

Abstract
CD200 induces immunosuppression in myeloid cells expressing its receptor CD200R, which may have consequences for tumor immunity. We found that human carcinoma tissues express not only full-length CD200 (CD200L) but also its truncated form, CD200S. Although CD200S is reported to antagonize the immunosuppressive actions of CD200L, the role of CD200S in tumor immunity has never been investigated. We established rat C6 glioma cell lines that expressed either CD200L or CD200S; the original C6 cell line did not express CD200 molecules. The cell lines showed no significant differences in growth. Upon transplantation into the neonatal Wistar rat forebrain parenchyma, rats transplanted with C6-CD200S cells survived for a significantly longer period than those transplanted with the original C6 and C6-CD200L cells. The C6-CD200S tumors were smaller than the C6-CD200L or C6-original tumors, and many apoptotic cells were found in the tumor cell aggregates. Tumor-associated macrophages (TAMs) in C6-CD200S tumors displayed dendritic cell (DC)-like morphology with multiple processes and CD86 expression. Furthermore, CD3+, CD4+ or CD8+ cells were more frequently found in C6-CD200S tumors, and the expression of DC markers, granzyme, and perforin was increased in C6-CD200S tumors. Isolated TAMs from original C6 tumors were co-cultured with C6-CD200S cells and showed increased expression of DC markers. These results suggest that CD200S activates TAMs to become DC-like antigen presenting cells, leading to the activation of CD8+ cytotoxic T lymphocytes, which induce apoptotic elimination of tumor cells. The findings on CD200S action may provide a novel therapeutic modality for the treatment of carcinomas.
Introduction
CD200 is a transmembrane glycoprotein belonging to the immunoglobulin superfamily, capable of exerting immunosuppressive effects on cells expressing its receptor CD200R [1], [2]. CD200 is expressed in many tissues and cell types, such as lymphocytes, kidney glomeruli, neurons and endothelial cells [3]. By contrast, CD200R is expressed mainly by myeloid cells such as granulocytes, monocytes and macrophages [2], [4]. In the brain, neurons express CD200, which has been implicated in the induction of immunologically inactive phenotypes of microglial cells, a resident macrophage in the brain [5].
Many recent studies have shown that CD200 possibly contributes to tumor outgrowth or aggravates outcomes by suppressing anti-tumor immune responses [4], [6]. Many kinds of malignant solid tumor cells [7], [8], [9] as well as leukemia [10], [11] express CD200, which is assumed to allow tumor cells to evade immune surveillance mainly through suppression of myeloid cell functions. However, there are conflicting hypotheses on the roles of CD200 in some solid tumor progression. Talebian et al. [12] reported that CD200 prevents melanoma cells from forming tumors or metastasizing into the lung. A recent report using CD200 transgenic and CD200R1 knock-out mice demonstrated that the enhancement of the CD200-CD200R interaction in some cases led to inhibition of metastasis and local growth of breast cancer [13].
Such contradictory data may be attributable to the presence of a splice variant or truncated form of CD200 (CD200S) with a shorter amino acid sequence [14], [15], because the truncated form exerts an antagonistic action on the immunosuppressive effects of CD200-CD200R interactions [16]. The expression of a splice variant of CD200 devoid of exons 1 and 2, but containing exon 3-derived sequences has been reported previously (see Figure 1A) [14], [15], [16]. A frame shift caused by the splicing of exon 2 resulted in the appearance of a stop codon within the sequence of exon 3, producing mRNA for CD200S. This alternative splicing of CD200 is regulated by an exonic splicing enhancer and the splicing regulatory protein SF2/ASF [17]. CD200S does not contain a C–C′ loop or complementarity determining region (CDR) 2, which is involved in binding to CD200R; however, it does contain an F–G loop and CDR3, which is also a binding domain for CD200R [18].
Figure 1.

Molecular structures of CD200L and CD200S (A), and constructed vectors for establishment of C6 rat glioma cell lines expressing either CD200L or CD200S (B). (A) Expression of CD200L (Aa) and CD200S (Ab). RNA splicing generates CD200S with a shorter amino acid sequence. CD200S contains F–G loop or CDR3 but not C–C′ loop or CDR2, all of which are responsible for the binding to CD200R. (B) Constructed vectors for transfection to original C6 glioma cells to generate C6-L or C6-S cell lines.
Various normal murine tissues and organs have been shown to express full-length CD200 (CD200L) and CD200S molecules [16]. In our previous study, macrophages were found to accumulate densely in ischemic rat brain lesions that expressed both molecules [15]. CD200 has been considered a candidate immune checkpoint allowing tumors to evade anti-tumor immunity [19]. However, CD200L expression seems to normally accompany the expression of CD200S. To determine the specific roles of CD200 in tumor immunity, it is necessary to distinguish the actions of CD200L and CD200S. Therefore, we established rat C6 glioma cell lines expressing either CD200L or CD200S and transplanted them into the forebrains of newborn Wistar rats, which do not reject transplanted cells owing to their immature immune system [20]. Consequently, CD200S expression in gliomas resulted in the prolonged survival of the transplanted rats compared with rats bearing CD200L-expressing tumors. Tumor-associated macrophages (TAMs) in the CD200S-expressing tumors displayed dendritic cell (DC)-like phenotypes. Our results suggest that CD200S may have its own specific effects on tumor immunity independently of CD200L.
Materials and Methods
Cloning of full-length CD200S cDNA, construction of retroviral expression vectors and establishment of C6 cells stably expressing CD200L or CD200S
Expression plasmids encoding rat CD200L and CD200S were constructed using the method described by Chen et al. [16]. cDNA fragments of CD200L or CD200S were amplified by PCR using CD200 cDNA purchased from Origene (Rockville, MD) with the following cloning primers containing BamHI and NotI respectively (see Figure 1): CD200L; forward 5′-GGA TCC ATG GGC AGT CCG GTA TTC AG-3′ and reverse 5′-ATG CGG CCG CTT ATT TCA TTC TTT GCA TCC CCT G-3′, CD200S; forward 5′-ACG GAT CCA TGA TCA CTT ACA GCA AAG CC-3′ and the same reverse primer as for CD200L. The PCR products were inserted into a BamHI–NotI site of pCX4-pur retrovirus vector (GenBank accession No.: AB086386.1, gift from Dr. Tsuyoshi Akagi, KAN Institute, Kobe, Japan). Virus particles were prepared in 293T cells by co-transfecting pCX4-pur vector with pGP and pE-ampho retrovirus packaging component vectors (Takara, Tokyo, Japan) using lipofectamine 2000 as described elsewhere [21], and applied to C6 cells. Infected cells were selected by incubation with 1 μg/ml of puromycin for 2 weeks. Expression of CD200 products was confirmed by immunocytochemistry and quantitative real-time RT-PCR (qPCR).
Human tissue analysis
Surgically resected human specimens of hepatocellular carcinoma (HCC) and cholangiocellular carcinoma (CCC) were obtained with informed consent and under approval by the local ethics committee at Ehime University Hospital, Japan. Part of the tumors and adjacent normal tissues were immediately frozen in liquid nitrogen after surgery and stored at − 80°C for subsequent extraction of RNA. The frozen samples were homogenized in ISOGEN (Nippon Gene, Tokyo, Japan) to prepare total RNA for RT-PCR as described previously [22]. The total-RNA-derived cDNA was amplified by PCR with primers (see Supplementary Table 1). PCR conditions were as follows: 94°C for 1 min, followed by 30 cycles of 94°C for 30 s, 52°C (CD200) or 58°C (GAPDH) for 30 s, and 72°C for 1 min.
