

Summary
The unmet clinical need for myocardial salvage during ischaemia–reperfusion injury requires the development of new techniques for myocardial protection. In this study the protective effect of different local ischaemic preconditioning (LIPC) and remote ischaemic preconditioning (RIPC) protocols was compared in the rat model of myocardial ischaemia–reperfusion, using infarct size and ischaemic tachyarrhythmias as end‐points. In addition, the hypothesis that there is involvement of reactive oxygen species (ROS) in the protective signalling by RIPC was tested, again in comparison with LIPC. The animals were subjected to 30‐min coronary occlusion and 90‐min reperfusion. RIPC protocol included either transient infrarenal aortic occlusion (for 5, 15 and 30 min followed by 15‐min reperfusion) or 15‐min mesenteric artery occlusion with 15‐min reperfusion. Ventricular tachyarrhythmias during test ischaemia were quantified according to Lambeth Conventions. It was found that the infarct‐limiting effect of RIPC critically depends on the duration of a single episode of remote ischaemia, which fails to protect the heart from infarction when it is too short or, instead, too prolonged. It was also shown that RIPC is ineffective in reducing the incidence and severity of ischaemia‐induced ventricular tachyarrhythmias. According to our data, the infarct‐limiting effect of LIPC could be partially eliminated by the administration of ROS scavenger N‐2‐mercaptopropionylglycine (90 mg/kg), whereas the same effect of RIPC seems to be independent of ROS signalling.
Keywords: infarct size, local ischaemic preconditioning, myocardial ischaemia–reperfusion, reactive oxygen species, remote ischaemic preconditioning, ventricular tachyarrhythmias
The unmet clinical need for myocardial salvage during ischaemia–reperfusion injury (IRI) requires the development of new techniques of myocardial protection. Local ischaemic preconditioning (LIPC), one of the most powerful endogenous cardioprotective responses known to date, is induced by several brief episodes of myocardial ischaemia–reperfusion preceding prolonged ischaemic insult (Murry et al. 1986). Myocardial tolerance to ischaemia could also be increased after the application of brief ischaemic stimulus to an organ remote from the heart, a phenomenon termed ‘remote ischaemic preconditioning' (RIPC). RIPC was first described by Przyklenk et al. (1993) who demonstrated in the open‐chest dog model that non‐lethal ischaemia in the circumflex vascular bed limited infarct size induced by a subsequent prolonged occlusion of the left anterior descending artery. Other experimental studies showed that the heart could be preconditioned by brief ischaemia of the small intestine (Gho et al. 1996), kidney (Gho et al. 1996; Pell et al. 1998) and limb (Birnbaum et al. 1997; Petrishchev et al. 2001). It should be noted that the RIPC procedure is generally more applicable to the clinical situation, primarily because of its non‐invasiveness in the case of cuff‐induced transient ischaemia of the lower extremity(ies). The interest in RIPC as a clinically feasible procedure has been reinforced by the observation that remote ischaemia can reduce infarct size when instituted not only before but also during test ischaemia (Schmidt et al. 2007). At present, there are clinical data demonstrating the limitation of myocardial IRI by RIPC in cardiac surgery patients (Cheung et al. 2006; Thielmann et al. 2010) and patients undergoing primary percutaneous coronary intervention (Botker et al. 2010), but several recent cardiac surgery trials have questioned this benefit (Jones et al. 2013; Hong et al. 2014). On the one hand, this discrepancy could be attributed to the differences in patient‐ and surgery‐related factors among the studies. On the other hand, the difference in the outcome could be due to the differences in RIPC protocol, including the number of RIPC cycles, duration of ischaemia and reperfusion phase(s), and the amount of tissue subjected to ischaemia. In this regard, it seems worthwhile to perform additional experimental studies on RIPC, particularly those aimed at the investigation of RIPC protocols and end‐points. For instance, while it is well established that RIPC results in infarct size limitation (Gho et al. 1996; Birnbaum et al. 1997; Pell et al. 1998) and the attenuation of reperfusion‐induced arrhythmias (Oxman et al. 1997; Dow et al. 2012), very little is known about the effect of RIPC on the incidence and severity of ischaemic tachyarrhythmias.
Another very important question is whether the mechanisms underlying LIPC‐ and RIPC‐mediated myocardial protection are similar. The intracellular signalling pathways and mediators of LIPC may at least partially overlap with those of RIPC because it is known that specific inhibitors of mitochondrial ATP‐sensitive potassium channels and protein kinase C can abolish the protective effect of both types of preconditioning (Auchampach et al. 1992; Liu et al. 1994; Wang et al. 2002). However, several studies demonstrated the important differences in the signalling pattern evoked by LIPC and RIPC (Heidbreder et al. 2008; Heinen et al. 2011). It is known that reactive oxygen species (ROS) play a key role in both triggering and mediating the protective effect of LIPC (Das et al. 1999). Much less is known about the involvement of ROS in the protective effect of RIPC.
In the present study, we were interested to compare the protective effect of different LIPC and RIPC protocols in the rat model of myocardial ischaemia–reperfusion using infarct size and ischaemic tachyarrhythmias as end‐points. In addition, the hypothesis on the involvement of ROS in the protective signalling by RIPC was tested, again in comparison with LIPC – a gold standard of cardiac protection.
Methods
Animals
Male Wistar rats weighting 220–260 g were used throughout the experiments. The animals were maintained on a 12‐h light/dark cycle and administered food and water ad libitum.
Myocardial ischaemia–reperfusion model
The animals were anaesthetized with sodium pentobarbital (60 mg/kg intraperitoneally), tracheotomized and ventilated (SAR‐830P; CWE, Inc., Ardmore, PA, USA) using room air, with a tidal volume of 2 ml/100 g and a rate of approximately 60 breaths per minute. Core body temperature was maintained at 37.0 ± 0.5°C by a feedback‐controlled heating pad (TCAT‐2LV controller; Physitemp Instruments Inc., Clifton, NJ, USA). The left carotid artery and right femoral vein were cannulated for the measurement of mean arterial pressure (MAP) and anaesthesia maintenance respectively. Lead II of the electrocardiogram was monitored for the registration of heart rate (HR) and arrhythmias. After 10 min of stabilization, a left thoracotomy was performed. A 6‐0 polypropylene thread was placed around a prominent branch of the left coronary artery, and the ends were passed through a polyethylene tube as an occluder. Exclusion criteria were MAP < 50 mmHg and/or HR < 300 at any time point during the experiment.
