

The tremendous rate of attrition during the process of metastasis implicates the existence of a rare and unique population of metastasis-initiating cells (MICs). In this review, Celià-Terrassa and Kang summarize the main tumor-intrinsic hallmarks of MICs and their dynamic interactions with the extrinsic environment to manifest their metastasis-forming activities and discuss the possible strategy of targeting MICs in cancer therapeutics.
Keywords: cancer metastasis, plasticity, epithelial–mesenchymal transition, metastasis-initiating cells, metastatic niche
Abstract
Primary tumors are known to constantly shed a large number of cancer cells into systemic dissemination, yet only a tiny fraction of these cells is capable of forming overt metastases. The tremendous rate of attrition during the process of metastasis implicates the existence of a rare and unique population of metastasis-initiating cells (MICs). MICs possess advantageous traits that may originate in the primary tumor but continue to evolve during dissemination and colonization, including cellular plasticity, metabolic reprogramming, the ability to enter and exit dormancy, resistance to apoptosis, immune evasion, and co-option of other tumor and stromal cells. Better understanding of the molecular and cellular hallmarks of MICs will facilitate the development and deployment of novel therapeutic strategies.
The majority of cancer deaths is caused by metastasis, when cancer cells manage to escape the primary tumor, survive the treacherous transit through the lymphovascular system, and eventually form secondary tumors in distant organs (Gupta and Massague 2006; Valastyan and Weinberg 2011; Wan et al. 2013). This is a highly challenging process with a tremendous rate of attrition; it is estimated that only <0.02% of disseminated tumor cells (DTCs) are able to successfully seed metastases (Luzzi et al. 1998; Cameron et al. 2000; Chambers et al. 2002). As a result, although tumor dissemination can occur relatively early in cancer progression (Husemann et al. 2008; Massard et al. 2011; Kang and Pantel 2013), sometimes even at the preneoplastic stage (Rhim et al. 2012), an extended gap time often exists between the formation of the primary tumor and clinical manifestations of metastasis (Yachida et al. 2010; Vanharanta and Massague 2013). Therefore, the capability to initiate metastatic growth is a major bottleneck during cancer progression and represents an ideal window for therapeutic intervention (Fig. 1A).
Figure 1.
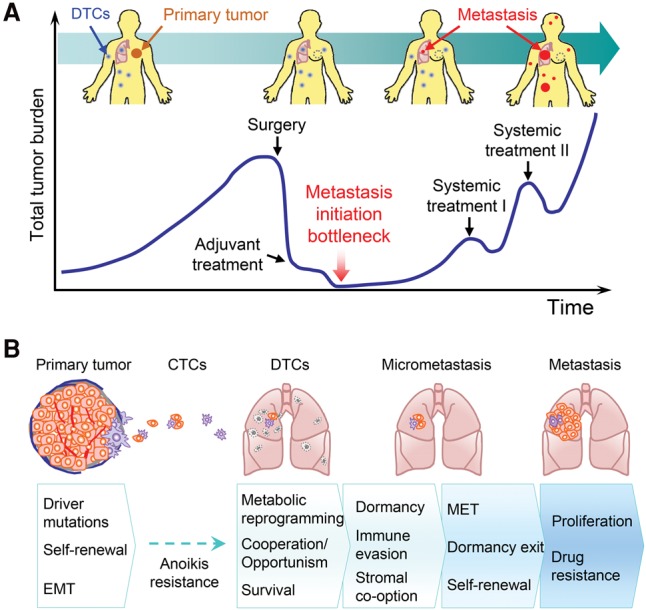
Metastasis-initiating cells (MICs) in cancer progression and metastasis. (A) Schematic depiction of the typical course of metastatic progression of an early-stage cancer. In many clinical cases, tumor dissemination precedes diagnosis of the primary tumor. Surgical debulking and systemic adjuvant treatment eliminate most of tumor cells at the primary site and throughout the body. However, a small proportion of DTCs survives the systemic treatment. After a period of dormancy with no clinical sign of cancer, which could last for months to decades, clinically detectable metastases start to emerge. The subsequent lines of systemic treatment often only temporarily reduce the tumor burden before metastatic lesions develop resistance and eventually overwhelm the patients. The ability to initiate metastatic outgrowth is therefore a major bottleneck in cancer progression. (B) Representation of the sequence of events leading to metastasis initiation and acquisition of MIC properties. At the primary tumor site, a tiny fraction of long-term self-renewing tumor-initiating cells (TICs) may represent early MICs with driver mutations and high cellular plasticity. During dissemination, the large majority of DTCs dies, except those with strong anoikis resistance. Further attrition occurs after DTCs infiltrate distant organs, and MICs need to acquire a series of properties to become fully competent in seeding overt metastases.
Metastasis-initiating cells (MICs), by definition, are cancer cells capable of seeding clinically significant metastatic colonies in secondary organs. Like their primary tumor counterparts, the tumor-initiating cells (TICs), MICs can hijack some of the normal stem cell pathways to increase cellular plasticity and stemness, which provide them with multiple malignant advantages. However, the MICs must possess additional capabilities that allow them to survive the metastatic cascade and function as TICs in an organ microenvironment distinctively different from the primary tumor. These cells form the link between the primary tumor and subsequent metastasis but are exceedingly difficult to identify, track, and characterize. Even the origin of MICs remains elusive; they might exist at the primary tumors or emerge during the journey through the metastatic cascade (where exposure to extreme stress conditions may select for MIC abilities) or may acquire such capabilities only after arriving at the distant site and engaging the stromal components (Fig. 1B). Such unique challenges in identifying and analyzing MICs demand research tools beyond what are commonly available and used in the study of TICs, such as in vitro tumorsphere assays, in vivo limited dilution tumor initiation studies, and analysis using cancer stem cell (CSC) surface markers. In the past few years, new and emerging technologies have begun to enable the study of MICs in animal and clinical models. Genomic sequencing studies have provided genome-wide comparisons between primary tumors and matched distant metastases from cancer patients and animal models (Campbell et al. 2010; Yachida et al. 2010; McFadden et al. 2014; Gundem et al. 2015). Gene expression analysis at the single-cell level has become a powerful tool to analyze the population dynamics of tumor cells during metastatic evolution (Lawson et al. 2015). In addition, lineage tracing and barcode sequencing studies have also been applied to study the interclonal interactions and population dynamics (Maddipati and Stanger 2015; Wagenblast et al. 2015). Advances in animal modeling of metastasis and detailed analysis of tumor-intrinsic pathways and tumor–stromal cross-talk further provided unprecedented insights into the molecular mechanism of metastasis initiation. Some consensus regarding the hallmarks of MICs has started to emerge from these studies, including the maintenance of TIC ability, the flexibility to undergo bidirectional transitions between the epithelial and mesenchymal states, resistance to anoikis and apoptosis, entry into and exit from dormancy, evasion of immune system attack, reprogramming of metabolic activities to adapt to the different nutrient and oxidative stresses, interclonal cooperations, and the ability to build or take advantage of a supportive stromal niche. Underlying all of these myriad properties of MICs is their remarkable cellular plasticity that allows them to survive and thrive against all odds. In this review, we summarize the main tumor-intrinsic hallmarks of MICs and their dynamic interactions with the extrinsic environment to manifest their metastasis-forming activities and discuss the possible strategy of targeting MICs in cancer therapeutics.
Genomic evolution of MIC traits
Cancer genome sequencing studies have shown that malignant tumors emerge from the sequential accumulation of mutations in driver genes involved in three core cellular processes during tumor initiation: cell fate regulation, genome maintenance, and cell survival (Vogelstein et al. 2013). These altered processes favor primary tumor initiation and may still be essential for MICs to seed metastases. However, it was previously unknown whether additional driver mutations are needed for metastasis to occur. Genome sequencing studies have shown high degrees of similarities among mutations in primary tumors and metastases (Yachida et al. 2010). The most remarkable finding of these studies is that no consistent metastasis-specific mutations have been found other than those that are already commonly found in primary tumors (Bozic et al. 2010; Campbell et al. 2010; Yachida et al. 2010), suggesting that important mutations for metastasis are already present in the primary tumor site. These studies frequently reveal a greater enrichment of clonal populations rather than an acquisition of new mutations, as observed in pancreatic cancer metastasis with amplifications of MYC, RASG13D, and CCDN1 (Campbell et al. 2010) and in lobular ER+ breast cancer with ERBB2 mutations (Shah et al. 2009). A recent study using whole-exome sequencing analysis of experimental metastasis models of multiple cancer types has shown that metastatic competence arises from the selection of pre-existing mutations, such as RASG13D and BRAFG464V, in heterogeneous populations without the need for additional mutations (Jacob et al. 2015). The selection of these oncogenic pathways favors their prevalence in metastasis, indicating that they are important contributors to metastatic fitness and thus may be required for MICs. Overall, these findings suggest that a large number of metastatic properties may be already forming in the primary tumor through enrichment of existing oncogenic mutations that favor metastasis initiation.
Beyond realignment of genomic mutations, epigenetic regulation might be a major source of MIC traits, especially in later steps of metastasis. After tumor cells escape the primary site, the epigenome is subjected to microenvironmental signal modulation, conferring cellular plasticity and adaptability to new and inhospitable conditions (Seftor et al. 2006; Hendrix et al. 2007; Tam and Weinberg 2013). Indeed, multiple studies have unveiled evidence of specific epigenetic pathways involved in the metastatic progression of different cancer types (Cunha et al. 2014; Gu et al. 2015; Okada et al. 2015; Tang et al. 2015). Therefore, the combination of genetic and epigenetic events during the course of metastasis likely determines the acquisition of MIC traits.
Cell fate determinants as regulators of MICs
Adult tissues are hierarchically organized and tightly controlled by lineage-specific transcription factors to regulate growth and differentiation and maintain the homeostasis of tissues and organs. During tumorigenesis, the metastatic potential of tumors with different cellular origins (adult stem cells, progenitor cells, or differentiated cells) may be shaped by the dominant lineage-specific cell fate regulators expressed in the originating cells. In addition, alteration or loss of differentiation control may result in dedifferentiation, acquisition of stem cell-like activities, and cellular plasticity that facilitate the development of metastatic traits (Reya et al. 2001; Ben-Porath et al. 2008).
Accumulating evidence supports the notion that loss of differentiation factors leads to dedifferentiation and acquisition of stem cell-like traits that are linked to metastasis initiation properties (Fig. 2; Cao et al. 2011). Mutation, epigenetic silencing, or reduced expression of luminal differentiation factors in the mammary gland (GATA3 and ELF5) has been shown to promote breast cancer metastasis (Kouros-Mehr et al. 2008; Chakrabarti et al. 2012). RARRES3, which is involved in retinoic acid-induced differentiation signaling, suppresses breast cancer lung metastasis initiation by promoting tumor differentiation (Morales et al. 2014). In lung adenocarcinoma, the loss of NKX2-1, a lung lineage-specific transcription factor, increases metastatic seeding (Winslow et al. 2011). In a recent follow-up study, NKX2-1 was found to work synergistically with other lineage-specific transcription factors (FOXA2 and CDX2) to suppress lung metastasis (Li et al. 2015). The simultaneous loss of these three lineage cell fate determinants induces dedifferentiation and stem cell-like properties to promote lung metastasis. Two other lung alveolar differentiation transcription factors (GATA6 and HOPX) also cooperatively limit the metastatic competence of lung adenocarcinoma (Cheung et al. 2013). Similarly, the loss of MITF, a melanocyte differentiation factor, is sufficient to increase metastasis of melanoma (Cheli et al. 2012).