Experimental rat glioma model
Rat C6-glioma cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Wako, Osaka, Japan), containing 10% fetal bovine serum (FBS; Wako), 4.5 g/L glucose, and a penicillin/streptomycin/amphotericin B mixture (Wako). We prepared a 2.5 × 107 cells/ml suspension of rat C6-glioma cells in phosphate-buffered saline (PBS) and transplanted percutaneously 20 μl (5.0 × 105 cells/body) into the right striatum of neonatal Wistar rats (within 24 h after birth) using syringes with 29 G needles (Terumo, Tokyo, Japan) at 2 mm lateral from the midline and 1 mm posterior from the bregma and 2 mm depth. A 1:1 mixture of C6-L and C6-S cells (5.0 × 105 cells in total/body) were transplanted in the same way in some experiments. The health of the transplanted rats was monitored every day and the survival periods were plotted by Kaplan-Meier method. Statistical analyses were performed with the use of a log-rank test.
Determination of the growth rates of the cell lines
To determine the growth rate of each cell line, the cells (3.0 × 104cells/well) were seeded on 4-well culture plates in DMEM containing 10% FS and 4.5 g/L glucose. One hour later, cells were collected using 0.025% Trypsin-EDTA in PBS (Wako) to determine the number of attached cells in a well. Cells in simultaneously prepared cultures were counted on days 2, 3, and 5. The growth rate (% of the data on day 1) was obtained by the formula; the growth rate (%) = (the cell number in a well − the number of cells on day 1)/the number of cells on day 1 × 100.
Isolation of TAMs from the tumor masses
TAMs were isolated from rat brain tumors 3 weeks after transplantation. Tumors were dissected out from rat brains and minced with scissors. Minced tumors were incubated in 0.1% Trypsin-EDTA (Sigma, St. Louis, MO, USA) while shaking for 15 min at room temperature. After addition of DMEM containing 10% FCS, the minced tumor suspensions were centrifuged at 1,000 rpm for 5 min at 4°C, and the pellets were recovered, resuspended in 4 ml DMEM containing 3% FS, and filtered through a nylon mesh bag with 160 μm pore. The filtrate was incubated in polystyrene dishes for suspension culture (Sumitomo, Akita, Japan) for 30 min in a CO2 incubator. TAMs but not the other cells preferentially attached to the dishes. The dishes were rinsed three times with warm DMEM containing 3% (v/v) FS to remove unattached cells. For qPCR and co-culture, purified TAMs were scraped off with a rubber scraper.
Quantitative real-time RT-PCR (qPCR)
Total RNA isolated from tissue or cell homogenate was performed as described [23]. cDNA was synthesized from RNA (0.5 μg) by reverse transcription using ReverTra Ace qPCR RT Master Mix with gDNA Remover (Toyobo, Osaka, Japan), according to the manufacturer's instructions. For qPCR analysis, cDNA was diluted at 1:3, and 1 μl was used for triplicate qPCR on a MJ mini instrument (Bio-Rad, Hercules, CA, USA) using Fast Start Universal SYBR Green (Roche Diagnostic Japan, Tokyo, Japan). All gene-specific mRNA expression values were normalized to GAPDH or Iba1 expression levels. Primer sequences are listed in Supplementary Table 1.
TUNEL staining
Apoptotic cells in rat experimental tumors were detected by TUNEL staining using a TACS 2 TdT-DAB In Situ Apoptosis Detection Kit (Trevigen, Gaithersburg, MD, USA) according to the manufacturer’s instructions [24]. Apoptotic cells in cryosections of rat tumors 18 d after transplantation were stained brown by the use of diaminobenzidine as a peroxidase substrate.
In vivo immunohistochemistry and morphometric analysis
Rats were sacrificed at 18 or 23 days after birth under deep anesthesia. The brains were dissected and 2-mm-thick coronal slices, which were immersed in 4% paraformaldehyde in PBS containing 2 mM MgCl2 for 2 h, and then immersed in 15% (w/v) sucrose in PBS at 4°C for 2 days. The fixed slices were rapidly frozen at − 80°C, and cryosectioned into 10 μm-thick coronal sections. Immunohistochemical observations were performed as previously described [23]. The cryosections were incubated overnight with the primary antibodies shown in Supplementary Table 2. After washing with Tris-buffered saline containing 0.02% Tween 20, sections were treated with Cy3-conjugated anti-rabbit IgG secondary antibody or Dylight488 labeled anti-mouse IgG secondary antibody (1:1,000; Jackson Immunoresearch, West Grove, PA, USA). Hoechst 33342 (Sigma) was used for nuclear staining. The immunostained sections were observed with a conventional microscope (BX-52, Olympus, Tokyo, Japan), a Nikon A1 confocal laser scan microscope (CLSM; Nikon, Tokyo, Japan) [25] or a scanning fluorescence microscope (BZ-9000; Keyence, Osaka, Japan). Brain areas containing tumor cells were morphometrically analyzed on Hoechst 33342-stained sections, after the fluorescently stained area was binarized generating black/white patterns using Adobe Photoshop CS5 Extended (Adobe Systems, San Jose, CA, USA). The white area was measured using ImageJ 1.43u as described elsewhere [26].
Co-culture of TAMs and established cells
Isolated TAMs (1.0×105 cells/well) from original C6 cells were co-cultured for 48 h either with C6-e, C6-L or C6-S cells (3.0×104 cells/well) in 4-well plates. Some of the co-culture was fixed and subjected to immunocytochemical staining according to the methods described above. As secondary antibodies and phalloidin the following agents were used; Cy3-conjugated anti-rabbit IgG secondary antibody, DyLight 649-labeled anti rabbit secondary antibody (Jackson Immunoresearch, West Grove, PA), and DY490-labeled phalloidin (DYNOMICS, Germany). Total RNA was prepared from other plates and the mRNA expression of DC markers was investigated by real-time PCR.
Statistics
Statistical analyses were conducted using one-way analysis of variance with Bonferroni's post-hoc test, or the log-rank test for evaluating the survival rate. The co-culture data were analyzed with the use of unpaired t-test. P<0.05 was considered significant.
Results
Expression of mRNA encoding CD200L and CD200S in human carcinoma tissues
Analysis of human carcinoma tissues revealed the expression of mRNA encoding both CD200L and CD200S (Figure 2). HCC and CCC tissues, as well as their surrounding normal tissues, were subjected to RT-PCR analyses. All of the tissues analyzed, including normal ones, expressed mRNA encoding both CD200L and CD200S, as has been described in mice elsewhere [16].
Figure 2.
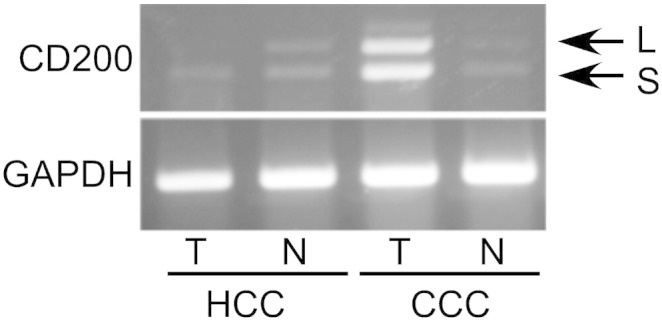
Human hepatocellular carcinoma (HCC) and cholangiocellular carcinoma (CCC) as well as surrounding normal tissues expressed mRNA encoding both CD200L and CD200S as revealed by RT-PCR. T, tumor tissues; N, normal tissues.