Experimental protocol
The present study included three experimental series. In Series 1, the infarct‐limiting effect of different durations of single RIPC stimulus was studied. In Series 2, we compared the effect of LIPC and RIPC on infarct size and ischaemic tachyarrhythmias (TA). Further, the role of ROS in the mechanism of LIPC and RIPC was assessed in Series 3 using the ROS scavenger N‐2‐mercaptopropionylglycine (MPG). A more detailed description of the experimental procedures and measurements in three series is provided below.
Series 1: Effect of remote ischaemia duration on myocardial infarct size
The following experimental groups were included in this series (Figure 1a): (i) controls (CON, n = 7) – after surgical preparation, the animals were subjected to 30‐min coronary artery occlusion (CAO) followed by 90‐min reperfusion; (ii) RIPC by 5‐min infrarenal aorta occlusion (RIPC IAO5, n = 7) – the abdominal aorta was dissected free from surrounding tissues under surgical microscope just below the origin of renal arteries, and 30‐min CAO was preceded by 5‐min infrarenal aortic occlusion (IAO) and 15‐min reperfusion; (iii) RIPC by 15‐min IAO (RIPC IAO15, n = 7) – the infrarenal aorta was occluded for 15 min; (iv) RIPC by 30‐min IAO (RIPC IAO30, n = 6) – the infrarenal aorta was occluded for 30 min. Haemodynamic parameters (MAP and HR) were registered at baseline, prior to CAO, at 15th and 30th minutes of ischaemia, as well as at 30th, 60th and 90th minutes of reperfusion. At the end of reperfusion, the animals were euthanized, followed by the determination of area at risk and infarct size.
Figure 1.
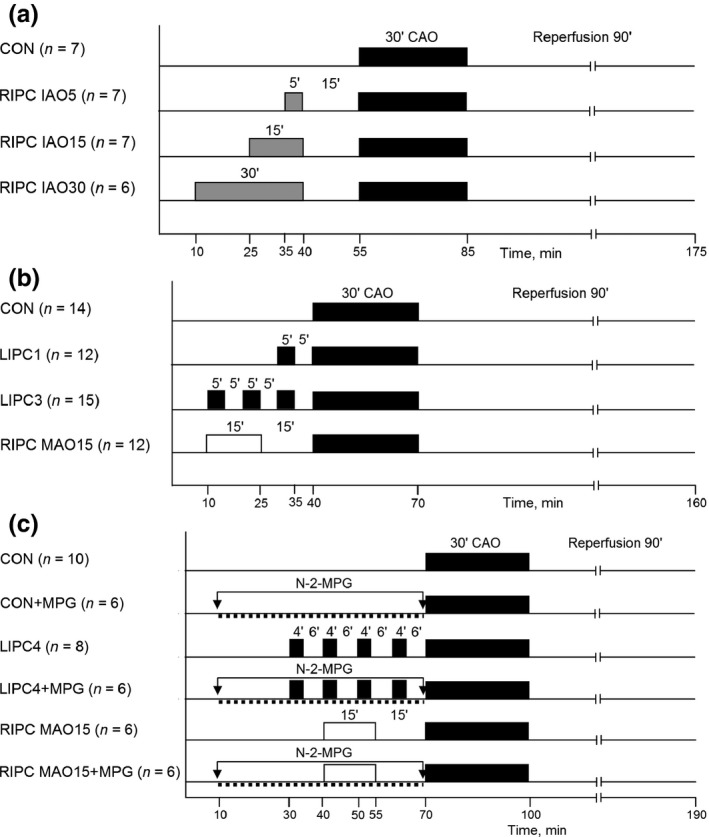
Experimental protocol. Series 1 (a), Series 2 (b), Series 3 (c). See text for details.
Series 2: Effect of LIPC and RIPC on infarct size and ischaemic tachyarrhythmias
The animals were randomly allocated into four groups (Figure 1b): (i) controls (CON, n = 14) underwent 30‐min CAO and 90‐min reperfusion; (ii) LIPC with a single ischaemic stimulus (LIPC1, n = 12) – 5‐min CAO with 5‐min reperfusion preceded 30‐min CAO; (iii) LIPC with three episodes of ischaemia (LIPC3, n = 15) – three 5‐min episodes of CAO interspersed with 5‐min bouts of reperfusion were used before test ischaemia; (iv) RIPC (RIPC MAO15, n = 12) – RIPC was induced by 15‐min mesenteric artery occlusion and 15‐min reperfusion prior to 30‐min CAO. Haemodynamic parameters were registered at baseline, prior to CAO, at 15th and 30th minutes of ischaemia, as well as at 30th, 60th and 90th minutes of reperfusion. End‐points were myocardial infarct size and test ischaemia‐induced tachyarrhythmias.
Series 3: The role of ROS in the mechanisms of LIPC and RIPC
The following experimental groups were included in this series (Figure 1c): (i) controls (CON, n = 10) – 30‐min CAO plus 90‐min reperfusion; (ii) controls treated with MPG (CON + MPG, n = 6) – intravenous infusion of MPG (MP Biomedicals, Solon, OH, USA) solution in normal saline (1 ml, pH = 7.36) was started 1 h prior to CAO. MPG was infused using a syringe pump (Razel, Saint Albans, VT, USA) at a total dose of 90 mg/kg; (iii) LIPC with four episodes of brief ischaemia (LIPC4, n = 8) – 30‐min CAO was preceded by four cycles of LIPC each consisting of 4‐min CAO and 6‐min reperfusion; (iv) LIPC4 + MPG (n = 6) – LIPC was combined with intravenous treatment with MPG, which was started 20 min prior to first episode of LIPC. The dose of MPG was the same as in the CON + MPG group; (v) RIPC MAO15 (n = 6) – RIPC was induced by 15‐min mesenteric artery occlusion and 15‐min reperfusion; (vi) RIPC MAO15 + MPG (n = 6) – MPG infusion was started 30 min prior to the induction of RIPC. Haemodynamic parameters were registered at baseline, 30 min prior to CAO, immediately prior to CAO, at 15th and 30th minutes of ischaemia, as well as at the end of reperfusion. At the end of the experiment, myocardial infarct size was determined histochemically.
Arrhythmia quantification
Ischaemic ventricular tachyarrhythmias were quantified according to Lambeth Conventions (Walker et al. 1988; Curtis et al. 2013) in Series 2. Ischaemia‐induced episodes of ventricular tachycardia (VT) and ventricular fibrillation (VF) were analysed separately. Mechanical or electrical cardioversion was not attempted to terminate VT/VF. The following characteristics of VT/VF were assessed: (i) the number of animals having at least one episode of VT/VF per group; (ii) the number of VT episodes per animal; (iii) the number of VF episodes per animal; (iv) time to onset of the first VT or VF episode; (v) total duration of VT plus VF episodes on the per animal basis (seconds); (vi) the number of animals with persistent forms of VF. Quantification of ischaemic ventricular tachyarrhythmias and data analyses were performed by an investigator blinded to the study groups.