Figure 2.

Cell fate determinants in development and their influence on MICs. (A) Embryonic and adult epithelial cell lineage transcription factors tightly control self-renewal and lineage-specific differentiation of normal adult tissue stem cells and embryonic stem cells. (OKSM) The Yamanaka factors Oct4, Klf4, Sox2, and Myc. (B) The same transcription factors also influence metastatic behavior of cancer cells and the formation of MICs. Differentiation factors of normal tissues, such as MITF, GATA3, FOXA2, and others, act as tumor suppressors and metastasis inhibitors. On the other hand, dedifferentiation factors, such as the Yamanaka factors or tissue-specific stem cell factors, drive dedifferentiation, plasticity, and metastasis of MICs. Interestingly, these transcription factors constitute a complex network of reciprocal regulation. For example, SOX2 antagonizes multiple tissue-specific differentiation factors. Other factors, such as MTDH, exclusively support TIC and MIC activities and have no known function in normal tissue development.
Opposing the function of lineage-specific differentiation factors, the increased activity of stem cell factors has been shown to promote metastasis. For example, the cooperation of mammary stem cell (MaSC) transcription factors SNAI2 and SOX9 induces luminal dedifferentiation toward a stem cell-like state with metastatic seeding abilities (Guo et al. 2012). In a similar fashion, ID1 (inhibitor of differentiation-1) increases breast cancer lung metastasis (Gupta et al. 2007), and the MaSC marker PROCR is also reported to be involved in self-renewal and metastasis (Spek and Arruda 2012; Wang et al. 2015a). Interestingly, other factors that support tumor initiation activity seem to work only in the malignant context and are not involved in the regulation of normal adult tissue stem cells. For example, MTDH, an essential factor to support tumor initiation and metastasis in breast, prostate, and liver cancers, is dispensable for embryonic and postnatal development (Robertson et al. 2014; Wan et al. 2014a,b). Such factors will be ideal candidates for therapeutic targeting to prevent metastasis initiation.
Not only are tissue-specific cell fate determinants critical in metastasis initiation, embryonic cell fate regulators also play important roles. With the discovery of the Yamanaka factors—Sox2, Myc, Klf4, Oct4, and others—as potent reprogramming factors, these genes have also garnered much attention in cancer research. Each of these factors has been linked to tumor aggressiveness and poor prognosis (Ben-Porath et al. 2008; Kim et al. 2013; Kareta et al. 2015). MYC is one of the most thoroughly studied oncogenes (Cole 1986), and KLF4 has also been classified as an oncogene (Leng et al. 2013). Recently, SOX2 was shown to maintain self-renewal and survival of CSCs in multiple tumor types, including squamous cell carcinoma (Boumahdi et al. 2014). In medulloblastoma, SOX2 drives the hierarchical organization of the tumors and promotes relapse (Vanner et al. 2014). Interestingly, during embryonic development, SOX2 specifies cell fate decisions by antagonizing tissue-specific factors involved in metastasis, such as NKX2-1, CDX2, MITF, and others mentioned above (Fig. 2B). In addition, SOX2 and NANOG have been reported to maintain quiescence programs in DTCs/residual cancer cells and may contribute to metastatic relapse (Sosa et al. 2015). Although SOX2, NANOG, OCT4, and KLF4 have been shown to increase metastasis of bladder cancer, breast cancer, lung cancer, and head and neck squamous carcinoma cells (Celia-Terrassa et al. 2012; Vaira et al. 2013; Lu et al. 2014; Habu et al. 2015), none of these factors has been specifically studied during metastasis initiation. Based on current knowledge, it is tempting to speculate that these factors may also facilitate metastatic initiation by promoting cell plasticity, adaptability, survival, and self-renewal as they do in primary tumors. Therefore, future research should be conducted to study these cell fate regulators during metastasis initiation.
Epithelial–mesenchymal plasticity and the acquisition of stem cell-like properties
Cancer cell plasticity is a dynamic state of dedifferentiation, with cells acquiring some characteristics of stem cells. Serious malignant advantages can be acquired when cancer cells hijack developmental processes such as epithelial–mesenchymal transition (EMT) to increase their cellular plasticity. EMT normally occurs during embryonic development and also in pathological conditions such as wound healing and metastasis (Thiery et al. 2009; Nieto 2013). During EMT, epithelial cells lose their polarity and cell–cell adhesions to gain mesenchymal-like properties, such as increased migratory abilities. Cancer cells often undergo EMT to escape from the primary tumor, and mounting experimental and clinical evidence suggests that a reversed process, mesenchymal-to-epithelial transition (MET), is required for the outgrowth of metastatic tumor cells in the secondary organ (Thiery et al. 2009; Korpal et al. 2011; Brabletz 2012; Tsai et al. 2012). Interestingly, besides promoting invasion, EMT can induce stem cell-like properties to promote initiation of primary tumors and accelerate metastasis (Mani et al. 2008; Thiery et al. 2009; Guo et al. 2012).
Whether EMT plays a crucial role in cancer metastasis in human patients and in some animal model systems is still under debate (Ledford 2011; Fischer et al. 2015; Zheng et al. 2015a), largely due to the lack of the ability to track the occurrence of EMT and follow the fate of cells undergoing EMT in clinical settings as well as the diversity of the EMT program that can elude detection using a single EMT marker or reporter in animal models (Li and Kang 2016). Nevertheless, a recent study used rigorous single-cell analysis of breast cancer-derived xenografts to show that MICs indeed display a stem cell program with EMT features at the early phase of metastasis development (Lawson et al. 2015). Metastatic cells from small metastatic lesions have increased expression of EMT and stem cell features and dormancy-associated genes, while such features are often attenuated and replaced with the expression of differentiation and proliferation markers in fully developed macrometastases (Lawson et al. 2015). This finding supports the notion that EMT is required for early seeding of metastasis, while MET is essential for metastatic outgrowth (Tsai et al. 2012). Indeed, other studies have shown that an extreme EMT can lock cancer cells into a terminally differentiated state, depriving them of stem cell-like properties and cell plasticity and reducing tumor growth (Tran et al. 2011, 2014; Celia-Terrassa et al. 2012).
It is thus important to note that EMT is not a binary process; instead, it represents a spectrum of transitional states that can display different degrees of epithelial and mesenchymal features depending on the driver genes and pathways that induce the EMT process. Indeed, distinct EMT programs have been shown to influence different cell populations, and it is proposed that SNAI1 has a stronger effect on TIC generation and metastasis progression than SNAI2, which is crucial for sustaining normal mammary gland stem cells (Ye et al. 2015). Therefore, it is important to consider distinct EMT drivers and target cell populations when analyzing results from EMT experiments. Further complicating the analysis, the reversion of EMT (MET) can also induce stem cell-like properties and increase metastasis initiation of epithelial-like CSCs, as has been documented in multiple recent studies (Celia-Terrassa et al. 2012; Ocana et al. 2012; Stankic et al. 2013; Beck et al. 2015; Schmidt et al. 2015). Based on these results, CSCs can exist in both an epithelial-like or a mesenchymal-like transitional state, while cells fixed at extreme epithelial or mesenchymal states lose plasticity and the associated stem cell activities (Nieto 2013; Oskarsson et al. 2014). It is also possible that loss of lineage-specific differentiation factors, as discussed above, can induce CSC properties without the involvement of EMTs.
To provide a unifying model to reconcile all of these experimental observations regarding the link between epithelial–mesenchymal plasticity with CSC properties, a bipotent or hybrid EMT/MET state has been proposed and mathematically modeled as the “window of stemness” (Fig. 3; Jolly et al. 2015a,b). According to this model, the bipotent state resides within a tiny transitory fraction of the tumor population with both epithelial and mesenchymal features. This “hybrid state” is bidirectional and displays a gradient of partial states toward either extreme EMT or extreme MET (Fig. 3). This model has been supported by a recent analysis of CSC markers in breast cancer (Liu et al. 2014). In this study, mesenchymal-like breast CSCs (BCSCs) characterized as CD24−CD44+ are primarily quiescent with high invasive ability, while epithelial-like BCSCs are ALDH+, highly proliferative, and less invasive. Breast cancer cells with dual expression of both sets of markers have the highest degrees of plasticity (Liu et al. 2014). However, more research is needed to evaluate the proposed hybrid state hypothesis in other cancer types and model systems and determine its importance for metastasis and MICs.
Figure 3.
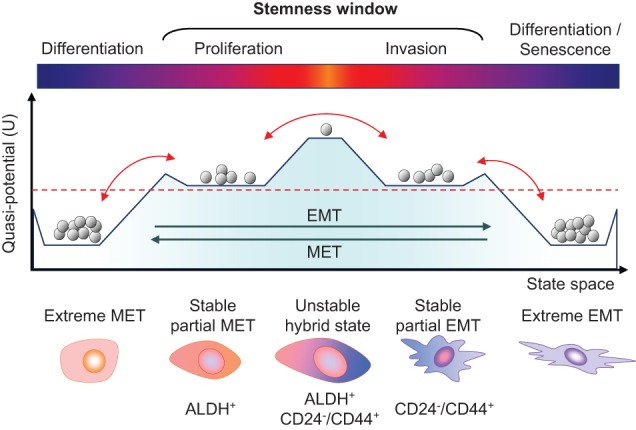
Epithelial–mesenchymal plasticity and the “stemness window.” The schematic graph shows the putative multistable quasipotential landscape of epithelial–mesenchymal plasticity and the possible window of stemness in the transitional states. The vertical axis represents potential energy (U) differences between cell states, with higher potential corresponding to greater plasticity and stemness. The horizontal axis represents the state space gradient of epithelial and mesenchymal phenotypes. Gray balls represent populations of cells falling into different levels of potential energy, being more stable at lower levels. The red dashed line denotes a hypothetical threshold of minimal cell plasticity required to generate CSC activity. EMT and MET can lead MICs over the threshold of required potential energy for cellular plasticity. In fact, bidirectional transitions above this threshold would maintain MICs’ plasticity within the window of stemness in either a partial mesenchymal-like or epithelial-like state. Transitions between both partial states may experience a transitory high peak of potential energy and stemness, but this may represent a state of high instability. Extreme EMT or MET leads to a differentiated state impoverished of potential energy; therefore, cells falling at these states may completely lose plasticity and would not be capable of becoming MICs.