Characterization of C6 rat glioma cell lines expressing CD200L or CD200S
To investigate the roles of CD200L and CD200S differentially, we established rat C6 glioma cells that specifically express either CD200L or CD200S using retrovirus vectors containing their cDNA (Figure 1B). Unaltered original rat C6 glioma cells did not express CD200L or CD200S (data not shown). We first investigated whether the established cell lines specifically expressed the transfected protein by immunocytochemical staining using antibodies recognizing either the N-terminal domain of CD200L (mouse monoclonal, clone OX2) or the C-terminal domain (goat polyclonal), of which specificities have been examined in our previous study (Figure 3, A–C) [15]. Our immunostaining results show that neither the C-terminal nor the N-terminal domain of CD200L was detected in C6 cells transfected with the empty vector (C6-e cells; Figure 3A). Both of these domains were detected in C6 cells transfected with the vector containing CD200L-cDNA (C6-L cells; Figure 3B), while only the C-terminal domain was detected in C6 cells transfected with the vector containing CD200S-cDNA (C6-S cells; Figure 3C). The intracellular localization of the C-terminal immunofluorescence was not the same in C6-L and C6-S cells, thus post-translational modification of the CD200 molecules may have occurred as has been suggested previously [15]. Furthermore, we have confirmed the specific expression of the CD200 species by qPCR using two specific primer sets; ‘CD200 common (com)’ primer sets amplified cDNA encoding both CD200L and CD200S, ‘CD200L (L)’ primer sets amplified only cDNA for CD200L (Figure 3D) [15]. C6-e cells did not express any of the mRNA. C6-L cells expressed mRNA that could be amplified with both primer sets, whereas C6-S cells expressed only mRNA that was amplified with ‘CD200 com’ primer sets. Thus, the three C6 cell lines, C6-e, C6-L, and C6-S were successfully established to express no CD200, CD200L, and CD200S, respectively.
Figure 3.

Characterization of established cell lines expressing CD200L and CD200S. (A–C) Immunocytochemical confirmation of CD200 expression with antibodies recognizing N- (OX2) or C-termini of CD200. The C6 cell line transfected with an empty vector (A, C6-e) did not display positive immunostaining against either antibody. (B) The C6-L cell line was positive for both antibodies. (C) The C6-S line was only reactive to the C-terminal antibody. (D) qPCR confirmed the specific expression of CD200L or CD200S. A primer set amplifying cDNA derived from both CD200L- and CD200S-mRNA was called common ‘Com’ (see Supplementary Table 1). The other primer set ‘L’ amplified cDNA only derived from CD200L-mRNA. C6-e cells expressed none of the cDNA, C6-L cells expressed both, and C6-S cells expressed cDNA amplified only by the Com primer set. (E) Growth curves of each cell line. Data from four independent cultures are expressed as means ± SD except for the data of Empty at 5 days, which is expressed as mean − SD.
Next, we compared the growth rate of the cell lines (Figure 3E) and the expression of factors that potentially affect tumorigenicity (Supplementary Figure 1). The established cell lines and the original C6 cells showed no significant differences in their growth rate for 5 d after seeding (Figure 3E). As revealed by qPCR analysis, there were no significant changes in expression of mRNA encoding factors such as chemokines, cytokines or an extracellular matrix protein, all of which contribute to the growth of malignant tumors (Supplementary Figure 1). mRNA level for proliferating cell nuclear antigen (PCNA) was also not different among the four cell lines. Thus, from the qPCR data it could not be predicted which cell line has the strongest tumorigenic potential.
Tumorigenicity of CD200 cell lines and prolonged survival of rats transplanted with C6-S cells
The four cell lines were transplanted into the right striatum through the crania of neonatal rats within 24 h after birth. As shown in Figure 4A, C6-e and C6-L cells formed apparent tumor masses in the center of the forebrain 18 days after transplantation; however, original C6 and C6-S cells did not form macroscopically apparent tumor masses. Fluorescent nuclear staining with Hoechst 33342 to identify the dense accumulation of cells revealed that tumors also developed in the rat brains transplanted with original C6 and C6-S cells (Figure 4B). To statistically analyze the growth of the brain tumors, the brains were dissected out at 18 and 23 days after transplantation and stained with Hoechst 33342 [26]. The ratio obtained by dividing the nuclei-accumulated white area by total brain area was used as an index for the tumor growth (Figure 4C). Tumor areas were found to be smaller in the C6-S-transplanted brains than those in brains transplanted with other cell lines.
Figure 4.

Tumor growth and survival of rats transplanted with the four lines of cells. (A) Macroscopic overviews of rat brains bearing tumors of the four cell lines. The brains were dissected and sliced on 18 days after transplantation. Apparent tumor masses (asterisks) were observed in the brains transplanted only with C6-e and C6-L lines. (B) Cryosections of the fixed brain were fluorescently stained with Hoechst 33342 to identify the spread of tumor cells. C6-e (Bb) and C6-L (Bc) cells formed large tumor masses. Original C6 cells (ori-C6) formed an apparent but smaller tumor mass (Ba, arrowheads). There was no apparent tumor mass in the brain transplanted with C6-S cells (Bd). (C) Ratios of tumor area/total brain area were obtained by measuring the area clearly stained with Hoechst 33342, where tumor cells densely accumulated. The brains were dissected on 18 and 23 days after transplantation. Tumor areas were smaller in the C6-S-transplanted brains. Data (n=4 at each time point) are expressed as means ± SD. (D) Kaplan-Meier survival plots of rats transplanted with each C6 cell line by 40 days after transplantation. As revealed by log-rank test, rats transplanted with C6-S cells were survived significantly longer (P<0.001) than rats bearing tumors from other cell lines. (E) Mean survival days were calculated, by assuming that all surviving rats die at 41 days after transplantation. C6-S-transplanted rats survived longer by 5.5 days versus rats bearing original C6 tumors and 11 days versus C6-e and C6-L tumors. *P < .01, **P < .001.
The survival of rats transplanted with the four cell lines was followed-up for 40 days after transplantation. Rats transplanted with C6-S cells survived for a significantly longer period than rats transplanted with the other lines of cells (Figure 4D). Rats transplanted with original C6 cells survived longer than those transplanted with C6-e or C6-L cells (P<0.001). There was no significant difference in the survival period of rats with C6-e and C6-L cells. Assuming that surviving rats at 40 days had died on the next day, mean survival days were calculated (Figure 4E). Rats transplanted with original C6 cells had a mean survival of 28.0 ± 6.7 (mean ± SD) days, compared with the mean survival with C6-e cells of 22.8 ± 5.9 days; C6-L cells of 22.1 ± 6.2 days; and C6-S cells of 33.6 ± 7.2 days. Thus, the rats bearing C6-S tumors survived for significantly longer periods compared with rats bearing tumors formed from the other cell types.