Infarct size measurement
At the end of the experiment, the left coronary artery was reoccluded, followed by the administration of 0.5 ml of 5% Evans Blue (MP Biomedicals) through the femoral vein for the identification of area at risk (AR). The hearts were excised and cut into five 2‐mm‐thick slices parallel to the atrioventricular groove. The basal surface of each slice was digitally photographed. The slices were immersed in 1% solution of 2,3,5‐triphenyltetrazolium chloride (MP Biomedicals) at 37°C (pH 7.4) for 15 min and photographed again for the identification of infarct area (IA). The images were digitized using Adobe Photoshop CS. The AR was expressed as percentage of the whole slice, and the IA was expressed as a percentage of AR. Values of AR and IA for each heart were obtained by summarizing data of the slices and calculating mean values. Animals with AR below 15% were excluded from the study. Infarct size measurement and data analyses were performed by an investigator blinded to the study groups.
Statistical analysis
All data are presented as mean ± SD. Statistical analysis was performed with the use of spss 12.0 software package. Differences in haemodynamic parameters over time in each group were analysed by Friedman repeated‐measures anova on ranks followed by Dunn multiple‐comparisons test. The post hoc tests were performed only if anova resulted in F < 0.05, and there was no variance in homogeneity. Fisher's exact test was utilized to determine whether there were any differences between the groups in number of rats with transient or persistent VT/VF. The differences in time to onset of VT/VF, number of episodes of VT or VF per animal and total duration of arrhythmias were analysed nonparametrically using Kruskal–Wallis test. Kruskal–Wallis test was used to determine the differences in infarct size, followed by pairwise intergroup comparisons made using nonparametric Mann–Whitney U‐test. The differences were considered significant at P < 0.05.
Ethical approval
All procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (NIH publication No. 85‐23, revised 1996), European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes, and were approved by the ethics committees at Federal Almazov North‐West Medical Research Centre and Pavlov First Saint‐Petersburg State Medical University, St. Petersburg, Russian Federation.
Results
Mortality and exclusions
At the end of the study, the results could be obtained from 111 of 122 rats (90.9%) (Figure 1). The mortality rates due to persistent arrhythmia were 1/7, 0/7, 1/7 and 0/6 in CON, RIPC IAO5, RIPC IAO15 and RIPC IAO30 groups, respectively (Series 1); 2/14, 0/12, 0/15 and 2/12 in CON, LIPC1, LIPC3 and RIPC MAO15 groups, respectively (Series 2); 2/10, 0/6, 0/8, 0/6, 0/6 and 0/6 in CON, CON + MPG, LIPC4, LIPC4 + MPG, RIPC MAO15 and RIPC MAO15 + MPG groups respectively (Series 3). One rat was excluded from the LIPC1 group due to the small AR size (12%), and two more animals were excluded because of severe bleeding during myocardial reperfusion (one in LIPC3 group and one in LIPC4 group). These animals were used for the quantification of ischaemic arrhythmias, but not for the analysis of infarct size or haemodynamic variables.
Haemodynamic variables
Haemodynamic data in Series 1–3 are summarized in Tables 1, 2, 3 respectively. MAP and HR values were not different among groups at baseline. A progressive decrease in both MAP and HR was evident in all groups over the course of the experiments. Thus, MAP values at the end of reperfusion were significantly lower in comparison with baseline values in all groups.
Table 1.
Haemodynamic data in different groups of experimental Series 1
| CON (n = 6) | RIPC IAO5 (n = 7) | RIPC IAO15 (n = 6) | RIPC IAO30 (n = 6) | |
|---|---|---|---|---|
| MAP, mmHg | ||||
| Baseline | 121 ± 18 | 119 ± 12 | 125 ± 16 | 118 ± 20 |
| Before CAO | 119 ± 11 | 123 ± 21 | 118 ± 24 | 68 ± 12* |
| 15‐min CAO | 115 ± 10 | 117 ± 17 | 117 ± 22 | 74 ± 13* |
| 30‐min CAO | 98 ± 16 | 103 ± 12 | 105 ± 17 | 72 ± 15* |
| 30‐min reperfusion | 86 ± 15* | 105 ± 18 | 108 ± 21 | 69 ± 17* |
| 60‐min reperfusion | 82 ± 19* | 91 ± 10* | 98 ± 25 | 68 ± 21* |
| 90‐min reperfusion | 79 ± 12* | 84 ± 14* | 86 ± 13* | 64 ± 15* |
| HR, beats/min | ||||
| Baseline | 412 ± 43 | 398 ± 36 | 405 ± 41 | 388 ± 38 |
| Before CAO | 408 ± 34 | 402 ± 27 | 394 ± 34 | 395 ± 26 |
| 15‐min CAO | 395 ± 48 | 388 ± 38 | 392 ± 39 | 379 ± 32 |
| 30‐min CAO | 402 ± 24 | 389 ± 47 | 381 ± 27 | 382 ± 34 |
| 30‐min reperfusion | 398 ± 28 | 378 ± 39 | 386 ± 30 | 385 ± 40 |
| 60‐min reperfusion | 388 ± 37 | 372 ± 32 | 378 ± 33 | 374 ± 41 |
| 90‐min reperfusion | 379 ± 45 | 369 ± 35 | 376 ± 46 | 366 ± 37 |
Data are mean ± SD. CAO, coronary artery occlusion. *P < 0.05 vs. baseline.
Table 2.