It is also possible that sequential transitions—first EMT and then MET—may be required to achieve an optimal CSC state in MICs. Recent studies using nonmalignant cells suggest that sequential EMT and MET events increase the pluripotent state in keratinocytes and fibroblasts (Liu et al. 2013; Unternaehrer et al. 2014). Other studies have validated this in mammary epithelial cells by transiently expressing TWIST1 (Schmidt et al. 2015). Although no experimental evidence supporting this hypothesis has been provided in the context of cancer metastasis, this model correlates with the natural sequence of events occurring during metastasis, where primary tumor cells undergo EMT to escape from the primary site and survive and later revert by MET to colonize distant tissues (Tsai et al. 2012). Taken together, how EMT contributes to the acquisition of stem cell properties in MICs is still not fully understood, and it is possible that different mechanisms may be at work in different cancer types or subtypes. Thorough analysis of the dynamics governing EMT is indispensible to further clarify this issue.
A more drastic transition observed in cancer is the transdifferentiation of tumor cells into other cell lineages mimicking stromal cells, which is enabled by the high plasticity of cancer stem-like cells (Huang et al. 2015). Although it is a rare and poorly studied phenomena, cancer cells can transdifferentiate into endothelial cells or pericytes to mimic components of the tumor microenvironment (Huang et al. 2015). Vascular mimicry has recently been reported to facilitate metastasis of 4T1 mouse mammary tumor cells (Wagenblast et al. 2015). Further research is needed to explore whether transdifferentiation events can also happen at the metastatic site to enable metastasis initiation and whether transdifferentiation is one of the cellular properties of some MICs.
Metabolic reprogramming
Different organs and tissues of the human body have specific metabolic characteristics. Therefore, it is likely that MICs may require a high metabolic adaptability and metabolic stress resistance. The aerobic glycolysis observed in the primary tumors—the Warburg effect—is frequently replaced in metastasis by other routes of energy acquisition. One of the main causes of this phenomenon may be the detachment of tumor cells from the extracellular matrix (ECM) during metastasis, which impairs glucose uptake and shuts down the Warburg effect, thereby requiring alternative ways to circumvent deficiencies by using mitochondrial metabolism and peroxide signaling (Weber 2016). Although EMT has been shown to induce aerobic glycolysis to promote the growth of basal-like breast cancer (Dong et al. 2013), another study showed that disseminated invasive cells do not use aerobic glycolysis. Instead, PPARGC1A (PGC1α) mediates mitochondrial oxidative phosphorylation and biogenesis to facilitate lung colonization (LeBleu et al. 2014). Accordingly, a recent study reported that extreme mesenchymal-like prostate cancer cells are dependent on the function of the mitochondrial respiratory chain function (Aguilar et al. 2016), which is typically associated with slowly proliferating cells (DeBerardinis et al. 2008). Therefore, the metabolic differences observed in cells undergoing EMT seem to be more dependent on their proliferation rates than the mesenchymal-like or epithelial-like phenotype. In addition, quiescent DTCs can sustain their metabolic fitness by autophagy as an alternative source of energy during nutrient deprivation (Liang et al. 2007; Sosa et al. 2014). However, to reinitiate metastatic growth and engage proliferation, MICs may display marked metabolic plasticity in order to obtain energy from multiple substrates and pathways, as has been shown for highly metastatic breast and prostate cancer cells (Chen et al. 2007; Aguilar et al. 2016).
Different host organs also have different nutrient and oxygen conditions. The brain and lungs have high levels of glucose and oxygen, which may grant easier colonization of metastatic cells using aerobic glycolysis (DeBerardinis et al. 2008) or oxidative phosphorylation (LeBleu et al. 2014; Weber 2016). An adaptive metabolic mechanism critical for brain metastatic cells is the ability to co-oxidize acetate and glucose in the citric acid cycle as main fuels to support the bioenergetic demands of rapidly proliferating cells (Mashimo et al. 2014). In contrast, the liver has lower levels of oxygen and irregular glucose availability, and therefore MICs in the liver need to adapt to such metabolic stresses. Due to low oxygen levels, liver metastatic breast cancer cells activate HIF-1α with a concomitant increase of the pyruvate dehydrogenase kinase-1 (PDK1), switching their metabolism to glycolysis (Fig. 4A). Such metabolic adaptation is essential for breast cancer cells to efficiently colonize the liver (Dupuy et al. 2015). Another recent study has demonstrated the necessity of scavenging energy from the extracellular environment to overcome liver metabolic stress (Loo et al. 2015). In this study, colon cancer cells, by down-regulating miR-483 and miR-551, derepress and secrete creatine kinase, brain-type (CKB) into the extracellular space to convert creatine and ATP into phosphocreatine. The phosphocreatine is then imported into the MICs to serve as an ATP source for growth functions (Fig. 4A; Loo et al. 2015). In metastatic ovarian cells, fatty acids secreted from adipocytes are imported by FABP4 in MICs to colonize the intra-abdominal fat (Nieman et al. 2011). Under nutrient deprivation conditions, mitochondrial HSP90 chaperones, including TRAP1, overcome metabolic stress and promote metastasis by limiting the activation of the nutrient sensor AMPK and preventing autophagy (Caino et al. 2013).
Figure 4.

Cross-talk between MICs and stromal microenvironment and niches at different organs. (A) Metabolic adaptation in the liver. Low oxygen levels in the liver microenvironment force tumor cells to adapt via HIF-1α/PKD1 induction of glycolytic metabolism, thereby enabling metastatic colonization. MICs of colon cancer secrete CKB, which phosphorylates extracellular creatine produced by hepatocytes using extracellular ATP to generate phosphocreatine. Extracellular phosphocreatine is then imported into metastatic cancer cells by the transporter SLC6A8 to regenerate the ATP as a source of energy for survival and metastatic colonization. (B) Vascular niche and brain MIC–astrocyte cross-talk. The perivascular niche provides nutrients and oxygen to the infiltrating tumor cells, which secrete anti-PA serpins to protect MICs from astrocyte-derived death signals. Astrocytes also express Jagged1, which activates Notch signaling in MICs to promote self-renewal. Furthermore, astrocytes secrete miR-19a-containing exosomes, which suppress PTEN expression and activate CCL2-dependent recruitment of myeloid cells to promote tumor growth and survival in the brain. (C) In the bone, MICs compete with the hematopoietic stem cells (HSCs) for the HSC niche. Furthermore, osteogenic cells form heterotypic adherens junction with MICs and induce mTOR signaling to promote outgrowth. MICs also use secreted and membrane-bound VCAM1 to recruit preosteoclasts (pre-Oc) and activate their differentiation to mature osteoclasts (Oc), which in turn promote bone degradation and the formation of the “vicious cycle in bone metastasis.” (D) In the lung, bone marrow-derived cells (BMDCs) facilitate the formation of the premetastatic niche. In addition, the secretion of ECM proteins—tenascin C secreted by tumor cells and periostin (POSTN) secreted by stromal cells such as cancer-associated fibroblasts (CAFs)—further establishes the metastatic niche and supports MIC self-renewal by inducing Notch and Wnt signaling, respectively.
Exposure to new inhospitable environments and drastic metabolic reprogramming cause high levels of metabolic stress. Indeed, redox signaling pathways are often up-regulated in metastasis (Pani et al. 2010). For example, it was recently shown that metastatic melanoma cells adopt detoxifying mechanisms, such as producing NADPH detoxifying enzymes of the folate pathway, including ALDH1L2 and MTHFD1, to withstand oxidative stress at the metastatic sites (Piskounova et al. 2015). This is in contrast to previous studies suggesting reactive oxygen species (ROS) as prometastatic effectors (Wu 2006; Ishikawa et al. 2008; Nishikawa 2008; Porporato et al. 2014). Such contradictory observations may be due to the fact that these earlier studies focused on the action of ROS on the primary tumor site, which may promote cancer progression by generating genomic instability. ALDH enzymes are well-established CSC markers in several cancer types (Ginestier et al. 2007; Medema 2013). Accumulating functional in vivo studies have also shown that different ALDH members can increase metastasis in breast and other cancers (Rodriguez-Torres and Allan 2016), which may be linked to their aldehyde and ROS detoxification functions.
In general, MICs can use oxidative phosphorylation in oxygenized organs and generate potent mechanisms to protect themselves from ROS and oxidative stress. In other organs with low oxygen and nutrients, such as the liver, MICs are able to use the creatine cycle to scavenge ATP or activate β-oxidation. Therefore, metabolic reprogramming is an essential hallmark for MICs to survive at distant sites.
Resistance to anoikis and apoptosis
Cancer cells often encounter multiple apoptotic death signals in the new environment. It is likely that DTCs already acquire anti-apoptotic mechanisms, such as elevated expression of caspase inhibitors and other anti-apoptotic genes, at the primary tumor sites (Su et al. 2015). Nevertheless, new extrinsic apoptotic signals are often present at the metastatic sites that present new challenges for MICs and coerce MICs to adopt different protective mechanisms. A good example is the protective action of anti-PA serpins to prevent proteolytic activation of the proapoptotic ligand FasL, produced by reactive astrocytes in brain metastasis (Fig. 4B; Valiente et al. 2014).
Another major apoptosis-inducing stress that cancer cells typically encounter during metastasis is detachment from ECM during dissemination, which often leads to anoikis. In a recent study, transcription factor ATF4 has been shown to be part of the integrated stress response (ISR) and protects MICs against death via anoikis (Dey et al. 2015). Another critical pathway promoting cell survival and anoikis resistance is the AKT signaling pathway (Vanharanta and Massague 2013; Wan et al. 2013). Various mechanisms converging on AKT activation have been reported to favor survival at different organs. In DTCs, elevated expression of neurotrophic receptor NTRK2 (TrkB) inhibits anoikis by activating the PI3K–AKT pathway (Douma et al. 2004). In breast cancer bone metastasis, SRC kinase mediates activation of AKT, which is required for CXCL12/SDF-mediated cell survival and resistance to TNFSF10 (TRAIL)-mediated apoptosis (Zhang et al. 2009). In lung metastasis, macrophages support the survival of breast tumor cells by activating the VCAM1–Ezrin–PI3K/Akt survival pathway via the counter-receptor α4-integrin (Chen et al. 2011). Pancreatic cancer metastasis has also been associated with STAT3-induced anoikis inhibition (Fofaria and Srivastava 2015). Another mechanism of anoikis resistance involves cell metabolic reprogramming to overcome an ATP deficit, as we discussed above (Schafer et al. 2009; Weber 2016).