Abundance of apoptotic cells in the C6-S tumor
We postulated that the smaller size of the C6-S tumors likely led to the prolonged survival of the rats, thus, we suspected an enhanced rate of cell death of the C6-S tumor cells. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining showed an increased number of apoptotic cells in the C6-S tumors than in the C6-L tumors (Figure 5A). Expression of cleaved caspase-3 (CC3), another marker of apoptosis [27], was also frequently found in C6-S tumors (Figure 5B). CD11b+ TAMs with multiple processes did not display CC3-immunoreactivity. The density of CC3+ cells was compared among the three tumor types (Figure 5, C–E), with denser accumulation of CC3+ cells found in the C6-S tumors. Dense tumor cell accumulation in C6-e (Figure 5Cd) and C6-L (Figure 5Dd) tumors was revealed by Hoechst 33342 staining, whereas C6-S tumors formed only small aggregates (Figure 5Ed). Closer observation revealed that only a few CC3+ cells were present in the regions with dense Hoechst 33342-staining in the C6-e (Figure 5Fa) and C6-L (Figure 5Fb) tumors. By contrast, many CC3+ cells were found in the tumor cell aggregates in the C6-S tumors (arrowheads in Figure 5Fc). The results suggest that apoptotic cell death occurred more frequently in the C6-S tumors and may be a likely cause for the smaller tumor sizes and prolonged survival of the rats.
Figure 5.

Apoptotic cell death in the tumor region of rat brains. (A) TUNEL staining detected more apoptotic cells in the brain tumor composed of C6-S cells (Ab) than in the tumor composed of C6-L cells (Aa). (B) Apoptotic cells identified by immunostaining with antibody to cleaved caspase 3 (CC3) in the C6-S tumor. CD11b+ TAMs in tumors. Ba shows a 2D image and Bb a 3D image. Note the morphology of CD11b+ TAMs displaying polygonal shape with multiple processes. (C–E) Double immunohistochemical observation of the tumors consisting of C6-e (C), C6-L (D), or C6-S (E) cells detecting CD11b and CC3. Densely accumulated tumor cells were present in the tumors of the C6-e and C6-L cells as revealed by Hoechst 33342 staining, whereas the tumor cells in the C6-S tumor formed only small aggregates (Ed). The C6-S tumor contained a number of CC3+ apoptotic cells that were scattered throughout the tumor region. (F) Enlarged micrographs of those shown in (C–E). Although many CC3+ apoptotic cells were found in C6-e (Fa) and C6-L (Fb)-tumors, most of those were found in non-tumor regions where were observed as less densely distributed Hoechst 33342+ nuclei. By contrast, apoptotic cells accumulated in the tumor region (arrowheads) in the C6-S tumor (Fc).
Dendritic cell morphology of TAMs in the C6-S tumor
TAMs in each tumor mass were stained with an antibody directed against Iba1, a monocyte/macrophage/microglia marker [28] (Figure 6A). Although all tumor types contained densely accumulated TAMs, TAMs in the C6-S tumors showed a distinct morphology characterized by multiple short processes, which resemble DCs (Figure 6Ac; also see Figure 9C). Furthermore, CD4+ cells were more frequently found in the C6-S tumors than in the C6-L tumors (Figure 6B). Many CD3+ lymphocytes were found in the C6-S tumors, some of which were CD4+ suggesting they were helper T cells (yellow arrowheads) (Figure 6C). CD3+/CD4- cells were likely CD8+ T cells (pink arrowheads). Indeed, CD8+ cells were often found in the C6-S tumors but not in the C6-L tumors (Figure 6D). Figure 6E shows the presence of what is likely a CD8+ lymphocyte surrounded by TAMs with processes; a probable evidence for cross-presentation by the TAMs in the C6-S tumors. Expression of the co-stimulatory factor CD86 was expressed by most TAMs in the C6-S tumors (Figure 6F). CD86 may be expressed by other cells than TAMs, taken its rather high expression in C6-ori, -e or L tumors (Supplementary Figure 2).
Figure 6.

DC-like morphology of TAMs and accumulation of CD4+ or CD8+ cells in the C6-S tumor. (A) Inclined 3D images on Iba1+ TAMs in the C6-ori (Aa), C6-e (Ab), C6-L (Ac), and C6-S (Ad) tumors. Compared with TAMs in other types of tumors, those in the C6-S tumor displayed DC-like ramified morphology characterized by multiple processes or spikes. (B) CD4+ cells and structures were more abundant in the C6-S tumor (Bb) than the C6-L tumor (Ba). (C) There were abundant CD4+ (Ca) and CD3+ (Cb) T cells in the C6-S tumor. Yellow arrowheads denote CD3+/CD4+ cells, pink ones CD3+/CD4- cells. Iba1+ TAMs with processes surrounded CD4+ lymphocyte-like round cells, as revealed by 3D images. (D) CD8+ cells and structures were frequently present in the CD200S-tumors (Db), whereas they were not in the C6-L tumor (Da). (E) CD8+ cells with round morphology were often intimately associated with process-bearing TAMs. (F) Most TAMs in the C6-S tumor were CD86+, since they displayed yellow or orange colors due to the mergence of Iba1 (red) and CD86 (green) immunofluorescence.
Figure 9.

Apoptotic cell death and the morphology of TAMs in the tumor prepared by transplanting mixture of C6-S and C6-L cells. (A) Most tumor cells of the mixed cell tumor underwent apoptotic changes as revealed by CC3-immunostaining. (B) Iba1+ TAMs in the mixed tumor (C6-S+L) displayed ramified morphology characterized with multiple processes. (C) Iba1+ TAMs in the C6-S tumor were observed in the same way as those show in (B). The TAMS displayed DC-like ramified morphology. (D) Iba1+ TAMs in C6-L tumor showed rather smooth cell surfaces devoid of ramified processes that were distinct from those of Tams in the C6-S and the C6-S+L tumors.
Characterization of the four types of tumors and their derived TAMs by qPCR
We dissected each tumor type to subject the tissues to qPCR analyses 21 days after transplantation (Figure 7A). ‘CD200 com’ primers amplified both CD200L and CD200S cDNA and detected a high level of expression of CD200 in the C6-L and C6-S tumors. ‘CD200L’ primers detected high expression of CD200L only in the C6-L tumors. However, low levels of CD200L mRNA were found in the other tumors, an indicative of expression of CD200 by other types of cells such as endothelial cells [15]. There are two CD200R isoforms; R1 and R2 [4], [15], [29]. No significant differences were found in the expression levels of mRNA encoding CD200Rs in each tumor type, suggesting that the density of CD200R+ TAMs was not significantly different in each tumor type (Supplementary Figure 2). This may correlate with the expression of Iba1-mRNA that was also at similar levels among the four types of tumors. mRNA encoding MHC class II α, CD11c, and CD80, all of which are DC markers, showed significantly higher expression in the C6-S tumor than in other types of tumors. F4/80 and CD68, known of their expression by DCs [30], [31], was highly expressed in the C6-S tumor. Expression of CD4-mRNA showed a tendency to be elevated in the C6-S tumors, but the change was not significant. mRNA encoding CD3δ, ε, and γ as well as CD8 α and β was highly expressed in the C6-S tumors, an indicative of more accumulation of T cells in the C6-S tumor than in the other tumors. Furthermore, the levels of mRNA encoding granzyme and perforin were elevated in the C6-S tumors. Interferon-γ (IFNγ) mRNA showed a tendency to be elevated in the C6-S tumors. NK cells express IFNγ as well as granzyme and perforin [32], [33], leading to apoptosis of tumor cells. The observation that CD56 expression was not marked in the C6-S tumor suggests that NK cells were not responsible for the apoptotic tumor cell death. Although natural killer T cells (NKT cells) may be another possible source of granzyme and perforin [33], the involvement of NKT cells was unlikely because of the high expression of CD16 mRNA in the C6-S tumor. C-type lectin domain family 9A (CLEC9A) was highly expressed in the C6-S tumor, which is expressed by myeloid DCs and involved in cross-presentation [34].
Figure 7.