Haemodynamic data in different groups of experimental Series 2
| CON (n = 12) | LIPC1 (n = 11) | LIPC3 (n = 14) | RIPC MAO15 (n = 10) | |
|---|---|---|---|---|
| MAP, mmHg | ||||
| Baseline | 115 ± 12 | 121 ± 15 | 118 ± 14 | 125 ± 16 |
| Before CAO | 118 ± 14 | 116 ± 17 | 109 ± 17 | 120 ± 10 |
| 15‐min CAO | 105 ± 13 | 108 ± 15 | 111 ± 8 | 117 ± 14 |
| 30‐min CAO | 117 ± 14 | 105 ± 11 | 102 ± 15 | 110 ± 21 |
| 30‐min reperfusion | 105 ± 16 | 95 ± 13* | 93 ± 12* | 101 ± 19 |
| 60‐min reperfusion | 96 ± 10* | 93 ± 14* | 91 ± 10* | 92 ± 13* |
| 90‐min reperfusion | 82 ± 9* | 87 ± 10* | 85 ± 18* | 90 ± 14* |
| HR, beats/min | ||||
| Baseline | 416 ± 25 | 421 ± 25 | 425 ± 32 | 402 ± 23 |
| Before CAO | 410 ± 31 | 406 ± 18 | 398 ± 36 | 388 ± 25 |
| 15‐min CAO | 405 ± 33 | 412 ± 34 | 396 ± 23 | 398 ± 19 |
| 30‐min CAO | 396 ± 19 | 398 ± 29 | 387 ± 31 | 406 ± 29 |
| 30‐min reperfusion | 398 ± 34 | 382 ± 32 | 389 ± 29 | 395 ± 31 |
| 60‐min reperfusion | 374 ± 26* | 379 ± 27* | 390 ± 38 | 391 ± 32 |
| 90‐min reperfusion | 361 ± 29* | 370 ± 28* | 378 ± 31* | 369 ± 24* |
Data are mean ± SD. CAO, coronary artery occlusion. *P < 0.05 vs. baseline.
Table 3.
Haemodynamic data in different groups of experimental Series 3
| CON (n = 8) | CON + MPG (n = 6) | LIPC4 (n = 7) | LIPC4 + MPG (n = 6) | RIPC MAO15 (n = 6) | RIPC MAO15 + MPG (n = 6) | |
|---|---|---|---|---|---|---|
| MAP, mmHg | ||||||
| Baseline | 117 ± 13 | 115 ± 14 | 118 ± 19 | 122 ± 17 | 116 ± 22 | 125 ± 16 |
| 30 min before CAO | 116 ± 15 | 111 ± 9 | 115 ± 21 | 120 ± 22 | 112 ± 24 | 119 ± 17 |
| Before CAO | 114 ± 28 | 109 ± 18 | 112 ± 16 | 110 ± 19 | 109 ± 17 | 118 ± 23 |
| 15‐min CAO | 110 ± 10 | 106 ± 13 | 107 ± 24 | 117 ± 14 | 106 ± 21 | 111 ± 19 |
| 30‐min CAO | 105 ± 16 | 102 ± 21 | 100 ± 16 | 104 ± 20 | 99 ± 19 | 108 ± 22 |
| 90‐min reperfusion | 92 ± 10* | 88 ± 15* | 83 ± 19* | 79 ± 12* | 85 ± 15* | 89 ± 19* |
| HR, beats/min | ||||||
| Baseline | 398 ± 37 | 401 ± 35 | 405 ± 29 | 388 ± 23 | 411 ± 22 | 384 ± 28 |
| 30 min before CAO | 395 ± 28 | 399 ± 32 | 398 ± 32 | 379 ± 25 | 404 ± 24 | 383 ± 32 |
| Before CAO | 392 ± 19 | 394 ± 21 | 385 ± 37 | 378 ± 28 | 396 ± 13 | 378 ± 33 |
| 15‐min CAO | 388 ± 24 | 391 ± 27 | 381 ± 29 | 372 ± 34 | 392 ± 24 | 376 ± 22 |
| 30‐min CAO | 380 ± 21 | 384 ± 30 | 376 ± 29 | 370 ± 16 | 387 ± 26 | 372 ± 25 |
| 90‐min reperfusion | 378 ± 30 | 382 ± 22 | 377 ± 33 | 369 ± 19 | 388 ± 29 | 370 ± 27 |
Data are mean ± SD. CAO, coronary artery occlusion. *P < 0.05 vs. baseline.
In Series 1, the haemodynamic response to RIPC procedure varied depending on the duration of RIPC stimulus (Figure 2). In all groups, IAO itself resulted in mild increase in MAP (approximately by 10–12%), which lasted for nearly 5 min and then normalized. Reperfusion of the hindquarter in RIPC IAO5 and RIPC IAO15 groups resulted in a transient MAP decrease (Figure 2a,b). Haemodynamic response to RIPC with mesenteric ischaemia was similar to that observed in RIPC IAO15 group. However, MAP was persistently decreased by approximately 60% in RIPC IAO30 group (Figure 2c, Table 1). No other intergroup differences in haemodynamic parameters were registered.
Figure 2.
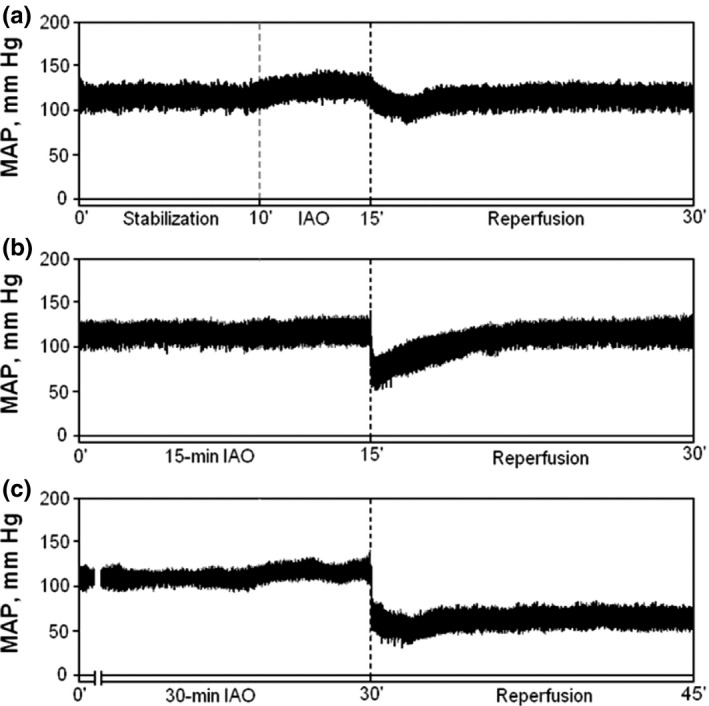
Representative samples of mean arterial pressure (MAP) recordings in anaesthetized rats subjected to infrarenal aortic occlusion–reperfusion in vivo. Ten‐minute stabilization was followed by 5‐, 15‐ and 30‐min occlusion of infrarenal aorta followed by 15‐min reperfusion (a–c).