Co-option of the metastatic niche
Distant tissues are normally hostile environments for the newly arriving tumor cells. Most of the DTCs will either die by anoikis/apoptosis due to lack of survival signals or energy resources or be killed by death signals from the incompatible stromal and immune cells of the host tissue (Hanahan and Coussens 2012; Quail and Joyce 2013; Piskounova et al. 2015). The ability of tumor cells to manage and re-educate their new stromal partners in the host microenvironment is a critical event in colonizing a distant tissue. Cancer cells can compete for, corrupt, or co-opt the existing normal tissue niches and recruit additional stromal cells to generate a supporting niche for MICs. Indeed, tumor cells and the host stroma coevolve during metastasis initiation and progression (Joyce and Pollard 2009; Polyak et al. 2009; Barcellos-Hoff et al. 2013). Additionally, emerging evidence shows that tumor–stromal interactions do not occur only after DTCs arrive at a distant tissue. Indeed, the primary tumor can systemically influence the metastasis milieu before and after MIC colonization (McAllister and Weinberg 2014). An example of systemic influence by the primary tumor is the instigation of a supportive stroma enriched in EGF and IGF1 to activate indolent breast cancer DTCs (Castano et al. 2013). In addition, primary tumors can secrete cytokines and exosomes and mobilize bone marrow-derived cells (BMDCs) to form premetastatic niches, which facilitate metastatic colonization by DTCs (Kaplan et al. 2005; Peinado et al. 2012; McAllister and Weinberg 2014; Zhou et al. 2014; Fong et al. 2015; Hoshino et al. 2015). The induction of a receptive microenvironment increases the metastatic efficiency of MICs and enriches the century-old “seed and soil” hypothesis formulated by Stephen Paget (Fidler 2003).
MICs may require a specific niche environment at the target organ to survive and eventually form overt lesions. Following this concept, many studies have focused on finding evidence of the metastatic niche. One of the first relevant studies showed that prostate cancer DTCs displace hematopoietic stem cells (HSCs) from their natural niche to colonize the bone marrow (Fig. 4C; Shiozawa et al. 2011). Recently, the osteogenic niche in bone has been proposed to induce mTOR signaling in DTCs via heterotypic adherens junctions and promote the initial outgrowth of micrometastases in bone (Fig. 4C; Wang et al. 2015b). To successfully form macrometastasis, cancer cells express VCAM1 to recruit preosteoclasts and stimulate osteoclast differentiation, leading to the eventual formation of a “vicious cycle” of metastatic tumor growth, osteoclast-mediated bone lysis, and the release of tumor-promoting growth factors (Fig. 4C; Lu et al. 2011; Sethi et al. 2011; Weilbaecher et al. 2011; Ell and Kang 2012).
Another important niche location in many organs is the vasculature. Endothelial cell sprouting abolishes inhibitory growth signals in dormant tumor cells to allow metastasis of breast cancer cells in different organs (Ghajar et al. 2013). Of note, in different types of cancers, certain microRNAs suppress or promote metastasis by influencing endothelial recruitment to form the metastatic niche (Pencheva and Tavazoie 2013). In the lung, bone marrow-derived endothelial progenitor cells drive the angiogenic switch to promote lung metastasis of luminal breast cancer cells (Gao et al. 2008). In the brain, vascular co-option of breast cancer cells through L1CAM-mediated adhesion facilitates MIC access to nutrients and oxygen, while tumor-derived anti-PA serpin protected MICs from FasL death signals from astrocytes (Fig. 4B; Valiente et al. 2014). MICs can also generate their own niche by building a supportive ECM in distant organs. For example, breast cancer cells secrete tenascin C, an ECM protein, in lungs to a stimulate stemness and favor metastasis (Fig. 4D; Oskarsson et al. 2011).
In order to cultivate a supporting “soil” at secondary organ sites, tumor cells can also activate other nonniche cells, such as fibroblasts in metastatic sites, and turn them into cancer-associated fibroblasts (CAFs) with metastasis-promoting functions (Kalluri and Zeisberg 2006), such as producing ECM niche components periostin (POSTN) and tenascin C (Fig. 4D; O'Connell et al. 2011; Malanchi et al. 2012). In addition, TGF-β released from colorectal cancer cells stimulated CAFs to secrete IL-11, which feeds back to tumor cells to activate STAT3 signaling, favoring the survival of metastatic cells in the liver (Calon et al. 2012). In the brain stroma, reactive astrocytes also mediate important cross-talks with MICs to enhance their proliferation, survival, and metastasis (Kodack et al. 2015). Astrocytes promote stem cell-like traits to breast cancer cells by activating Notch signaling in the brain (Fig. 4B; Xing et al. 2013). A recent study demonstrated how PTEN expression is suppressed in MICs by the interaction with astrocytes. In this study, astrocyte-derived exosomes transfer the PTEN targeting miR-19a to the MICs. PTEN repression increases NFκB-dependent CCL2 secretion and recruitment of myeloid cells to promote survival and growth of MICs in the brain (Fig. 4C; Zhang et al. 2015).
A main threat to MICs is the immune cells present at the new organ sites. The immune system is believed to prevent the formation of >80% of all primary tumors (Hanahan and Weinberg 2011). Even if DTCs have successfully evaded the immune system at the primary tumor site, they are likely to encounter new, hostile immune cells with the ability to recognize and kill them in the circulation and at metastatic sites. Indeed, the plasticity of MICs to readily fluctuate between EMT–MET states might facilitate the immune evasion during metastasis. EMT transcription factors have been shown to have immunosuppressive functions. SNAI1 induces CD4+ CD25− Treg immune-suppressive cells and impairs dendritic cell activity (Kudo-Saito et al. 2009). Moreover, ZEB1 repression of miR-200s up-regulates its target PD-L1, a known immune checkpoint regulator of CD8+ T cells (Chen et al. 2014). The secretion of TGF-β from tumor cells can repress the production of cytolytic and proapoptotic factors by CD8+ CTLs (Thomas and Massague 2005). Therefore, mesenchymal-like DTCs, which often have elevated expression of TGF-β, may escape attack by CTLs upon arrival in distant tissues. In contrast, BMP4, another member of the TGF-β family, functions as a metastasis suppressor in breast cancer by blocking G-CSF-induced expansion of myeloid-derived suppressor cells (MDSCs) (Cao et al. 2014). As another mechanism to compromise the function of innate immune cells during metastasis, melanoma cells express FcγRIIb that negatively regulates B-cell recognition and humoral immunity to promote liver metastasis (Cohen-Solal et al. 2010).
Alternatively, some immune cells can be subverted by DTCs to promote their metastatic growth. For example, activated M2 macrophages can promote metastatic colonization of different cancers by supporting growth, survival, and vascularization while impairing immunogenicity (Qian and Pollard 2010; Quail and Joyce 2013). In the lungs, breast cancer cells can interact with macrophages to activate the PI3K–AKT pathway and protect the cancer cells from apoptotic signals (Chen et al. 2011). In fact, ablation of macrophage activation by blocking CSF-1R or CCR2 is a promising strategy to prevent macrophage instigation of metastasis outgrowth (Quail and Joyce 2013).
Taken together, MICs have evolved multiple mechanisms to turn a potentially hostile environment in a secondary organ into a supportive niche. This can be achieved by releasing systemic growth and survival signals from the primary tumor to foster a premetastatic niche, competing for existing normal stem cell niches, and engaging and converting the stromal cells to thwart death signals and immune attack.
Exit from dormancy
Metastatic dormancy is a frequent occurrence in many cancer types, with distant relapse occurring many years after the successful treatment of an early-stage primary tumor and initial complete remission. Dormant DTCs have been defined with three main features: growth arrest, survival, and therapy resistance (Ghajar 2015). Furthermore, their entry into dormancy and reactivation not only is triggered by intrinsic programs but is also dependent on specialized microenvironmental niches, extrinsic signals, and immune effects (Giancotti 2013; Quail and Joyce 2013; Sosa et al. 2014).
Due to technical limitations, it is impractical to follow a single cell for years and witness its awakening from dormancy to initiate metastatic outgrowth, especially in clinical settings. Consequently, little has been known about how dormant cells escape growth arrest to initiate metastasis. Some studies propose different mechanisms for different organ-specific metastases (Sosa et al. 2014). In bone metastasis, elevated expression of VCAM1 induced by inflammatory pathways in tumor cells promotes the transition from indolent micrometastasis to overt metastasis (Lu et al. 2011). In lung metastasis, BMP signaling from the parenchyma restrains breast DTCs from exiting a dormant state by repressing self-renewal and inducing differentiation (Gao et al. 2012). Production of BMP inhibitors, such as Coco, by tumor cells can release them from latency, prevent differentiation, and promote metastasis initiation. Thus, the ability of dormant DTCs to overcome such anti-growth signals is what turns them into active MICs. Other signals from the stromal niche can also induce the reactivation of growth and self-renewal pathways, such as ERK, Wnt, and Notch (Giancotti 2013). For example, an ECM component of the metastatic niche, tenascin C, can activate Notch and β-catenin signaling (Oskarsson et al. 2011), while POSTN can present Wnt ligands to activate WNT/TCF signaling (Fig. 4D; Malanchi et al. 2012). Moreover, the perivascular niche can reactivate metastatic growth of dormant DTCs by endothelial sprouting and secretion of POSTN and TGF-β (Ghajar et al. 2013). Therefore, MICs can overcome dormancy by activating self-renewal and stem cell-related pathways, such as Wnt, Notch, and TGF-β.
We discussed above how EMT or MET can generate stem cell properties in cancer cells and how mesenchymal-like cancer cells are less proliferative than epithelial-like cancer cells (Brabletz 2012; Liu et al. 2014). According to paradigm, it has been proposed, but not yet proven, that mesenchymal-like TICs remain in a dormant state upon arrival in a distant organ and need to undergo MET in order to reactivate and initiate metastasis (Giancotti 2013). In this scenario, both processes of EMT and MET would be critical for metastasis: EMT for entering dormancy, promoting survival, and drug resistance and MET as the mechanism to reactivate proliferation and self-renewal to initiate metastasis. This could also explain the pathological observation that metastases display epithelial traits rather than mesenchymal characters (Chaffer et al. 2007; Korpal et al. 2011; Tsai et al. 2012; Chui 2013).
Drug resistance of MICs
A close correlation between metastasis and treatment resistance is frequently observed. Metastatic tumors are invariably more chemoresistant than primary tumors, as evidenced by the marked decrease of chemotherapy response rate in metastatic settings as compared with neoadjuvant settings (Gonzalez-Angulo et al. 2007). Conversely, poor response to neoadjuvant chemotherapy often correlates with earlier metastatic recurrence and shorter survival, indicating that chemoresistant tumors are prone to metastasize (Gonzalez-Angulo et al. 2007). Therefore, the generation of MIC properties may be phenotypically linked to enhanced drug resistance capacities. MICs enriched with CSC-like features may benefit from resistant mechanisms of CSCs, such as a stronger DNA damage response (Wang 2015), elevated expression of efflux drug pumps (Schinkel et al. 1994; Zhou et al. 2001; Dean et al. 2005), and ALDH detoxifying enzymes (Honoki et al. 2010; Rausch et al. 2010). Therefore, inhibitors of pathways involved in CSC regulation, such as antibodies against NOTCH, FZD, IL6R, and other relevant signaling pathway receptors, may also have a therapeutic impact on MICs (Brooks et al. 2015).