Expression levels in tumors and isolated TAMs of mRNA encoding molecules related to CD200, DCs, lymphocytes and T cells, as revealed by qPCR. Data are shown in an alphabetical order. (A) cDNA was prepared from tumor masses dissected out 21 days after transplantation. CD200-mRNA amplified with ‘CD200 com’ primers was highly expressed by C6-L and C6-S tumors, whereas the CD200L-mRNA level was high only in the C6-L-tumors. The C6-S-tumors expressed DC markers, MHC class II α, CD11c, and CD80 at significantly higher levels than other tumors. Expression of mRNA encoding lymphocyte markers CD8α and β, CTL-related apoptosis-inducing molecules, granzyme, and perforin was significantly higher in the C6-S tumors. Despite not showing statistical significance, CD4 and IFNγ-mRNA were also highly expressed in the C6-S tumors. CLEC9A, which is expressed by myeloid DCs and responsible for cross-presentation, and chemokines (CCL12, CXCL10 and CXCL16), which are invovlved in recruitment of monocytes and lymphocytes, are also highly expressed in the C6-S tumor. (B) Shortly after isolation from the tumor mass, TAMs were subjected to qPCR analysis. TAMs from the C6-S-tumors expressed MHC class II α-, and CD11c-mRNA was expressed at significantly higher levels than in TAMs from other tumors. CD86-mRNA expression in TAMs from the C6-S tumors was also high. Data from 3 tumors of each cell lines are expressed as means ± SEM. *P < .05, **P < .01, ***P < .001 versus CD200S; #P < .05, ##P < .01 versus CD200L.
In this series of experiments, we investigated whether CD200S induces a M1-like phenotype in TAMs, which may originally have M2-like properties, which support tumor growth [35]. Therefore, we investigated the expression of M1 and M2 markers such as arginase-1 (Arg-1), CD163, inducible nitric oxide synthase (iNOS), interleukin-10 (IL-10), IL-12 [15], [36], [37], tumor necrosis factor α (TNFα), transforming growth factor (TGF) β1 as shown in Supplementary Figure 2. However, there were no significant changes in expression in these factors among the tumor types. Factors affecting apoptotic tumor cell death such as Bcl-xL, Bax, Fas, or FasL expression were not significantly different among the tumors in their mRNA levels (Supplementary Figure 2). Among these, FasL expression appeared to be elevated in C6-S, but it was not a significant change.
Some chemokines were highly expressed in primary rat microglial cells (data not shown). Their expression in the tumors was investigated (Figure 7 and Supplementary Figure 2). CCL12, CXCL10 and CXCL16 mRNA were highly expressed in the C6-S tumor. CCL12 may play a role in recruitment of lymphocytes and monocytes [38]. CXCL10 is expressed by a variety of cells and a chemoattractant for monocytes, T cells and NK cells [39]. CXCL16 may be expressed by the DC-like TAMs in the C6-S tumor, while recruiting activated T cells based on notions described elsewhere [40]. These three chemokines may play a role in recruitment of T cells. On the other hand, there were no significant differences in expression levels of mRNA encoding CCL6, CCL7, CCL9, CXCL1, or CXCL2.
Shortly after isolation from each type of tumor, TAMs were subjected to qPCR analyses (Figure 7B). The isolated TAMs from the C6-S tumors expressed mRNA encoding major histocompatibility complex (MHC) class II α and CD11c at higher levels than those from other types of tumors. The TAMs from the C6-S tumors also showed a tendency to have elevated CD86 mRNA expression.
Expression of DC markers in co-culture with TAMs and C6-S cells
TAMs isolated from original C6 tumors were co-cultured with C6-L or C6-S cells for 48 h, followed by collection of total RNA for immunocytochemical staining and qPCR analyses. There were no CD80+ TAMs found in co-culture with C6-L cells (Figure 8A), whereas some of the CD11b+ TAMs were CD80+ following co-culture with C6-S cells (Figure 8B). Note that C6 cells did not show a damaged morphology, indicating that TAMs did not have cytotoxic activity against co-cultured tumor cells. MHC class II α, CD11c and CD80 mRNA expression was significantly elevated following co-culture with C6-S cells compared with C6-L cells (Figure 8C).
Figure 8.

DC markers on TAMs were induced by co-culturing with C6-CD200S. TAMs isolated from original C6 brain tumors were cocultured with C6-L- and C6-S-cells. (A and B) The cells were fixed and immunostained with antibodies to CD11b and CD80. FITC-labeled phalloidin was used to identify cocultured C6 cells by staining actin filaments. The white cells in Aa or Bb are TAMs and green cells are C6 cells. (A) TAMs were cocultured with C6-CD200L cells. (B) TAMs were cocultured with C6-CD200S cells. There were some CD80+/Iba1+ TAMs only in the coculture with C6-CD200S cells (Bc). (C) qPCR experiments showed that coculture with TAMs and C6-S cells significantly enhanced expression of mRNA encoding MHC class II α, CD11c, and CD80. The data (n = 5) were normalized by dividing the % GAPDH data obtained with C6-L or C6-S cells by the data with C6-e cells and are expressed as means ± SEM. *P < .05.
A mixed cell tumor
To further investigate the tumor-suppressive effects of CD200S in vivo, a 1:1 mixture of C6-L and C6-S cells were transplanted into the brain of neonatal rats. CD200L and CD200S mRNA expression in the mixed tumor dissected 21 days after transplantation was evaluated by qPCR using the two primer sets; CD200L and CD200 common (see Figure 3). The mRNA expressions of CD200L and CD200 common were 674 ± 169 % and 411 ± 80.8 %, respectively (data from 3 rats are expressed as % of GAPDH mRNA and as means ± SEM). Thus, the two CD200 molecular species were actually expressed in the mixed tumor. The brain sections were subjected to immunohistochemical staining with antibodies to CC3 and Iba1. As shown in Figure 9, CC3+ apoptotic cells were abundantly present in the tumor regions that were identified with densely accumulated nuclei (Figure 9A). Note that the small tumor mass that resembled that of C6-S alone tumor. Furthermore, DC-like ramified morphology of TAMs in the mixed tumor was not distinguishable from that in the C6-S tumor (Figure 9C). TAMs in C6-L tumor displayed amoeboid shape (Figure 9D).
Discussion
The present study revealed that C6-S cells formed intracranial tumors in rats, the sizes of which were smaller than those consisting of C6-L or control cells. Furthermore, rats bearing C6-S tumors survived for longer than those with other tumors. The smaller size of C6-S tumors may have been caused by enhanced apoptotic cell death as revealed by TUNEL staining and immunohistochemistry for CC3. C6-S tumor cells induced a DC-like phenotype with multiple short processes in TAMs. Morphological observations demonstrated the presence of CD3+ T cells and the intimate attachment of DC-like TAMs to CD4+ or CD8+ cells, suggesting antigen presentation by TAMs to T cells. qPCR studies showed significantly elevated expression of DC markers, T cell markers, perforin and granzyme in C6-S tumors. When co-cultured with C6-S cells, TAMs from the original C6 tumors expressed DC markers.