Infarct size
There were no differences in AR among groups (Figure 3). In Series 1, IA was significantly lower in RIPC IAO15 group only (35 ± 4.5 vs. 58 ± 5.2% in controls, P < 0.05; Figure 3a). Both too short (RIPC IAO5) and prolonged (RIPC IAO30) episodes of remote ischaemia accompanied by the same duration of reperfusion failed to protect the heart from infarction (55 ± 7.1 and 62 ± 8.3%, respectively, P > 0.05 vs. CON). On the basis of these data, the optimal duration of remote ischaemic stimulus, that is 15 min, was used in other experiments.
Figure 3.
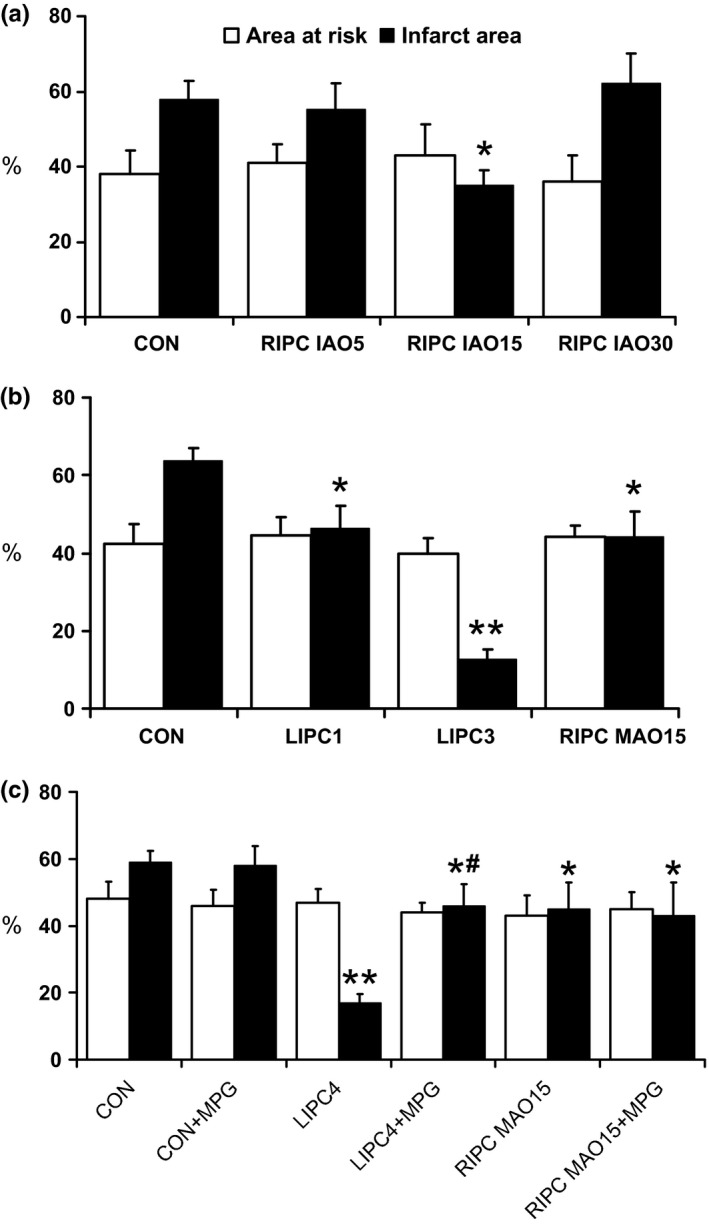
Area at risk and infarct area size in experimental Series 1–3 (a–c respectively). *P < 0.05 vs. control; **P < 0.01 vs. control; # P < 0.05 vs. LIPC4. For group legends, see text.
In Series 2, both LIPC and RIPC showed a significant limitation in infarct size, although the most prominent infarct‐limiting effect was observed in LIPC3 group (13 ± 2.5 vs. 64 ± 3.3% in CON, P < 0.01; Figure 3b). Infarct size in RIPC MAO15 group was comparable to that in LIPC1 group (44 ± 6.4 and 46 ± 5.8%, P > 0.05).
In Series 3, MPG administration itself had no effect on myocardial infarct size (58 ± 5.8 and 59 ± 3.3% in CON + MPG and CON groups, respectively, P > 0.05; Figure 3c). LIPC with four episodes of ischaemia (LIPC4 group) provided a robust reduction in infarct size (17 ± 2.5%, P < 0.01 vs. CON), which, however, has not exceeded the effect of three cycles of brief ischaemia–reperfusion applied in Series 2 (LIPC3 group). The infarct‐limiting effect of LIPC was partially abolished with MPG infusion in LIPC4 + MPG group (46 ± 6.4%, P < 0.05 vs. both LIPC4 and CON groups). In contrast, infarct size‐limiting effect of RIPC remained unchanged when RIPC was performed in conjunction with MPG infusion (43 ± 10.2 and 45 ± 8.1% in RIPC MAO15 + MPG and RIPC MAO15 groups, respectively, P > 0.05).
Ischaemic tachyarrhythmias
Quantitative characteristics of VT and VF occurring during 30‐min regional myocardial ischaemia in experimental Series 2 are summarized in Table 4. The number of episodes of ventricular tachycardia and ventricular fibrillation in the individual animals in different groups of experimental Series 2 is provided in Table S1. As follows from Table 4, a single episode of LIPC resulted in a significant reduction in both incidence and severity of ischaemic VT/VF, while three episodes of LIPC completely abolished both VT and VF. The characteristics of ischaemic VT/VF in RIPC MAO15 group generally did no differ from those in controls. The only significant difference was longer time to onset of the first episode of arrhythmia in RIPC MAO15 group (475 ± 63 s vs. 274 ± 43 s in controls, P < 0.05). Of note, despite the fact that the extent of infarct size limitation was comparable in LIPC1 and RIPC MAO15 group, the latter procedure failed to protect the heart from ventricular tachyarrhythmias.
Table 4.
Quantity of ventricular tachycardia and ventricular fibrillation in different groups of experimental Series 2
| CON (n = 14) | LIPC1 (n = 12) | LIPC3 (n = 15) | RIPC MAO15 (n = 12) | |
|---|---|---|---|---|
| Number of animals having at least one episode of VT/VF | 13 (93%) | 3 (25%)** | 0** | 11 (92%) |
| Number of VT episodes per animal | 1.9 ± 1.0 | 1.0 ± 1.0 | – | 2.3 ± 1.0 |
| Number of VF episodes per animal | 0.8 ± 0.8 | 0.3 ± 0.6 | – | 0.8 ± 0.9 |
| Time to onset of the first VT or VF episode, s | 274 ± 43 | 408 ± 69* | – | 475 ± 63* |
| Total duration of VT + VF episodes on the per animal basis, s | 48 ± 27 | 16 ± 13* | – | 56 ± 29 |
| Number of animals with persistent forms of VF | 2 (14%) | 0 | 0 | 2 (17%) |
Data are mean ± SD. VT, ventricular tachycardia, VF, ventricular fibrillation. *P < 0.05 vs. control; **P < 0.01 vs. control.
