. [DOI] [PubMed] [Google Scholar]
- 27.Kharchenko PV, Silberstein L, Scadden DT. Bayesian approach to single-cell differential expression analysis. Nat Methods. 2014;11:740–742. doi: 10.1038/nmeth.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang X, Wang G, Lee AJ, Stornetta RL, Zhu JJ. The organization of two new cortical interneuronal circuits. Nat Neurosci. 2013;16:210–218. doi: 10.1038/nn.3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oláh S, et al. Regulation of cortical microcircuits by unitary GABA-mediated volume transmission. Nature. 2009;461:1278–1281. doi: 10.1038/nature08503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalashnikova E, et al. SynDIG1: an activity-regulated, AMPA- receptor-interacting transmembrane protein that regulates excitatory synapse development. Neuron. 2010;65:80–93. doi: 10.1016/j.neuron.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macintyre G, et al. Association of NPAS3 exonic variation with schizophrenia. Schizophr Res. 2010;120:143–149. doi: 10.1016/j.schres.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 32.Stanco A, et al. NPAS1 represses the generation of specific subtypes of cortical interneurons. Neuron. 2014;84:940–953. doi: 10.1016/j.neuron.2014.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin L, et al. DPP6 regulation of dendritic morphogenesis impacts hippocampal synaptic development. Nat Commun. 2013;4:2270. doi: 10.1038/ncomms3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brose N. For better or for worse: complexins regulate SNARE function and vesicle fusion. Traffic. 2008;9:1403–1413. doi: 10.1111/j.1600-0854.2008.00758.x. [DOI] [PubMed] [Google Scholar]
- 35.Chao HT, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ting JT, Daigle TL, Chen Q, Feng G. Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol Biol. 2014;1183:221–242. doi: 10.1007/978-1-4939-1096-0_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sucher NJ, Deitcher DL. PCR and patch-clamp analysis of single neurons. Neuron. 1995;14:1095–1100. doi: 10.1016/0896-6273(95)90257-0. [DOI] [PubMed] [Google Scholar]
- 38.Picelli S, et al. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 39.Picelli S, et al. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 2014;24:2033–2040. doi: 10.1101/gr.177881.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramsköld D, Wang ET, Burge CB, Sandberg R. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput Biol. 2009;5:e1000598. doi: 10.1371/journal.pcbi.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brennecke P, et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat Methods. 2013;10:1093–1095. doi: 10.1038/nmeth.2645. [DOI] [PubMed] [Google Scholar]
- 43.Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 44.Clopper C, Pearson ES. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika. 1934;26:404–413. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.