Collectively, these results suggest the following processes to induce apoptotic elimination of tumor cells: C6-S cells induce TAMs to acquire a DC-like nature leading to the presentation of tumor cell antigens to T cells. In turn, antigen presentation primes CD4+ T cells to stimulate CD8+ CTLs to eliminate tumor cells by releasing perforin and granzyme. Direct activation of CTLs in C6-S tumors through cross-presentation by DC-like TAMs may primarily occur [41], as expression of mRNA encoding CD4 and IFNγ was not markedly increased in C6-S tumors, whereas mRNA expression for CD8, granzyme, and perforin was much more evident compared with expression levels in the tumors formed by other cell lines. High-level expression of CLEC9A mRNA in C6-S tumor may also be a suggestive of cross-presentation [34]. CD3 and CD16 mRNA was also highly expressed only in the C6-S tumor, indicating that the source of granzyme and perforin is CTLs rather than NKT or NK cells.
To date, CD200 has been considered as an immunosuppressive molecule that partly resembles the well-studied immune checkpoints, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) or programed cell death protein 1 (PD-1) [42], [43]. Humanized monoclonal antibodies ipilimumab and nivolumab, which target CTLA-4 and PD-1, respectively, have been clinically introduced and proven to be effective for several kinds of cancers. Similarly, inhibitory agents for the CD200-CD200R interactions have been investigated [44], [45]. CD200-derived short peptides [44] or an anti-CD200 antibody [46], [47] have been shown to prolong survival of tumor bearing animals or to suppress tumor growth. However, the effects of the CD200-CD200R interaction on tumor immunity have been controversial [12], [13], [19]. Some studies have shown that CD200 inhibits tumor growth and metastases in melanoma [12] and breast cancer [13] models. Such a tumor-suppressing effect of CD200 has been attributed to its suppressive effects on myeloid cells, which can potentially cause pro-tumorigenic inflammation [19]. These ideas may suggest that inhibitors of the CD200-CD200R interaction have less significant anti-tumor effects than the immune checkpoint inhibitors against CTLA-4 or PD-1. In this study, we could not find any effects of C6-L cell lines that were distinct from those of C6-e tumors in terms of tumor growth or on the survival periods of tumor-bearing rats. This observation suggests that CD200L does not have marked effects on tumor immunity at least in the present rat glioma model.
CD200+ macrophages have been identified in the ischemic core region of a rat stroke model [15], [48]. These are not activated microglia but bone marrow-derived cells [49]. CD200+ macrophages in an ischemic brain lesion expressed both CD200L and CD200S in addition to CD200R [15]. Since the cells were densely accumulated with intimate attachment to each other, it is likely that both CD200L and CD200S can affect their own functions through binding to CD200R. The CD200L+/CD200S+ macrophages displayed M1-like phenotypes with expression of IL-1β and iNOS, suggesting that immune activating effects of CD200S precede immunosuppression by CD200L. As shown in the present study (Figure 2) and the previous study [16], CD200L appears to be simultaneously expressed with CD200S. Thus, the actions of a suppressor CD200L and an activator CD200S may compete in the regulation of tumor immunity. Although the outcome of the competition might be altered by the context of the tumor pathophysiological processes as suggested by others [13], CD200S, at least in some cases, may play a more critical role in tumor immunity than CD200L. The different actions of CD200L and CD200S might be attributable to the different binding affinity to the CD200R isoforms; there are two known isoforms in humans and four in mice [4], [29]. In rats, CD200R1 and R2 isoforms may be expressed as shown in Supplementary Figure 2. It remains to be elucidated whether CD200L and CD200S have different affinities to CD200R1 or R2 causing the distinct actions on tumor growth or apoptosis.
The present study proposes the novel possibility that molecules mimicking CD200S functions may be used as an anti-tumor intervention. CD200S can alter the nature of TAMs to display a DC-like phenotype causing activation of CD4+ or CD8+ T lymphocytes, leading to apoptotic glioma cell death. Despite that CD200S is thought to have antagonistic effects on CD200L-CD200R interactions [16], the present data suggest that CD200S directly affects CD200R+ TAMs. Normally, only negligible numbers of CD200L+ cells are found in recipient rats compared with the huge number of CD200L-/CD200S+ tumor cells. Some peptides mapping to the presumptive CDR3 or F–G loop of CD200 are reported to overcome immunosuppressive effects of CD200Fc or soluble CD200L peptide in the induction of CTLs and the release of TNFα in mixed lymphocyte cultures [45]. Interestingly, CD200S contained these peptide sequences. By contrast, peptides mapping to the CDR2 region of CD200 are reported to exhibit a similar effect to that of CD200Fc. CD200S does not have the CDR2 region. Furthermore, some CDR3-derived peptides consisting of a CD200R-binding domain showed curative effects in a murine breast cancer model [44]. Our present experiment using the co-culture of TAMs from an original C6 tumor with C6-L or C6-S cells showed the direct effect of CD200S on CD200R+ TAMs. Collectively, these data suggest that CD200S not only antagonizes the CD200-CD200R interaction but also directly exhibits activating effects on CD200R+ myeloid cells including TAMs. Furthermore, it has been speculated that CDR3 or F–G loop-derived peptides with antagonistic effects on the CD200-CD200R interaction act directly on DCs, increasing both the number of CD8+ CTLs and the release of TNFα [44], [45]. However, the peptides have not been shown to upregulate expression of DC markers by TAMs. Therefore, the action of the peptides with the partial sequence of CD200S may not be completely the same as that of CD200S. If some sequences in CD200S critical for the action of inducing DC-like phenotypes are determined, a peptide containing the relevant sequences might be a novel intervention to treat carcinomas.
When a mixture of C6-L and C6-S cells were transplanted into the neonatal rat brains, the formed tumor displayed indistinguishable histological features from those of C6-S tumor, in terms of the presence of DC-like TAMs and abundant apoptotic cells. This observation suggests that the effect of CD200S overcame that of CD200L on the immune system. Specific actions of solely expressed CD200S on tumor immunity have not been reported, although malignant tumors express both CD200 species as shown in Figure 2. It is necessary to determine whether the solely expressed CD200S in a single cell devoid of CD200L is critical for induction of DC-like cells in the tumor. Nevertheless, the finding in the mixed tumor would be a hint to develop a novel curative intervention using CD200S itself or its related molecules.
In conclusion, we first demonstrated that CD200S with only one of the two domains of CD200L that binds to CD200R exhibit unique actions on TAMs inducing them to differentiate into DC-like cells. The TAMs activate CTLs to induce apoptotic tumor cell death. The anti-tumor action of CD200S appeared to be more active than that of the CD200-derived peptides that antagonize CD200-CD200R interactions, because CD200S exhibits its actions on TAMs virtually in the absence of CD200L. The results shown here may provide a novel strategy to develop a therapy for malignancies that evade anti-tumor immunity.
Acknowledgment
The authors are grateful to staff in the Animal Center of INCS of Ehime University for their gentle care of experimental animals.
Footnotes
Grant sponsor: Ministry of Education, Ministry of Education, Culture, Sports, Science, and Technology, Japan (#815K143510, 22390037 to JT; #80127894 to YK).
There are no actual or potential conflicts of interest, including any financial, personal, or other relationships with people or organizations regarding this work.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2016.02.006.
Appendix A. Supplementary data
Supplementary material.