Discussion
The present study demonstrated that infarct‐limiting effect of RIPC critically depends on the duration of a single episode of remote ischaemia, which fails to protect the heart from infarction when it is too short or, instead, too prolonged. It was also shown that RIPC is ineffective in reducing the incidence and severity of ischaemia‐induced arrhythmias. According to our data, the infarct‐limiting effect of LIPC could be partially eliminated by the administration of ROS scavenger MPG, whereas the same effect of RIPC seems to be independent on ROS signalling.
The effect of RIPC protocol on the extent of cardiac protection
While the earlier studies on RIPC usually explored the effect of a single 10‐ to 15‐min episode of remote ischaemia on myocardial tolerance to IRI (Gho et al. 1996; Birnbaum et al. 1997; Pell et al. 1998; Weinbrenner et al. 2002), most of the recent experimental and also clinical studies used repeated 5‐min cycles of RIPC (Heinen et al. 2011; Ahmed et al. 2012; Jones et al. 2013; Hong et al. 2014). Does it matter whether a single or multiple cycles of RIPC are used and to what extent the protocol of RIPC affects the outcome? The data on this issue are still limited. Ahmed et al. (2012) found in the rat model that myocardial injury was significantly reduced only when three episodes of 5‐min femoral artery occlusion were used. No protection was evident with one, two or four episodes of RIPC. Weinbrenner et al. (2002) investigated the effect of 5‐, 10‐ and 15‐min IAO followed by 10‐min reperfusion on myocardial infarct size and found that only 10‐ and 15‐min IAO resulted in a significant infarct size limitation. These data fit well with our findings demonstrating that 5‐min IAO had no effect of infarct size. It might be hypothesized that such a brief episode of remote ischaemia is not sufficient for the accumulation of threshold concentration of metabolites and/or signalling molecules in the remote from the heart tissue. Therefore, the washout of these signalling agents upon reperfusion does not result in a systemic concentration required to trigger cardioprotective response in the heart. On the other hand, the data obtained in the present study showed that extended duration of remote ischaemia was not associated with myocardial protection. In our experiments, 30‐min IAO failed to protect the heart from infarction. This protocol of RIPC was associated with a significant haemodynamic instability, possibly diminishing myocardial perfusion and negating the protective effect of RIPC. From the clinical viewpoint, it is more feasible to use shorter episodes of limb ischaemia. However, it is important to realize that the overall reduction in infarct size due to RIPC is usually less pronounced than that due to LIPC (Weinbrenner et al. 2002; Ahmed et al. 2012). The amount of tissue subjected to ischaemia during RIPC is another very important determinant of RIPC effectiveness. Together with published evidence, the results of the present study indicate that the amount of ischaemic tissue should be significant enough to evoke cardiac protection.
Anti‐arrhythmic effect of LIPC and RIPC
At present, it is well established that LIPC results in the attenuation of ventricular arrhythmias developing during the first 10 min of ischaemia (phase 1A) (Liu & Downey 1992; Lu et al. 1993; Vegh et al. 1993). Moreover, LIPC is known to suppress reperfusion‐induced arrhythmias (Hagar et al. 1991; Liu & Downey 1992). The latter effect has been also shown in the experimental models of RIPC (Oxman et al. 1997; Dow et al. 2012). Much less is known about the effects of RIPC on early phase of ischaemic arrhythmogenesis. The recent study by Ahmed et al. (2012) demonstrated that RIPC induced by several 5‐min episodes of limb ischaemia in rats had no significant effect on arrhythmia score. Our data corroborate these findings because RIPC induced by 15‐min MAO and 15‐min reperfusion prior to test ischaemia had no influence on early ischaemic arrhythmias. The only significant difference found between control and RIPC group was delayed onset of the first episode of arrhythmias in RIPC. It seems that this effect of RIPC could be explained by the retardation of ischaemic injury development. One important conclusion stemming from these observations is that infarct‐limiting and anti‐arrhythmic effects of preconditioning are not necessarily correlated. For instance, RIPC could cause infarct size reduction but it is not resulting in arrhythmia suppression. Consistent with this idea, some studies indicated that the mechanisms of infarct‐limiting and anti‐arrhythmic effect of LIPC may in fact be different. For example, the anti‐arrhythmic effect of LIPC was shown to be independent of B2 bradykinin receptor activation (Sun & Wainwright 1994), nitric oxide synthase function (Lu et al. 1995) and mitochondrial ATP‐sensitive potassium channels (mKATP) (Lu et al. 1993; Vegh et al. 1993).
The role of ROS in LIPC‐ and RIPC‐mediated myocardial protection
One of the most intriguing questions as to the mechanisms of RIPC is how the protective signal is transferred from the remote organ to the heart. At present, there are three main hypotheses explaining this phenomenon (for review, see Kanoria et al. 2007; Galagudza et al. 2008; Przyklenk & Whittaker 2013 and references therein). First, the humoral protective signal(s) could be released into the systemic circulation upon reperfusion, reach the heart and activate corresponding receptors of cardiac myocytes (Weinbrenner et al. 2002, 2004). While it has been originally suggested that humoral mediators of RIPC are circulating in plasma, recent data demonstrated that they could be packed into extracellular vesicles (Giricz et al. 2014). Second, visceral afferent nerves could become activated by the accumulating adenosine, bradykinin and opioids in the remote organ, resulting in the activation of visceral reflex and release of catecholamines from cardiac adrenergic nerve endings (Gho et al. 1996; Tang et al. 1999). Third, autacoids released during remote ischaemia may modulate the function of circulating monocytes, neutrophils and regulatory T cells involved in the pathogenesis of lethal myocardial IRI (Weber 2010). In the present study, we were interested to investigate the role of ROS in LIPC‐ and RIPC‐mediated cardiac protection. Previous studies have shown that ROS are involved in both trigger and mediator stage of LIPC signalling (Das et al. 1999; Yue et al. 2002). It was suggested that small amounts of ROS produced during brief episodes of ischaemia–reperfusion are able to cause the phosphorylation of protein kinase C, mitogen‐activated protein kinases and tyrosine kinases (Das et al. 1999). The activation of kinases, in turn, results in the opening of mKATP and generation of additional amount of intracellular ROS playing a pivotal signalling function (Yue et al. 2002). The final end‐effector of LIPC still remains elusive, but it has been hypothesized that mitochondrial ROS‐mediated protection may arise from the phosphorylation of heat shock protein 27 and stabilization of stress fibres (Yue et al. 2002).