Importantly, EMT induction is well known to increase chemoresistance (Thiery et al. 2009; Yu et al. 2013; Zheng et al. 2015b) and recently has been shown to induce chemoresistance in lung metastases using an EMT lineage tracing system in breast cancer (Fischer et al. 2015). These studies help explain why conventional treatments like gemcitabine or cyclophosphamide usually do not affect mesenchymal-like cells. Therefore, the existence of dormant mesenchymal-like clones at a distant site could resist many conventional treatments (Giancotti 2013; Kang and Pantel 2013) and require novel therapeutic strategies targeting EMT-related pathways and features. For example, tumor cells undergoing EMT become resistant to EGFR inhibitors due to the activation of AXL kinase, which may be blocked with specific kinase inhibitors (Zhang et al. 2012). The inhibition of PKCα and FRA1 can suppress tumor initiation by mesenchymal-like CSCs and is therefore a potential target for mesenchymal-like MICs (Tam et al. 2013). However, dormant cancer cells can also escape existing cancer treatments because of their quiescent status or niche protection (Braun et al. 2000; Naumov et al. 2003). Therefore, dormancy-specific treatment strategies should be designed to target the dormant cells (Sosa et al. 2014; Ghajar 2015). Furthermore, other MIC-associated features, such as metabolic reprogramming and activation of survival pathways, are additional candidates for developing new treatment options (Holohan et al. 2013; Loo et al. 2015).
Besides these MIC-intrinsic properties, tumor-associated stroma has also been found to severely increase resistance to traditional cancer therapies (Gilbert and Hemann 2010; Sun et al. 2012). In metastasis, primary tumor-associated endothelial cells produce TNFα to increase CXCL1/2 in cancer cells. These attract myeloid cells at the metastatic site, which produces S100A8/9 to feed back to metastatic cells and stimulate increased chemoresistance (Acharyya et al. 2012). The tumor–stromal niche interactions discussed earlier provide additional opportunities to disrupt a prosurvival niche for MICs and sensitize them to anti-cancer agents (Wan et al. 2013).
MIC heterogeneity: clonal or polyclonal metastasis
Primary tumors are heterogeneous masses of cells containing multiple subclones that are genetically and epigenetically different (Marusyk et al. 2012). Primary tumors are considered to arise from single TICs capable of both self-renewing and producing heterogeneity (Hanahan and Weinberg 2011; Greaves and Maley 2012). In metastasis, the classical view also considers a single tumor cell as the origin of metastases, based on chromosomal analysis (Talmadge et al. 1982). However, circulating tumor cells (CTCs) have been found to be genetically and phenotypically heterogeneous (Stoecklein et al. 2008; Kang and Pantel 2013; Yu et al. 2013), raising the possibility of polyclonal seeding and metastases. Until now, little was known about the clonal population dynamics throughout the different steps of metastasis leading into the formation of overt metastases. However, recent studies using lineage tracing, barcode sequencing, and whole-genome sequencing are shedding light on this question and have demonstrated a mostly polyclonal nature of metastasis (Fig. 5; McFadden et al. 2014; Gundem et al. 2015; Maddipati and Stanger 2015; Wagenblast et al. 2015).
Figure 5.

Clonal cooperation in metastasis. (A) Representation of different macrometastasis outputs from initial polyclonal dissemination and seeding. Polyclonal seeding and micrometastasis may develop polyclonal (left) or monoclonal (middle) macrometastasis depending on the clonal and tumor–stroma interaction dynamics in the target organ. (Right) In addition, metastasis heterogeneity can result from the generation of multiple phenotypes from a single metastatic clone. (B, left) Mesenchymal-like secretory cells can induce invasive phenotypes in epithelial-like TICs through secreted factors such as SPARC, facilitating their escape from the primary site. (Middle) In addition, noninvasive TICs/MICs can opportunistically follow trailblazer invasive cells to escape from the primary site or extravasate and infiltrate a distant tissue. (Right) Polyclonal seeding of a distant organ as a result of clonal cooperation.
Mutation analysis between the primary tumor and the metastatic lesions indicated polyclonal metastatic spread in the lymph nodes but not in the liver (McFadden et al. 2014). Another whole-genome sequencing study analyzed 51 tumors of 10 prostate cancer patients, including primary tumors and multiple metastases in the same patients, and revealed the coexistence of multiple clones in the metastases, including those from metastasis-to-metastasis spreads (Gundem et al. 2015). Molecular barcoding offers another effective method to track clonal populations in experimental animal models of metastasis, and this approach has recently been used to analyze metastasis heterogeneity generated by the 4T1 mouse mammary tumor cell line (Wagenblast et al. 2015). In this study, orthotopic injection of barcoded cells generated metastases composed of multiple, different clones in various tissues, although it cannot be ruled out that independent metastatic nodules in the same organ might be seeded monoclonally.
Lineage tracing using fluorescence markers is another robust method to study polyclonal metastases in animal models. Combining the multicolor “confetti” mouse model for multiclonal tracking with the K-rasLSL.G12D/+; p53R172H/+; PdxCre (KPC) mouse model of pancreatic ductal adenocarcinoma (PDAC), a high frequency of polyclonal metastasis was revealed, including 11%–14% in the lungs and liver and 80% in the peritoneum and diaphragm. Interestingly, during the metastatic outgrowth to overt lesions, the clonal diversity usually decreases, leading to formation of monoclonal or polyclonal expansions that appear to depend on the metastatic site (Maddipati and Stanger 2015). Taken together, emerging evidence suggests that MICs are heterogeneous, and different clones are often involved in seeding and forming overt metastasis. Depending on the interaction of MIC clones and the conditions of the host organ, the metastatic outgrowth can remain polyclonal or become monoclonal (Fig. 5A). Additionally, the demonstration of polyclonal metastasis suggests the contribution of different heterotypic interactions among different tumor clonal subpopulations to initiate metastasis.
Clonal cooperation
The importance of tumor heterogeneity in cancer evolution has led to the idea that tumors may function as ecosystems of interactive populations within an environment. Consequently, several studies have started to focus on the ecological cooperation or competitive interactions between tumor populations (Merlo et al. 2006; Moreno 2008; Neelakantan et al. 2015; Tabassum and Polyak 2015). In primary breast tumors, recent research using mouse models has characterized the polyclonal origin of certain tumor types and the interclonal cooperation between multiple subclones (Cleary et al. 2014). Small subpopulations can drive the growth of other non-cell-autonomous clones through paracrine interaction and microenvironment modulation (Marusyk et al. 2014). In the metastatic context, this phenomenon was first reported by coinjecting nonmetastatic cells with metastatic cells to increase the metastasis of the former (Miller 1983). Cooperation can be promoted by endocrine and exosome signaling between different clones (Martorana et al. 1998; Neelakantan et al. 2015). ECM proteins, such as SPARC, also serve as messengers of cooperation to enhance invasion and metastasis (Fig. 5B; Mateo et al. 2014). In addition, heterotypic interactions among EMT and non-EMT cells have also been demonstrated to increase metastasis progression of hamster cheek pouch carcinoma cells (Tsuji et al. 2008) as well as in xenograft models of prostate cancer metastasis (Celia-Terrassa et al. 2012). In the latter study, both clonal populations seeded distant organs, although only the non-EMT clonal population—enriched in epithelial-like CSCs—expanded to overt metastases. Interestingly, the different clones presented complementary essential properties for metastasis, invasion in the mesenchymal-like clones and self-renewal/proliferation in the epithelial-like clones. As a result, the combination of clones enhanced metastasis, including new organs never colonized by either clone alone. This kind of cooperation has been further characterized in other models proposing a leading invasive cell followed by “opportunistic” cells (Fig. 5B; Chapman et al. 2014; Westcott et al. 2015).
This synergy is in agreement with the observation that CTC clusters have dramatically increased metastatic potential compared with single CTCs (Aceto et al. 2014) and the polyclonal nature of metastases (Gundem et al. 2015; Maddipati and Stanger 2015). Therefore, MICs might be “opportunistic” cells, which benefit from the establishment of heterotypic interactions with other DTCs with complementary abilities that can be exploited by MICs (Fig. 5B).
The polyclonal origin of metastases and cooperative interactions between different clonal populations in metastasis may be highly relevant to the understanding of the so-called polygenic drug resistance (Holohan et al. 2013). Heterogeneous polyclonal metastases confer a great diversity of drug resistance. Furthermore, hierarchically organized populations are reported to stochastically transition between phenotypic states to balance cancer cell populations in any direction, even from non-stem-like cells to stem-like cells (Gupta et al. 2011). Such a high degree of plasticity of MICs, combined with the polyclonal nature of metastasis, presents a major challenge for conventional therapy.
Concluding remarks and perspectives
In this review, we enumerate a compendium of relevant hallmarks of MICs. Many of these hallmarks are represented within the same MIC population; however, it should be kept in mind that not all of them need to be possessed by the same tumor cell to initiate metastasis. A great degree of diversity, including the requirement of different assortments of MIC hallmarks, exists among MICs in different cancer types and subtypes and in different metastatic organ sites. Furthermore, many of the MIC features are required only at specific windows of metastatic progression. Importantly, the discovery of polyclonal metastasis introduces an additional layer of variability, as different clonal populations may cooperate to collectively seed metastasis, and even non-MIC populations can provide important functions to complement MICs in enhancing their metastatic competency. Therefore, the ability to form a metastatic lesion may not be the privilege of a specific tumor cell population with the requisite molecular and functional hallmarks of MICs. Instead, metastasis initiation may be the culmination of a highly fluid process involving multiple iterations of transitional cellular states, dynamic interactions between clonal tumor populations, and both short-distance and long-range interactions between tumor cells and the host organs. Despite all of these ambiguities in defining MICs, a central core property of MICs is their cellular plasticity, which underlies almost all other MIC hallmarks (Fig. 6). This most fundamental hallmark of MICs may therefore represent a potential Achilles’ heel of cancer that can be exploited in developing new treatments. Future research should address these key questions: Is cellular plasticity really crucial for metastatic initiation, and how can we target cellular plasticity? Cellular plasticity targeting treatments are unlikely to be cytotoxic when applied as single agents in treatments and therefore are likely to fail standard clinical trials that often rely on reduced tumor burden as an indication of effectiveness. Instead, such treatment may demonstrate efficacy only when combined with other treatments to induce stress on potential MICs and in adjuvant settings. These will be major hurdles to advance cellular plasticity targeting treatment through the traditional drug development and clinical trial pipelines. Nevertheless, as technical innovations continue to bring about major breakthroughs in the study of MICs in clinical settings and experimental models, our newly developed insights into the mysterious process of metastasis initiation will undoubtedly lead to improved prevention and treatment of metastatic diseases.
Figure 6.