References
- 1.Wright GJ, Puklavec MJ, Willis AC, Hoek RM, Sedgwick JD, Brown MH, Barclay AN. Lymphoid/neuronal cell surface OX2 glycoprotein recognizes a novel receptor on macrophages implicated in the control of their function. Immunity. 2000;13:233–242. doi: 10.1016/s1074-7613(00)00023-6. [DOI] [PubMed] [Google Scholar]
- 2.Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, Blom B, Homola ME, Streit WJ, Brown MH. Down-regulation of the macrophage lineage through interaction with OX2 (CD200) Science. 2000;290:1768–1771. doi: 10.1126/science.290.5497.1768. [DOI] [PubMed] [Google Scholar]
- 3.Wright GJ, Jones M, Puklavec MJ, Brown MH, Barclay AN. The unusual distribution of the neuronal/lymphoid cell surface CD200 (OX2) glycoprotein is conserved in humans. Immunology. 2001;102:173–179. doi: 10.1046/j.1365-2567.2001.01163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wright GJ, Cherwinski H, Foster-Cuevas M, Brooke G, Puklavec MJ, Bigler M, Song Y, Jenmalm M, Gorman D, McClanahan T. Characterization of the CD200 receptor family in mice and humans and their interactions with CD200. J Immunol. 2003;171:3034–3046. doi: 10.4049/jimmunol.171.6.3034. [DOI] [PubMed] [Google Scholar]
- 5.Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. 2010;468:253–262. doi: 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- 6.Kretz-Rommel A, Qin F, Dakappagari N, Ravey EP, McWhirter J, Oltean D, Frederickson S, Maruyama T, Wild MA, Nolan MJ. CD200 expression on tumor cells suppresses antitumor immunity: new approaches to cancer immunotherapy. J Immunol. 2007;178:5595–5605. doi: 10.4049/jimmunol.178.9.5595. [DOI] [PubMed] [Google Scholar]
- 7.Stumpfova M, Ratner D, Desciak EB, Eliezri YD, Owens DM. The immunosuppressive surface ligand CD200 augments the metastatic capacity of squamous cell carcinoma. Cancer Res. 2010;70:2962–2972. doi: 10.1158/0008-5472.CAN-09-4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petermann KB, Rozenberg GI, Zedek D, Groben P, McKinnon K, Buehler C, Kim WY, Shields JM, Penland S, Bear JE. CD200 is induced by ERK and is a potential therapeutic target in melanoma. J Clin Invest. 2007;117:3922–3929. doi: 10.1172/JCI32163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siva A, Xin H, Qin F, Oltean D, Bowdish KS, Kretz-Rommel A. Immune modulation by melanoma and ovarian tumor cells through expression of the immunosuppressive molecule CD200. Cancer Immunol Immunother. 2008;57:987–996. doi: 10.1007/s00262-007-0429-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tonks A, Hills R, White P, Rosie B, Mills KI, Burnett AK, Darley RL. CD200 as a prognostic factor in acute myeloid leukaemia. Leukemia. 2007;21:566–568. doi: 10.1038/sj.leu.2404559. [DOI] [PubMed] [Google Scholar]
- 11.Coles SJ, Wang EC, Man S, Hills RK, Burnett AK, Tonks A, Darley RL. CD200 expression suppresses natural killer cell function and directly inhibits patient anti-tumor response in acute myeloid leukemia. Leukemia. 2011;25:792–799. doi: 10.1038/leu.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Talebian F, Liu JQ, Liu Z, Khattabi M, He Y, Ganju R, Bai XF. Melanoma cell expression of CD200 inhibits tumor formation and lung metastasis via inhibition of myeloid cell functions. PLoS One. 2012;7:e31442. doi: 10.1371/journal.pone.0031442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erin N, Podnos A, Tanriover G, Duymus O, Cote E, Khatri I, Gorczynski RM. Bidirectional effect of CD200 on breast cancer development and metastasis, with ultimate outcome determined by tumor aggressiveness and a cancer-induced inflammatory response. Oncogene. 2015;34:3860–3870. doi: 10.1038/onc.2014.317. [DOI] [PubMed] [Google Scholar]
- 14.Borriello F, Tizard R, Rue E, Reeves R. Characterization and localization of Mox2, the gene encoding the murine homolog of the rat MRC OX-2 membrane glycoprotein. Mamm Genome. 1998;9:114–118. doi: 10.1007/s003359900700. [DOI] [PubMed] [Google Scholar]
- 15.Matsumoto S, Tanaka J, Yano H, Takahashi H, Sugimoto K, Ohue S, Inoue A, Aono H, Kusakawa A, Watanabe H. CD200 + and CD200- macrophages accumulated in ischemic lesions of rat brain: the two populations cannot be classified as either M1 or M2 macrophages. J Neuroimmunol. 2015;282:7–20. doi: 10.1016/j.jneuroim.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 16.Chen Z, Chen DX, Kai Y, Khatri I, Lamptey B, Gorczynski RM. Identification of an expressed truncated form of CD200, CD200tr, which is a physiologic antagonist of CD200-induced suppression. Transplantation. 2008;86:1116–1124. doi: 10.1097/TP.0b013e318186fec2. [DOI] [PubMed] [Google Scholar]
- 17.Chen Z, Ma X, Zhang J, Hu J, Gorczynski RM. Alternative splicing of CD200 is regulated by an exonic splicing enhancer and SF2/ASF. Nucleic Acids Res. 2010;38:6684–6696. doi: 10.1093/nar/gkq554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hatherley D, Lea SM, Johnson S, Barclay AN. Structures of CD200/CD200 receptor family and implications for topology, regulation, and evolution. Structure. 2013;21:820–832. doi: 10.1016/j.str.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rygiel TP, Meyaard L. CD200R signaling in tumor tolerance and inflammation: A tricky balance. Curr Opin Immunol. 2012;24:233–238. doi: 10.1016/j.coi.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez-Martin A, Munoz-Espin D, Alonso AM, Izquierdo M. Parent phenotype and age dependence, on rat glioma tumor rejection. J Neurooncol. 2004;70:29–34. doi: 10.1023/b:neon.0000040838.40115.f5. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi K, Takahashi H, Inoue A, Harada H, Toshimori S, Kobayashi Y, Goto K, Sugimoto K, Yano H, Ohnishi T. Oct-3/4 promotes migration and invasion of glioblastoma cells. J Cell Biochem. 2012;113:508–517. doi: 10.1002/jcb.23374. [DOI] [PubMed] [Google Scholar]
- 22.Matsumoto H, Kumon Y, Watanabe H, Ohnishi T, Shudou M, Chuai M, Imai Y, Takahashi H, Tanaka J. Accumulation of macrophage-like cells expressing NG2 proteoglycan and Iba1 in ischemic core of rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2008;28:149–163. doi: 10.1038/sj.jcbfm.9600519. [DOI] [PubMed] [Google Scholar]
- 23.Sugimoto K, Nishioka R, Ikeda A, Mise A, Takahashi H, Yano H, Kumon Y, Ohnishi T, Tanaka J. Activated microglia in a rat stroke model express NG2 proteoglycan in peri-infarct tissue through the involvement of TGF-beta1. Glia. 2014;62:185–198. doi: 10.1002/glia.22598. [DOI] [PubMed] [Google Scholar]
- 24.Tei N, Tanaka J, Sugimoto K, Nishihara T, Nishioka R, Takahashi H, Yano H, Matsumoto S, Ohue S, Watanabe H. Expression of MCP-1 and fractalkine on endothelial cells and astrocytes may contribute to the invasion and migration of brain macrophages in ischemic rat brain lesions. J Neurosci Res. 2013;91:681–693. doi: 10.1002/jnr.23202. [DOI] [PubMed] [Google Scholar]