In this study, we used MPG as a pharmacological tool to investigate the role of ROS in LIPC and RIPC. MPG is a low molecular weight ROS scavenger, which readily diffuses through the membrane and predominantly quenches hydroxyl radical and peroxynitrite (Tang et al. 2002). MPG at a dose of 90 mg/kg/h has been previously shown to completely abolish the infarct‐limiting effect of LIPC in rat model (Yamashita et al. 1998). In our experiments, the same dose of MPG resulted in incomplete elimination of LIPC‐induced infarct limitation, which might point to the conclusion that signalling pathways of LIPC are highly redundant. The role of ROS in IAO‐induced RIPC has been previously investigated in the rat model (Weinbrenner et al. 2004). In contrast to our results, MPG at a dose of 20 mg/kg blocked the infarct‐limiting effect of RIPC in this study. The difference observed could be attributed to different doses of MPG and experimental protocols. As was mentioned before, the mechanisms of RIPC might differ depending on the organ, which is subjected to ischaemia. It seems that the neurogenic pathway is more important in the organs with abundant sensory innervation like small intestine (Gho et al. 1996; Tang et al. 1999), while the humoral route has been predominantly found in RIPC induced by the ischaemia of skeletal muscle and kidney (Weinbrenner et al. 2002).
In conclusion, RIPC effectiveness heavily depends on the duration of remote organ ischaemia and is lost when preconditioning ischaemia is too long. In contrast to LIPC, RIPC does not decrease the quantity of early ischaemic tachyarrhythmias, at least in the model used in the present study. ROS are unlikely to be involved in triggering or mediating the infarct‐limiting effect of RIPC.
Conflict of interest
The authors declare that they have no conflict of interest.
Funding source
This study was supported by the Russian Scientific Foundation (project 14‐15‐00473) and by Government of Russian Federation, Grant 074‐U01.
Supporting information
Table S1. The number of episodes of ventricular tachycardia and ventricular fibrillation in the individual animals in different groups of experimental Series 2. For group legends, see text.
References
- Ahmed L.A., Salem H.A., Attia A.S. et al (2012) Comparative study of the cardioprotective effects of local and remote preconditioning in ischemia/reperfusion injury. Life Sci. 90, 249–256. [DOI] [PubMed] [Google Scholar]
- Auchampach J.A., Grover G.J. & Gross G.J. (1992) Blockade of ischaemic preconditioning in dogs by the novel ATP dependent potassium channel antagonist sodium 5‐hydroxydecanoate. Cardiovasc. Res. 26, 1054–1062. [DOI] [PubMed] [Google Scholar]
- Birnbaum Y., Hale S.L. & Kloner R.A. (1997) Ischemic preconditioning at a distance: reduction of myocardial infarct size by partial reduction of blood supply combined with rapid stimulation of the gastrocnemius muscle in the rabbit. Circulation 96, 1641–1646. [DOI] [PubMed] [Google Scholar]
- Botker H.E., Kharbanda R., Schmidt M.R. et al (2010) Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: a randomised trial. Lancet 375, 727–734. [DOI] [PubMed] [Google Scholar]
- Cheung M.M., Kharbanda R.K., Konstantinov I.E. et al (2006) Randomized controlled trial of the effects of remote ischemic preconditioning on children undergoing cardiac surgery: first clinical application in humans. J. Am. Coll. Cardiol. 47, 2277–2282. [DOI] [PubMed] [Google Scholar]
- Curtis M.J., Hancox J.C., Farkas A. et al (2013) The Lambeth Conventions (II): guidelines for the study of animal and human ventricular and supraventricular arrhythmias. Pharmacol. Ther. 139, 213–248. [DOI] [PubMed] [Google Scholar]
- Das D.K., Engelman R.M. & Maulik N. (1999) Oxygen free radical signaling in ischemic preconditioning. Ann. N. Y. Acad. Sci. 874, 49–65. [DOI] [PubMed] [Google Scholar]
- Dow J., Bhandari A., Simkhovich B.Z. et al (2012) The effect of acute versus delayed remote ischemic preconditioning on reperfusion induced ventricular arrhythmias. J. Cardiovasc. Electrophysiol. 23, 1374–1383. [DOI] [PubMed] [Google Scholar]
- Galagudza M.M., Blokhin I.O., Shmonin A.A. et al (2008) Reduction of myocardial ischemia‐reperfusion injury with pre‐ and postconditioning: molecular mechanisms and therapeutic targets. Cardiovasc. Hematol. Disord. Drug Targets 8, 47–65. [DOI] [PubMed] [Google Scholar]
- Gho B.C., Schoemaker R.G., van den Doel M.A. et al (1996) Myocardial protection by brief ischemia in noncardiac tissue. Circulation 94, 2193–2200. [DOI] [PubMed] [Google Scholar]
- Giricz Z., Varga Z.V., Baranyai T. et al (2014) Cardioprotection by remote ischemic preconditioning of the rat heart is mediated by extracellular vesicles. J. Mol. Cell. Cardiol. 68, 75–78. [DOI] [PubMed] [Google Scholar]
- Hagar J.M., Hale S.L. & Kloner R.A. (1991) Effect of preconditioning ischemia on reperfusion arrhythmias after coronary artery occlusion and reperfusion in the rat. Circ. Res. 68, 61–68. [DOI] [PubMed] [Google Scholar]
- Heidbreder M., Naumann A., Tempel K. et al (2008) Remote vs. ischaemic preconditioning: the differential role of mitogen‐activated protein kinase pathways. Cardiovasc. Res. 78, 108–115. [DOI] [PubMed] [Google Scholar]
- Heinen N.M., Putz V.E., Gorgens J.I. et al (2011) Cardioprotection by remote ischemic preconditioning exhibits a signaling pattern different from local ischemic preconditioning. Shock 36, 45–53. [DOI] [PubMed] [Google Scholar]
- Hong D.M., Lee E.H., Kim H.J. et al (2014) Does remote ischaemic preconditioning with postconditioning improve clinical outcomes of patients undergoing cardiac surgery? Remote Ischaemic Preconditioning with Postconditioning Outcome Trial. Eur. Heart J. 35, 176–183. [DOI] [PubMed] [Google Scholar]