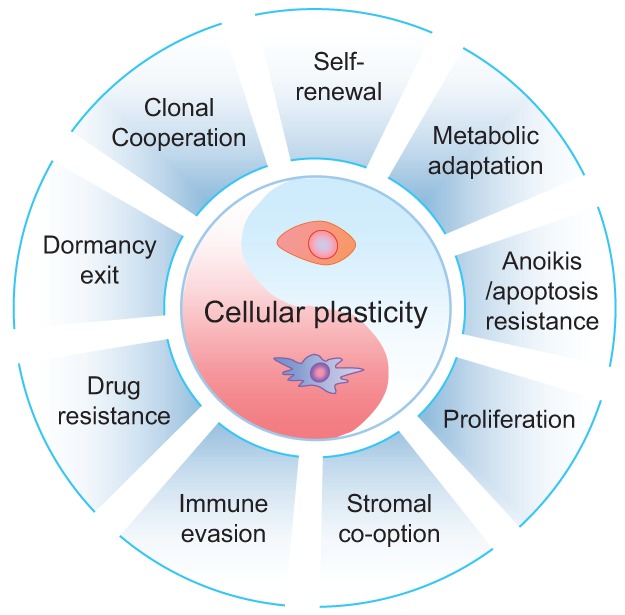
Cellular plasticity as the core characteristic of MICs. This diagram lists nine characteristics commonly observed in MICs. These abilities are often enabled by cancer cell plasticity, represented as the core of MIC properties. Not all of these properties may need to be acquired simultaneously by MICs to grow metastases. Instead, multiple different combinations may influence the emergence of MICs in the affected organs.
Acknowledgments
We thank members of our laboratories for helpful discussions, and, in particular, H.A. Smith and D. Liu for critical reading of the manuscript. We also apologize to the many investigators whose important studies could not be cited directly here owing to space limitations. The work was supported by a Susan G. Komen Fellowship to T.C.-T. (PDF15332075), and grants from the Brewster Foundation, the Breast Cancer Research Foundation, the Department of Defense (BC123187), and the National Institutes of Health (R01CA141062) to Y.K.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.277681.116.
References
- Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, et al. 2014. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158: 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, Manova-Todorova K, Leversha M, Hogg N, Seshan VE, et al. 2012. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 150: 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar E, Marin de Mas I, Zodda E, Marin S, Morrish F, Selivanov V, Meca-Cortés O, Delowar H, Pons M, Izquierdo I, et al. 2016. Metabolic reprogramming and dependencies associated with epithelial cancer stem cells independent of the epithelial–mesenchymal transition program. Stem Cells 10.1002/stem.2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcellos-Hoff MH, Lyden D, Wang TC. 2013. The evolution of the cancer niche during multistage carcinogenesis. Nat Rev Cancer 13: 511–518. [DOI] [PubMed] [Google Scholar]
- Beck B, Lapouge G, Rorive S, Drogat B, Desaedelaere K, Delafaille S, Dubois C, Salmon I, Willekens K, Marine JC, et al. 2015. Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell 16: 67–79. [DOI] [PubMed] [Google Scholar]
- Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. 2008. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 40: 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, Delatte B, Caauwe A, Lenglez S, Nkusi E, et al. 2014. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 511: 246–250. [DOI] [PubMed] [Google Scholar]
- Bozic I, Antal T, Ohtsuki H, Carter H, Kim D, Chen S, Karchin R, Kinzler KW, Vogelstein B, Nowak MA. 2010. Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci 107: 18545–18550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T. 2012. To differentiate or not—routes towards metastasis. Nat Rev Cancer 12: 425–436. [DOI] [PubMed] [Google Scholar]
- Braun S, Kentenich C, Janni W, Hepp F, de Waal J, Willgeroth F, Sommer H, Pantel K. 2000. Lack of effect of adjuvant chemotherapy on the elimination of single dormant tumor cells in bone marrow of high-risk breast cancer patients. J Clin Oncol 18: 80–86. [DOI] [PubMed] [Google Scholar]
- Brooks MD, Burness ML, Wicha MS. 2015. Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell 17: 260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caino MC, Chae YC, Vaira V, Ferrero S, Nosotti M, Martin NM, Weeraratna A, O'Connell M, Jernigan D, Fatatis A, et al. 2013. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J Clin Invest 123: 2907–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Cespedes MV, Sevillano M, Nadal C, Jung P, Zhang XH, et al. 2012. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 22: 571–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron MD, Schmidt EE, Kerkvliet N, Nadkarni KV, Morris VL, Groom AC, Chambers AF, MacDonald IC. 2000. Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res 60: 2541–2546. [PubMed] [Google Scholar]
- Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, et al. 2010. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467: 1109–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao PD, Cheung WK, Nguyen DX. 2011. Cell lineage specification in tumor progression and metastasis. Discov Med 12: 329–340. [PubMed] [Google Scholar]
- Cao Y, Slaney CY, Bidwell BN, Parker BS, Johnstone CN, Rautela J, Eckhardt BL, Anderson RL. 2014. BMP4 inhibits breast cancer metastasis by blocking myeloid-derived suppressor cell activity. Cancer Res 74: 5091–5102. [DOI] [PubMed] [Google Scholar]
- Castano Z, Marsh T, Tadipatri R, Kuznetsov HS, Al-Shahrour F, Paktinat M, Greene-Colozzi A, Nilsson B, Richardson AL, McAllister SS. 2013. Stromal EGF and IGF-I together modulate plasticity of disseminated triple-negative breast tumors. Cancer Discov 3: 922–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celia-Terrassa T, Meca-Cortes O, Mateo F, de Paz AM, Rubio N, Arnal-Estape A, Ell BJ, Bermudo R, Diaz A, Guerra-Rebollo M, et al. 2012. Epithelial–mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest 122: 1849–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, Thompson EW, Williams ED. 2007. Mesenchymal to epithelial transition in development and disease. Cells Tissues Organs 185: 7–19. [DOI] [PubMed] [Google Scholar]
- Chakrabarti R, Hwang J, Andres Blanco M, Wei Y, Lukacisin M, Romano RA, Smalley K, Liu S, Yang Q, Ibrahim T, et al. 2012. Elf5 inhibits the epithelial–mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2. Nat Cell Biol 14: 1212–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers AF, Groom AC, MacDonald IC. 2002. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2: 563–572. [DOI] [PubMed] [Google Scholar]
- Chapman A, Fernandez del Ama L, Ferguson J, Kamarashev J, Wellbrock C, Hurlstone A. 2014. Heterogeneous tumor subpopulations cooperate to drive invasion. Cell Rep 8: 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheli Y, Giuliano S, Fenouille N, Allegra M, Hofman V, Hofman P, Bahadoran P, Lacour JP, Tartare-Deckert S, Bertolotto C, et al. 2012. Hypoxia and MITF control metastatic behaviour in mouse and human melanoma cells. Oncogene 31: 2461–2470. [DOI] [PubMed] [Google Scholar]
- Chen EI, Hewel J, Krueger JS, Tiraby C, Weber MR, Kralli A, Becker K, Yates JR III, Felding-Habermann B. 2007. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res 67: 1472–1486. [DOI] [PubMed] [Google Scholar]
- Chen Q, Zhang XH, Massague J. 2011. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 20: 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W, et al. 2014. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun 5: 5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung WK, Zhao M, Liu Z, Stevens LE, Cao PD, Fang JE, Westbrook TF, Nguyen DX. 2013. Control of alveolar differentiation by the lineage transcription factors GATA6 and HOPX inhibits lung adenocarcinoma metastasis. Cancer Cell 23: 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chui MH. 2013. Insights into cancer metastasis from a clinicopathologic perspective: epithelial–mesenchymal transition is not a necessary step. Int J Cancer 132: 1487–1495. [DOI] [PubMed] [Google Scholar]
- Cleary AS, Leonard TL, Gestl SA, Gunther EJ. 2014. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature 508: 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Solal JF, Cassard L, Fournier EM, Loncar SM, Fridman WH, Sautes-Fridman C. 2010. Metastatic melanomas express inhibitory low affinity Fcγ receptor and escape humoral immunity. Dermatol Res Pract 2010: 657406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole MD. 1986. The myc oncogene: its role in transformation and differentiation. Annu Rev Genet 20: 361–384. [DOI] [PubMed] [Google Scholar]
- Cunha S, Lin YC, Goossen EA, DeVette CI, Albertella MR, Thomson S, Mulvihill MJ, Welm AL. 2014. The RON receptor tyrosine kinase promotes metastasis by triggering MBD4-dependent DNA methylation reprogramming. Cell Rep 6: 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M, Fojo T, Bates S. 2005. Tumour stem cells and drug resistance. Nat Rev Cancer 5: 275–284. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. 2008. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7: 11–20. [DOI] [PubMed] [Google Scholar]
- Dey S, Sayers CM, Verginadis II, Lehman SL, Cheng Y, Cerniglia GJ, Tuttle SW, Feldman MD, Zhang PJ, Fuchs SY, et al. 2015. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J Clin Invest 125: 2592–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, Lin Y, Yao J, Shi J, Kang T, et al. 2013. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 23: 316–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douma S, Van Laar T, Zevenhoven J, Meuwissen R, Van Garderen E, Peeper DS. 2004. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature 430: 1034–1039. [DOI] [PubMed] [Google Scholar]
- Dupuy F, Tabaries S, Andrzejewski S, Dong Z, Blagih J, Annis MG, Omeroglu A, Gao D, Leung S, Amir E, et al. 2015. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab 22: 577–589. [DOI] [PubMed] [Google Scholar]
- Ell B, Kang Y. 2012. SnapShot: bone metastasis. Cell 151: 690–690.e1. [DOI] [PubMed] [Google Scholar]
- Fidler IJ. 2003. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 3: 453–458. [DOI] [PubMed] [Google Scholar]
- Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al. 2015. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527: 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fofaria NM, Srivastava SK. 2015. STAT3 induces anoikis resistance, promotes cell invasion and metastatic potential in pancreatic cancer cells. Carcinogenesis 36: 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong MY, Zhou W, Liu L, Alontaga AY, Chandra M, Ashby J, Chow A, O'Connor ST, Li S, Chin AR, et al. 2015. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol 17: 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Nolan DJ, Mellick AS, Bambino K, McDonnell K, Mittal V. 2008. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science 319: 195–198. [DOI] [PubMed] [Google Scholar]
- Gao H, Chakraborty G, Lee-Lim AP, Mo Q, Decker M, Vonica A, Shen R, Brogi E, Brivanlou AH, Giancotti FG. 2012. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 150: 764–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghajar CM. 2015. Metastasis prevention by targeting the dormant niche. Nat Rev Cancer 15: 238–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, Almeida D, Koller A, Hajjar KA, Stainier DY, et al. 2013. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 15: 807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti FG. 2013. Mechanisms governing metastatic dormancy and reactivation. Cell 155: 750–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Hemann MT. 2010. DNA damage-mediated induction of a chemoresistant niche. Cell 143: 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, et al. 2007. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1: 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. 2007. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol 608: 1–22. [DOI] [PubMed] [Google Scholar]
- Greaves M, Maley CC. 2012. Clonal evolution in cancer. Nature 481: 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Frommel SC, Oakes CC, Simon R, Grupp K, Gerig CY, Bar D, Robinson MD, Baer C, Weiss M, et al. 2015. BAZ2A (TIP5) is involved in epigenetic alterations in prostate cancer and its overexpression predicts disease recurrence. Nat Genet 47: 22–30. [DOI] [PubMed] [Google Scholar]
- Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JM, Papaemmanuil E, Brewer DS, Kallio HM, Hognas G, Annala M, et al. 2015. The evolutionary history of lethal metastatic prostate cancer. Nature 520: 353–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zurrer-Hardi U, Bell G, et al. 2012. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 148: 1015–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta GP, Massague J. 2006. Cancer metastasis: building a framework. Cell 127: 679–695. [DOI] [PubMed] [Google Scholar]