- 25.Inoue A, Tanaka J, Takahashi H, Kohno S, Ohue S, Umakoshi A, Gotoh K, Ohnishi T. Blood vessels expressing CD90 in human and rat brain tumors. Neuropathology. 2016 doi: 10.1111/neup.12244. [in press] [DOI] [PubMed] [Google Scholar]
- 26.Choudhury ME, Sugimoto K, Kubo M, Nagai M, Nomoto M, Takahashi H, Yano H, Tanaka J. A cytokine mixture of GM-CSF and IL-3 that induces a neuroprotective phenotype of microglia leading to amelioration of (6-OHDA)-induced Parkinsonism of rats. Brain Behav. 2011;1:26–43. doi: 10.1002/brb3.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arai M, Sasaki A, Saito N, Nakazato Y. Immunohistochemical analysis of cleaved caspase-3 detects high level of apoptosis frequently in diffuse large B-cell lymphomas of the central nervous system. Pathol Int. 2005;55:122–129. doi: 10.1111/j.1440-1827.2005.01808.x. [DOI] [PubMed] [Google Scholar]
- 28.Matsumoto H, Kumon Y, Watanabe H, Ohnishi T, Shudou M, Ii C, Takahashi H, Imai Y, Tanaka J. Antibodies to CD11b, CD68, and lectin label neutrophils rather than microglia in traumatic and ischemic brain lesions. J Neurosci Res. 2007;85:994–1009. doi: 10.1002/jnr.21198. [DOI] [PubMed] [Google Scholar]
- 29.Gorczynski RM, Chen Z, Clark DA, Kai Y, Lee L, Nachman J, Wong S, Marsden P. Structural and functional heterogeneity in the CD200R family of immunoregulatory molecules and their expression at the feto-maternal interface. Am J Reprod Immunol. 2004;52:147–163. doi: 10.1111/j.1600-0897.2004.00192.x. [DOI] [PubMed] [Google Scholar]
- 30.Makala LH, Reyes JC, Nishikawa Y, Tsushima Y, Xuan X, Huang X, Battsetseg B, Matsuo T, Nagasawa H. Phenotype and function of murine discrete Peyer's patch macrophage derived - dendritic cells. J Vet Med Sci. 2003;65:491–499. doi: 10.1292/jvms.65.491. [DOI] [PubMed] [Google Scholar]
- 31.Ferenbach D, Hughes J. Macrophages and dendritic cells: what is the difference? Kidney Int. 2008;74:5–7. doi: 10.1038/ki.2008.189. [DOI] [PubMed] [Google Scholar]
- 32.Hodge G, Barnawi J, Jurisevic C, Moffat D, Holmes M, Reynolds PN, Jersmann H, Hodge S. Lung cancer is associated with decreased expression of perforin, granzyme B and interferon (IFN)-gamma by infiltrating lung tissue T cells, natural killer (NK) T-like and NK cells. Clin Exp Immunol. 2014;178:79–85. doi: 10.1111/cei.12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bade B, Boettcher HE, Lohrmann J, Hink-Schauer C, Bratke K, Jenne DE, Virchow JC, Jr., Luttmann W. Differential expression of the granzymes A, K and M and perforin in human peripheral blood lymphocytes. Int Immunol. 2005;17:1419–1428. doi: 10.1093/intimm/dxh320. [DOI] [PubMed] [Google Scholar]
- 34.Schreibelt G, Klinkenberg LJ, Cruz LJ, Tacken PJ, Tel J, Kreutz M, Adema GJ, Brown GD, Figdor CG, de Vries IJ. The C-type lectin receptor CLEC9A mediates antigen uptake and (cross-)presentation by human blood BDCA3 + myeloid dendritic cells. Blood. 2012;119:2284–2292. doi: 10.1182/blood-2011-08-373944. [DOI] [PubMed] [Google Scholar]
- 35.Zarif JC, Taichman RS, Pienta KJ. TAM macrophages promote growth and metastasis within the cancer ecosystem. Oncoimmunology. 2014;3:e941734. doi: 10.4161/21624011.2014.941734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S, Guerau-de-Arellano M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS One. 2015;10:e0145342. doi: 10.1371/journal.pone.0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Durafourt BA, Moore CS, Zammit DA, Johnson TA, Zaguia F, Guiot MC, Bar-Or A, Antel JP. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia. 2012;60:717–727. doi: 10.1002/glia.22298. [DOI] [PubMed] [Google Scholar]
- 38.Jia GQ, Gonzalo JA, Lloyd C, Kremer L, Lu L, Martinez AC, Wershil BK, Gutierrez-Ramos JC. Distinct expression and function of the novel mouse chemokine monocyte chemotactic protein-5 in lung allergic inflammation. J Exp Med. 1996;184:1939–1951. doi: 10.1084/jem.184.5.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu M, Guo S, Stiles JK. The emerging role of CXCL10 in cancer (Review) Oncol Lett. 2011;2:583–589. doi: 10.3892/ol.2011.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shimaoka T, Nakayama T, Fukumoto N, Kume N, Takahashi S, Yamaguchi J, Minami M, Hayashida K, Kita T, Ohsumi J. Cell surface-anchored SR-PSOX/CXC chemokine ligand 16 mediates firm adhesion of CXC chemokine receptor 6-expressing cells. J Leukoc Biol. 2004;75:267–274. doi: 10.1189/jlb.1003465. [DOI] [PubMed] [Google Scholar]
- 41.Bevan MJ. Cross-priming. Nat Immunol. 2006;7:363–365. doi: 10.1038/ni0406-363. [DOI] [PubMed] [Google Scholar]
- 42.Callahan MK, Wolchok JD. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J Leukoc Biol. 2013;94:41–53. doi: 10.1189/jlb.1212631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moertel CL, Xia J, LaRue R, Waldron NN, Andersen BM, Prins RM, Okada H, Donson AM, Foreman NK, Hunt MA. CD200 in CNS tumor-induced immunosuppression: the role for CD200 pathway blockade in targeted immunotherapy. J Immunother Cancer. 2014;2:46. doi: 10.1186/s40425-014-0046-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gorczynski R, Boudakov I, Khatri I. Peptides of CD200 modulate LPS-induced TNF-alpha induction and mortality in vivo. J Surg Res. 2008;145:87–96. doi: 10.1016/j.jss.2007.04.043. [DOI] [PubMed] [Google Scholar]
- 46.Kretz-Rommel A, Qin F, Dakappagari N, Cofiell R, Faas SJ, Bowdish KS. Blockade of CD200 in the presence or absence of antibody effector function: implications for anti-CD200 therapy. J Immunol. 2008;180:699–705. doi: 10.4049/jimmunol.180.2.699. [DOI] [PubMed] [Google Scholar]
- 47.Gorczynski RM, Chen Z, Diao J, Khatri I, Wong K, Yu K, Behnke J. Breast cancer cell CD200 expression regulates immune response to EMT6 tumor cells in mice. Breast Cancer Res Treat. 2010;123:405–415. doi: 10.1007/s10549-009-0667-8. [DOI] [PubMed] [Google Scholar]
- 48.Matsumoto H, Kumon Y, Watanabe H, Ohnishi T, Takahashi H, Imai Y, Tanaka J. Expression of CD200 by macrophage-like cells in ischemic core of rat brain after transient middle cerebral artery occlusion. Neurosci Lett. 2007;418:44–48. doi: 10.1016/j.neulet.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 49.Smirkin A, Matsumoto H, Takahashi H, Inoue A, Tagawa M, Ohue S, Watanabe H, Yano H, Kumon Y, Ohnishi T. Iba1(+)/NG2(+) macrophage-like cells expressing a variety of neuroprotective factors ameliorate ischemic damage of the brain. J Cereb Blood Flow Metab. 2010;30:603–615. doi: 10.1038/jcbfm.2009.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.