- Jones B.O., Pepe S., Sheeran F.L. et al (2013) Remote ischemic preconditioning in cyanosed neonates undergoing cardiopulmonary bypass: a randomized controlled trial. J. Thorac. Cardiovasc. Surg. 146, 1334–1340. [DOI] [PubMed] [Google Scholar]
- Kanoria S., Jalan R., Seifalian A.M. et al (2007) Protocols and mechanisms for remote ischemic preconditioning: a novel method for reducing ischemia reperfusion injury. Transplantation 84, 445–458. [DOI] [PubMed] [Google Scholar]
- Liu Y. & Downey J.M. (1992) Ischemic preconditioning protects against infarction in rat heart. Am. J. Physiol. 263, H1107–H1112. [DOI] [PubMed] [Google Scholar]
- Liu Y., Cohen M.V. & Downey J.M. (1994) Chelerythrine, a highly selective protein kinase C inhibitor, blocks the anti‐infarct effect of ischemic preconditioning in rabbit hearts. Cardiovasc. Drugs Ther. 8, 881–882. [DOI] [PubMed] [Google Scholar]
- Lu H., Remeysen P. & De Clerck F. (1993) The protection by ischemic preconditioning against myocardial ischemia‐ and reperfusion‐induced arrhythmias is not mediated by ATP‐sensitive potassium channels in rats. Coron. Artery Dis. 4, 649–657. [DOI] [PubMed] [Google Scholar]
- Lu H.R., Remeysen P. & De Clerck F. (1995) Does the antiarrhythmic effect of ischemic preconditioning in rats involve the L‐arginine nitric oxide pathway? J. Cardiovasc. Pharmacol. 25, 524–530. [DOI] [PubMed] [Google Scholar]
- Murry C.E., Jennings R.B. & Reimer K.A. (1986) Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74, 1124–1136. [DOI] [PubMed] [Google Scholar]
- Oxman T., Arad M., Klein R. et al (1997) Limb ischemia preconditions the heart against reperfusion tachyarrhythmia. Am. J. Physiol. 273, H1707–H1712. [DOI] [PubMed] [Google Scholar]
- Pell T.J., Baxter G.F., Yellon D.M. et al (1998) Renal ischemia preconditions myocardium: role of adenosine receptors and ATP‐sensitive potassium channels. Am. J. Physiol. 275, H1542–H1547. [DOI] [PubMed] [Google Scholar]
- Petrishchev N.N., Vlasov T.D., Sipovsky V.G. et al (2001) Does nitric oxide generation contribute to the mechanism of remote ischemic preconditioning? Pathophysiology 7, 271–274. [DOI] [PubMed] [Google Scholar]
- Przyklenk K. & Whittaker P. (2013) Genesis of remote conditioning: action at a distance – “hypotheses non fingo”? J. Cardiovasc. Med. (Hagerstown) 14, 180–186. [DOI] [PubMed] [Google Scholar]
- Przyklenk K., Bauer B., Ovize M. et al (1993) Regional ischemic “preconditioning” protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 87, 893–899. [DOI] [PubMed] [Google Scholar]
- Schmidt M.R., Smerup M., Konstantinov I.E. et al (2007) Intermittent peripheral tissue ischemia during coronary ischemia reduces myocardial infarction through a KATP‐dependent mechanism: first demonstration of remote ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 292, H1883–H1890. [DOI] [PubMed] [Google Scholar]
- Sun W. & Wainwright C.L. (1994) The potential antiarrhythmic effects of exogenous and endogenous bradykinin in the ischaemic rat heart in vivo . Coron. Artery Dis. 5, 541–550. [PubMed] [Google Scholar]
- Tang Z.L., Dai W., Li Y.J. et al (1999) Involvement of capsaicin‐sensitive sensory nerves in early and delayed cardioprotection induced by a brief ischaemia of the small intestine. Naunyn Schmiedebergs Arch. Pharmacol. 359, 243–247. [DOI] [PubMed] [Google Scholar]
- Tang X.L., Takano H., Rizvi A. et al (2002) Oxidant species trigger late preconditioning against myocardial stunning in conscious rabbits. Am. J. Physiol. Heart Circ. Physiol. 282, H281–H291. [DOI] [PubMed] [Google Scholar]
- Thielmann M., Kottenberg E., Boengler K. et al (2010) Remote ischemic preconditioning reduces myocardial injury after coronary artery bypass surgery with crystalloid cardioplegic arrest. Basic Res. Cardiol. 105, 657–664. [DOI] [PubMed] [Google Scholar]
- Vegh A., Papp J.G., Szekeres L. et al (1993) Are ATP‐sensitive potassium channels involved in the pronounced antiarrhythmic effects of preconditioning? Cardiovasc. Res. 27, 638–643. [DOI] [PubMed] [Google Scholar]
- Walker M.J., Curtis M.J., Hearse D.J. et al (1988) The Lambeth Conventions: guidelines for the study of arrhythmias in ischaemia infarction, and reperfusion. Cardiovasc. Res. 22, 447–455. [DOI] [PubMed] [Google Scholar]
- Wang Y.P., Maeta H., Mizoguchi K. et al (2002) Intestinal ischemia preconditions myocardium: role of protein kinase C and mitochondrial K(ATP) channel. Cardiovasc. Res. 55, 576–582. [DOI] [PubMed] [Google Scholar]
- Weber C. (2010) Far from the heart: receptor cross‐talk in remote conditioning. Nat. Med. 16, 760–762. [DOI] [PubMed] [Google Scholar]
- Weinbrenner C., Nelles M., Herzog N. et al (2002) Remote preconditioning by infrarenal occlusion of the aorta protects the heart from infarction: a newly identified non‐neuronal but PKC‐dependent pathway. Cardiovasc. Res. 55, 590–601. [DOI] [PubMed] [Google Scholar]
- Weinbrenner C., Schulze F., Sarvary L. et al (2004) Remote preconditioning by infrarenal aortic occlusion is operative via delta1‐opioid receptors and free radicals in vivo in the rat heart. Cardiovasc. Res. 61, 591–599. [DOI] [PubMed] [Google Scholar]
- Yamashita N., Hoshida S., Taniguchi N. et al (1998) A “second window of protection” occurs 24 h after ischemic preconditioning in the rat heart. J. Mol. Cell. Cardiol. 30, 1181–1189. [DOI] [PubMed] [Google Scholar]
- Yue Y., Qin Q., Cohen M.V. et al (2002) The relative order of mK(ATP) channels, free radicals and p38 MAPK in preconditioning's protective pathway in rat heart. Cardiovasc. Res. 55, 681–689. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The number of episodes of ventricular tachycardia and ventricular fibrillation in the individual animals in different groups of experimental Series 2. For group legends, see text.