- Gupta GP, Perk J, Acharyya S, de Candia P, Mittal V, Todorova-Manova K, Gerald WL, Brogi E, Benezra R, Massague J. 2007. ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc Natl Acad Sci 104: 19506–19511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES. 2011. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 146: 633–644. [DOI] [PubMed] [Google Scholar]
- Habu N, Imanishi Y, Kameyama K, Shimoda M, Tokumaru Y, Sakamoto K, Fujii R, Shigetomi S, Otsuka K, Sato Y, et al. 2015. Expression of Oct3/4 and Nanog in the head and neck squamous carcinoma cells and its clinical implications for delayed neck metastasis in stage I/II oral tongue squamous cell carcinoma. BMC Cancer 15: 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Coussens LM. 2012. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21: 309–322. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- Hendrix MJ, Seftor EA, Seftor RE, Kasemeier-Kulesa J, Kulesa PM, Postovit LM. 2007. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer 7: 246–255. [DOI] [PubMed] [Google Scholar]
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. 2013. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 13: 714–726. [DOI] [PubMed] [Google Scholar]
- Honoki K, Fujii H, Kubo A, Kido A, Mori T, Tanaka Y, Tsujiuchi T. 2010. Possible involvement of stem-like populations with elevated ALDH1 in sarcomas for chemotherapeutic drug resistance. Oncol Rep 24: 501–505. [DOI] [PubMed] [Google Scholar]
- Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, et al. 2015. Tumour exosome integrins determine organotropic metastasis. Nature 527: 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Wu T, Liu AY, Ouyang G. 2015. Differentiation and transdifferentiation potentials of cancer stem cells. Oncotarget 6: 39550–39563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, Forni G, Eils R, Fehm T, Riethmuller G, et al. 2008. Systemic spread is an early step in breast cancer. Cancer Cell 13: 58–68. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J. 2008. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 320: 661–664. [DOI] [PubMed] [Google Scholar]
- Jacob LS, Vanharanta S, Obenauf AC, Pirun M, Viale A, Socci ND, Massague J. 2015. Metastatic competence can emerge with selection of preexisting oncogenic alleles without a need of new mutations. Cancer Res 75: 3713–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly MK, Boareto M, Huang B, Jia D, Lu M, Ben-Jacob E, Onuchic JN, Levine H. 2015a. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front Oncol 5: 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly MK, Jia D, Boareto M, Mani SA, Pienta KJ, Ben-Jacob E, Levine H. 2015b. Coupling the modules of EMT and stemness: a tunable ‘stemness window’ model. Oncotarget 6: 25161–25174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce JA, Pollard JW. 2009. Microenvironmental regulation of metastasis. Nat Rev Cancer 9: 239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Zeisberg M. 2006. Fibroblasts in cancer. Nat Rev Cancer 6: 392–401. [DOI] [PubMed] [Google Scholar]
- Kang Y, Pantel K. 2013. Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell 23: 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et al. 2005. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438: 820–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kareta MS, Gorges LL, Hafeez S, Benayoun BA, Marro S, Zmoos AF, Cecchini MJ, Spacek D, Batista LF, O'Brien M, et al. 2015. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 16: 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Hoffman JP, Alpaugh RK, Rhim AD, Reichert M, Stanger BZ, Furth EE, Sepulveda AR, Yuan CX, Won KJ, et al. 2013. An iPSC line from human pancreatic ductal adenocarcinoma undergoes early to invasive stages of pancreatic cancer progression. Cell Rep 3: 2088–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodack DP, Askoxylakis V, Ferraro GB, Fukumura D, Jain RK. 2015. Emerging strategies for treating brain metastases from breast cancer. Cancer Cell 27: 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celia-Terrassa T, Mercatali L, Khan Z, Goodarzi H, Hua Y, et al. 2011. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med 17: 1101–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouros-Mehr H, Bechis SK, Slorach EM, Littlepage LE, Egeblad M, Ewald AJ, Pai SY, Ho IC, Werb Z. 2008. GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell 13: 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. 2009. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 15: 195–206. [DOI] [PubMed] [Google Scholar]
- Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang CY, et al. 2015. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 526: 131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu VS, O'Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, et al. 2014. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 16: 992–1003, 1001–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledford H. 2011. Cancer theory faces doubts. Nature 472: 273. [DOI] [PubMed] [Google Scholar]
- Leng Z, Tao K, Xia Q, Tan J, Yue Z, Chen J, Xi H, Li J, Zheng H. 2013. Kruppel-like factor 4 acts as an oncogene in colon cancer stem cell-enriched spheroid cells. PLoS One 8: e56082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Kang Y. 2016. Probing the fifty shades of EMT in metastasis. Trends Cancer 2: 65–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CM, Gocheva V, Oudin MJ, Bhutkar A, Wang SY, Date SR, Ng SR, Whittaker CA, Bronson RT, Snyder EL, et al. 2015. Foxa2 and Cdx2 cooperate with Nkx2-1 to inhibit lung adenocarcinoma metastasis. Genes Dev 29: 1850–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, et al. 2007. The energy sensing LKB1–AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol 9: 218–224. [DOI] [PubMed] [Google Scholar]
- Liu X, Sun H, Qi J, Wang L, He S, Liu J, Feng C, Chen C, Li W, Guo Y, et al. 2013. Sequential introduction of reprogramming factors reveals a time-sensitive requirement for individual factors and a sequential EMT–MET mechanism for optimal reprogramming. Nat Cell Biol 15: 829–838. [DOI] [PubMed] [Google Scholar]
- Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, Martin-Trevino R, Shang L, McDermott SP, Landis MD, et al. 2014. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports 2: 78–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo JM, Scherl A, Nguyen A, Man FY, Weinberg E, Zeng Z, Saltz L, Paty PB, Tavazoie SF. 2015. Extracellular metabolic energetics can promote cancer progression. Cell 160: 393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, Yan J, Hua Y, Tiede BJ, Lu X, et al. 2011. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging α4β1-positive osteoclast progenitors. Cancer Cell 20: 701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Mazur SJ, Lin T, Appella E, Xu Y. 2014. The pluripotency factor nanog promotes breast cancer tumorigenesis and metastasis. Oncogene 33: 2655–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, Groom AC. 1998. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol 153: 865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddipati R, Stanger BZ. 2015. Pancreatic cancer metastases harbor evidence of polyclonality. Cancer Discov 5: 1086–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF, Huelsken J. 2012. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 481: 85–89. [DOI] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. 2008. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell 133: 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martorana AM, Zheng G, Crowe TC, O'Grady RL, Lyons JG. 1998. Epithelial cells up-regulate matrix metalloproteinases in cells within the same mammary carcinoma that have undergone an epithelial–mesenchymal transition. Cancer Res 58: 4970–4979. [PubMed] [Google Scholar]
- Marusyk A, Almendro V, Polyak K. 2012. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer 12: 323–334. [DOI] [PubMed] [Google Scholar]
- Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, Polyak K. 2014. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 514: 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, et al. 2014. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159: 1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massard C, Loriot Y, Fizazi K. 2011. Carcinomas of an unknown primary origin—diagnosis and treatment. Nat Rev Clin Oncol 8: 701–710. [DOI] [PubMed] [Google Scholar]
- Mateo F, Meca-Cortes O, Celia-Terrassa T, Fernandez Y, Abasolo I, Sanchez-Cid L, Bermudo R, Sagasta A, Rodriguez-Carunchio L, Pons M, et al. 2014. SPARC mediates metastatic cooperation between CSC and non-CSC prostate cancer cell subpopulations. Mol Cancer 13: 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister SS, Weinberg RA. 2014. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol 16: 717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden DG, Papagiannakopoulos T, Taylor-Weiner A, Stewart C, Carter SL, Cibulskis K, Bhutkar A, McKenna A, Dooley A, Vernon A, et al. 2014. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 156: 1298–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema JP. 2013. Cancer stem cells: the challenges ahead. Nat Cell Biol 15: 338–344. [DOI] [PubMed] [Google Scholar]
- Merlo LM, Pepper JW, Reid BJ, Maley CC. 2006. Cancer as an evolutionary and ecological process. Nat Rev Cancer 6: 924–935. [DOI] [PubMed] [Google Scholar]
- Miller FR. 1983. Tumor subpopulation interactions in metastasis. Invasion Metastasis 3: 234–242. [PubMed] [Google Scholar]
- Morales M, Arenas EJ, Urosevic J, Guiu M, Fernandez E, Planet E, Fenwick RB, Fernandez-Ruiz S, Salvatella X, Reverter D, et al. 2014. RARRES3 suppresses breast cancer lung metastasis by regulating adhesion and differentiation. EMBO Mol Med 6: 865–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno E. 2008. Is cell competition relevant to cancer? Nat Rev Cancer 8: 141–147. [DOI] [PubMed] [Google Scholar]
- Naumov GN, Townson JL, MacDonald IC, Wilson SM, Bramwell VH, Groom AC, Chambers AF. 2003. Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metastases. Breast Cancer Res Treat 82: 199–206. [DOI] [PubMed] [Google Scholar]
- Neelakantan D, Drasin DJ, Ford HL. 2015. Intratumoral heterogeneity: clonal cooperation in epithelial-to-mesenchymal transition and metastasis. Cell Adh Migr 9: 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, et al. 2011. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 17: 1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA. 2013. Epithelial plasticity: a common theme in embryonic and cancer cells. Science 342: 1234850. [DOI] [PubMed] [Google Scholar]
- Nishikawa M. 2008. Reactive oxygen species in tumor metastasis. Cancer Lett 266: 53–59. [DOI] [PubMed] [Google Scholar]
- Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A, Nieto MA. 2012. Metastatic colonization requires the repression of the epithelial–mesenchymal transition inducer Prrx1. Cancer Cell 22: 709–724. [DOI] [PubMed] [Google Scholar]
- O'Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, Dewar R, Rocha RM, Brentani RR, Resnick MB, et al. 2011. VEGF-A and tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc Natl Acad Sci 108: 16002–16007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Sinha S, Esposito I, Schiavon G, Lopez-Lago MA, Su W, Pratilas CA, Abele C, Hernandez JM, Ohara M, et al. 2015. The Rho GTPase Rnd1 suppresses mammary tumorigenesis and EMT by restraining Ras–MAPK signalling. Nat Cell Biol 17: 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, Downey RJ, Manova-Todorova K, Brogi E, Massague J. 2011. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med 17: 867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsson T, Batlle E, Massague J. 2014. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell 14: 306–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pani G, Galeotti T, Chiarugi P. 2010. Metastasis: cancer cell's escape from oxidative stress. Cancer Metastasis Rev 29: 351–378. [DOI] [PubMed] [Google Scholar]
- Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar C, et al. 2012. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 18: 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pencheva N, Tavazoie SF. 2013. Control of metastatic progression by microRNA regulatory networks. Nat Cell Biol 15: 546–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, Morrison SJ. 2015. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527: 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Haviv I, Campbell IG. 2009. Co-evolution of tumor cells and their microenvironment. Trends Genet 25: 30–38. [DOI] [PubMed] [Google Scholar]
- Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C, et al. 2014. A mitochondrial switch promotes tumor metastasis. Cell Rep 8: 754–766. [DOI] [PubMed] [Google Scholar]
- Qian BZ, Pollard JW. 2010. Macrophage diversity enhances tumor progression and metastasis. Cell 141: 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Joyce JA. 2013. Microenvironmental regulation of tumor progression and metastasis. Nat Med 19: 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch V, Liu L, Kallifatidis G, Baumann B, Mattern J, Gladkich J, Wirth T, Schemmer P, Buchler MW, Zoller M, et al. 2010. Synergistic activity of sorafenib and sulforaphane abolishes pancreatic cancer stem cell characteristics. Cancer Res 70: 5004–5013. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. 2001. Stem cells, cancer, and cancer stem cells. Nature 414: 105–111. [DOI] [PubMed] [Google Scholar]
- Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, et al. 2012. EMT and dissemination precede pancreatic tumor formation. Cell 148: 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CL, Srivastava J, Siddiq A, Gredler R, Emdad L, Rajasekaran D, Akiel M, Shen XN, Guo C, Giashuddin S, et al. 2014. Genetic deletion of AEG-1 prevents hepatocarcinogenesis. Cancer Res 74: 6184–6193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Torres M, Allan AL. 2016. Aldehyde dehydrogenase as a marker and functional mediator of metastasis in solid tumors. Clin Exp Metastasis 33: 97–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, Brugge JS. 2009. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461: 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, Mol CA, van der Valk MA, Robanus-Maandag EC, te Riele HP, et al. 1994. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 77: 491–502. [DOI] [PubMed] [Google Scholar]
- Schmidt JM, Panzilius E, Bartsch HS, Irmler M, Beckers J, Kari V, Linnemann JR, Dragoi D, Hirschi B, Kloos UJ, et al. 2015. Stem-cell-like properties and epithelial plasticity arise as stable traits after transient Twist1 activation. Cell Rep 10: 131–139. [DOI] [PubMed] [Google Scholar]
- Seftor EA, Meltzer PS, Kirschmann DA, Margaryan NV, Seftor RE, Hendrix MJ. 2006. The epigenetic reprogramming of poorly aggressive melanoma cells by a metastatic microenvironment. J Cell Mol Med 10: 174–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi N, Dai X, Winter CG, Kang Y. 2011. Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell 19: 192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, et al. 2009. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 461: 809–813. [DOI] [PubMed] [Google Scholar]
- Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, Kim JK, Patel LR, Ying C, Ziegler AM, et al. 2011. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest 121: 1298–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa MS, Bragado P, Aguirre-Ghiso JA. 2014. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 14: 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa MS, Parikh F, Maia AG, Estrada Y, Bosch A, Bragado P, Ekpin E, George A, Zheng Y, Lam HM, et al. 2015. NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat Commun 6: 6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spek CA, Arruda VR. 2012. The protein C pathway in cancer metastasis. Thromb Res 129: S80–S84. [DOI] [PubMed] [Google Scholar]
- Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, Massague J, Benezra R. 2013. TGF-β–Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep 5: 1228–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklein NH, Hosch SB, Bezler M, Stern F, Hartmann CH, Vay C, Siegmund A, Scheunemann P, Schurr P, Knoefel WT, et al. 2008. Direct genetic analysis of single disseminated cancer cells for prediction of outcome and therapy selection in esophageal cancer. Cancer Cell 13: 441–453. [DOI] [PubMed] [Google Scholar]
- Su Z, Yang Z, Xu Y, Chen Y, Yu Q. 2015. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer 14: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Campisi J, Higano C, Beer TM, Porter P, Coleman I, True L, Nelson PS. 2012. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med 18: 1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabassum DP, Polyak K. 2015. Tumorigenesis: it takes a village. Nat Rev Cancer 15: 473–483. [DOI] [PubMed] [Google Scholar]
- Talmadge JE, Wolman SR, Fidler IJ. 1982. Evidence for the clonal origin of spontaneous metastases. Science 217: 361–363. [DOI] [PubMed] [Google Scholar]
- Tam WL, Weinberg RA. 2013. The epigenetics of epithelial–mesenchymal plasticity in cancer. Nat Med 19: 1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, Wu ZJ, Krall JA, Bierie B, Guo W, et al. 2013. Protein kinase Cα is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 24: 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YN, Ding WQ, Guo XJ, Yuan XW, Wang DM, Song JG. 2015. Epigenetic regulation of Smad2 and Smad3 by profilin-2 promotes lung cancer growth and metastasis. Nat Commun 6: 8230. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. 2009. Epithelial–mesenchymal transitions in development and disease. Cell 139: 871–890. [DOI] [PubMed] [Google Scholar]
- Thomas DA, Massague J. 2005. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8: 369–380. [DOI] [PubMed] [Google Scholar]
- Tran DD, Corsa CA, Biswas H, Aft RL, Longmore GD. 2011. Temporal and spatial cooperation of Snail1 and Twist1 during epithelial–mesenchymal transition predicts for human breast cancer recurrence. Mol Cancer Res 9: 1644–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HD, Luitel K, Kim M, Zhang K, Longmore GD, Tran DD. 2014. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res 74: 6330–6340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. 2012. Spatiotemporal regulation of epithelial–mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22: 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji T, Ibaragi S, Shima K, Hu MG, Katsurano M, Sasaki A, Hu GF. 2008. Epithelial–mesenchymal transition induced by growth suppressor p12CDK2–AP1 promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res 68: 10377–10386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unternaehrer JJ, Zhao R, Kim K, Cesana M, Powers JT, Ratanasirintrawoot S, Onder T, Shibue T, Weinberg RA, Daley GQ. 2014. The epithelial–mesenchymal transition factor SNAIL paradoxically enhances reprogramming. Stem Cell Rep 3: 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaira V, Faversani A, Martin NM, Garlick DS, Ferrero S, Nosotti M, Kissil JL, Bosari S, Altieri DC. 2013. Regulation of lung cancer metastasis by Klf4–Numb-like signaling. Cancer Res 73: 2695–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valastyan S, Weinberg RA. 2011. Tumor metastasis: molecular insights and evolving paradigms. Cell 147: 275–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ, Chaft JE, Kris MG, Huse JT, Brogi E, et al. 2014. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 156: 1002–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanharanta S, Massague J. 2013. Origins of metastatic traits. Cancer Cell 24: 410–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanner RJ, Remke M, Gallo M, Selvadurai HJ, Coutinho F, Lee L, Kushida M, Head R, Morrissy S, Zhu X, et al. 2014. Quiescent sox2+ cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 26: 33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. 2013. Cancer genome landscapes. Science 339: 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenblast E, Soto M, Gutierrez-Angel S, Hartl CA, Gable AL, Maceli AR, Erard N, Williams AM, Kim SY, Dickopf S, et al. 2015. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature 520: 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Pantel K, Kang Y. 2013. Tumor metastasis: moving new biological insights into the clinic. Nat Med 19: 1450–1464. [DOI] [PubMed] [Google Scholar]
- Wan L, Hu G, Wei Y, Yuan M, Bronson RT, Yang Q, Siddiqui J, Pienta KJ, Kang Y. 2014a. Genetic ablation of metadherin inhibits autochthonous prostate cancer progression and metastasis. Cancer Res 74: 5336–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Lu X, Yuan S, Wei Y, Guo F, Shen M, Yuan M, Chakrabarti R, Hua Y, Smith HA, et al. 2014b. MTDH–SND1 interaction is crucial for expansion and activity of tumor-initiating cells in diverse oncogene- and carcinogen-induced mammary tumors. Cancer Cell 26: 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QE. 2015. DNA damage responses in cancer stem cells: implications for cancer therapeutic strategies. World J Biol Chem 6: 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Cai C, Dong X, Yu QC, Zhang XO, Yang L, Zeng YA. 2015a. Identification of multipotent mammary stem cells by protein C receptor expression. Nature 517: 81–84. [DOI] [PubMed] [Google Scholar]
- Wang H, Yu C, Gao X, Welte T, Muscarella AM, Tian L, Zhao H, Zhao Z, Du S, Tao J, et al. 2015b. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 27: 193–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber GF. 2016. Metabolism in cancer metastasis. Int J Cancer 138: 2061–2066. [DOI] [PubMed] [Google Scholar]
- Weilbaecher KN, Guise TA, McCauley LK. 2011. Cancer to bone: a fatal attraction. Nat Rev Cancer 11: 411–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westcott JM, Prechtl AM, Maine EA, Dang TT, Esparza MA, Sun H, Zhou Y, Xie Y, Pearson GW. 2015. An epigenetically distinct breast cancer cell subpopulation promotes collective invasion. J Clin Invest 125: 1927–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow MM, Dayton TL, Verhaak RG, Kim-Kiselak C, Snyder EL, Feldser DM, Hubbard DD, DuPage MJ, Whittaker CA, Hoersch S, et al. 2011. Suppression of lung adenocarcinoma progression by Nkx2-1. Nature 473: 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WS. 2006. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev 25: 695–705. [DOI] [PubMed] [Google Scholar]
- Xing F, Kobayashi A, Okuda H, Watabe M, Pai SK, Pandey PR, Hirota S, Wilber A, Mo YY, Moore BE, et al. 2013. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol Med 5: 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, et al. 2010. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467: 1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E, Weinberg RA. 2015. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 525: 256–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, et al. 2013. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339: 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, Foekens JA, Massague J. 2009. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 16: 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, et al. 2012. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 44: 852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang S, Yao J, Lowery FJ, Zhang Q, Huang WC, Li P, Li M, Wang X, Zhang C, et al. 2015. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 527: 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R. 2015a. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527: 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R. 2015b. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527: 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M, Nakauchi H, et al. 2001. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med 7: 1028–1034. [DOI] [PubMed] [Google Scholar]
- Zhou W, Fong MY, Min Y, Somlo G, Liu L, Palomares MR, Yu Y, Chow A, O'Connor ST, Chin AR, et al. 2014. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 25: 501–515. [DOI] [PMC free article] [PubMed] [Google Scholar]