

Here, Horigome et al. used imaging and in vivo targeting tools to dissect the mechanistic interactions of SUMO, SMC5/6, and Slx5/8 at double-strand breaks (DSBs) for the relocation of these breaks to nuclear pores. They show that DSB relocation to the nuclear envelope depends on the nature of SUMOylation deposited by the E3 ligases Siz2 and Mms21 and link break relocation to some of the most intensely studied modulators of DNA repair.
Keywords: SUMO, Siz2, Mms21, Slx5, DNA damage, nuclear pores, nuclear organization
Abstract
High-resolution imaging shows that persistent DNA damage in budding yeast localizes in distinct perinuclear foci for repair. The signals that trigger DNA double-strand break (DSB) relocation or determine their destination are unknown. We show here that DSB relocation to the nuclear envelope depends on SUMOylation mediated by the E3 ligases Siz2 and Mms21. In G1, a polySUMOylation signal deposited coordinately by Mms21 and Siz2 recruits the SUMO targeted ubiquitin ligase Slx5/Slx8 to persistent breaks. Both Slx5 and Slx8 are necessary for damage relocation to nuclear pores. When targeted to an undamaged locus, however, Slx5 alone can mediate relocation in G1-phase cells, bypassing the requirement for polySUMOylation. In contrast, in S-phase cells, monoSUMOylation mediated by the Rtt107-stabilized SMC5/6–Mms21 E3 complex drives DSBs to the SUN domain protein Mps3 in a manner independent of Slx5. Slx5/Slx8 and binding to pores favor repair by ectopic break-induced replication and imprecise end-joining.
The accurate repair of DNA double-strand breaks (DSBs) is crucial for genome integrity. Nonhomologous end-joining (NHEJ) and repair by homologous recombination (HR) are common and highly conserved pathways of DSB repair. Breaks that are difficult to repair due to a lack of homologous donor sequences or conditions that impair end-to-end ligation must be repaired by alternative pathways, which include imprecise or microhomology-mediated end-joining or break-induced replication (BIR) (for review, see Ceccaldi et al. 2016). Repair pathway choice is influenced by both the cell cycle (which in turn impacts resection at the break site) and the chromatin context of the damage (Nagai et al. 2010; Geli and Lisby 2015).
Certain types of DNA repair appear to be favored by specific subnuclear compartments. Compartmentation can either stem from the chromatin context of the damage (Therizols et al. 2006; Khadaroo et al. 2009; Agmon et al. 2013) or arise through recruitment of the damage to specific subnuclear sites (for review, see Nagai et al. 2010; Geli and Lisby 2015). For example, DSBs in the repetitive ribosomal DNA locus of yeast or in heterochromatic satellite repeats of flies and mammalian cells shift away from the repetitive sequence domain prior to repair by HR (Torres-Rosell et al. 2007; Chiolo et al. 2011; Lemaître et al. 2014). This is thought to avoid unequal crossover events in cis and/or chromosomal translocations that arise from strand invasion into other chromosomes. In budding yeast, DSBs that occur in unique sequences also shift their subnuclear position if they lack an intact donor for HR or if repeated cleavage/ligation cycles occur (Nagai et al. 2008). Within 2 h of their induction, such persistent DNA breaks accumulate at the nuclear envelope (NE) (Nagai et al. 2008). Similarly, collapsed replication forks and those stalled at expanded triplet repeats (Su et al. 2015) were shown to shift to nuclear pores, as do uncapped telomeres that arise from telomere erosion in telomerase-deficient cells (for review, see Geli and Lisby 2015). It is noteworthy that the repair of these three types of damage requires mechanisms other than precise end-joining or canonical HR.
The perinuclear sites at which yeast damage accumulates were initially identified as nuclear pores, and, in several instances, sequestration was shown to require the Nup84 complex, which, in budding yeast, includes Nup84, Nup120, Nup133, and the associated Nup60 (Nagai et al. 2008; Khadaroo et al. 2009). Subsequent studies identified a second binding site at the NE, the Saccharomyces cerevisiae SUN domain protein Mps3 (Kalocsay et al. 2009; Oza et al. 2009). Mps3 sequesters repair intermediates containing ssDNA complexed with Rad51 and reduces promiscuous recombination events (Kalocsay et al. 2009; Oza et al. 2009; Ferreira et al. 2011). The loss of the Nup84 complex or its associated Slx5/Slx8 SUMO targeted ubiquitin ligase (STUbL), on the other hand, compromised survival after replication fork collapse and enhanced ectopic recombination (Nagai et al. 2008; Oza et al. 2009; Horigome et al. 2014). The two sites of sequestration not only affect repair outcome differentially but have distinct requirements for damage recruitment. Binding to Mps3 appears to be restricted to S and G2 phases of the cell cycle and requires extensive resection at DSBs (Kalocsay et al. 2009; Oza et al. 2009), unlike relocation to nuclear pores, which occurs in G1 without extensive end resection (Horigome et al. 2014). We note that in otherwise unperturbed cells, a transient pore association of triplet repeat stalled replication forks was scored in late S/G2 phase, while they could not be recovered with Mps3 (Su et al. 2015). Intriguingly, ablation of the relevant Nup84 pore subcomplex or of the STUbL subunits Slx5/Slx8 increased the frequency of triplet repeat expansion/contraction events during stalled fork recovery (Su et al. 2015).
Early genetic screens in budding yeast implicated nuclear pore proteins in DNA repair. Screens for survival of ionizing radiation showed that the yeast Nup84 complex contributes to cell survival (Bennett et al. 2001; Loeillet et al. 2005), and loss of Nup84, Nup120, or Nup133 led to pleiotropic DNA damage sensitivities and synthetic lethality with components of the Rad52 pathway of HR (Chang et al. 2002; Loeillet et al. 2005; Nagai et al. 2008). This sensitivity is consistent with the notion that pores mediate repair pathways distinct from canonical HR. Indeed, sites of active HR, visualized as Rad52 foci, are found in the nuclear interior and are specifically excluded from the NE and the nucleolus (Bystricky et al. 2009; Dion et al. 2013). Besides conferring sensitivity to exogenous agents (Bennett et al. 2001), nuclear pore mutants showed impaired replication fork restart (for review, see Bukata et al. 2013; Geli and Lisby 2015) and have recently been implicated in the repair of subtelomeric DSBs by strand invasion events (Chung et al. 2015). In human cells, components in the Nup84 complex were found to suppress elevated levels of H2AX phosphorylation in cells exposed to aphidicolin (Paulsen et al. 2009), and, in flies, the loss of the corresponding complex enhanced the appearance of damage foci provoked by ionizing irradiation (Ryu et al. 2015).
In both yeast and flies, Nup84-linked damage sensitivity and the closely associated nuclear pore basket proteins Nup60 and Mlp1/Mlp2 have been linked to enzymes that control SUMO metabolism (Zhao et al. 2004; Palancade et al. 2007; Nagai et al. 2008; Ryu et al. 2015). Indeed, DSBs that occur in the ribosomal DNA in budding yeast (Torres-Rosell et al. 2007) and in heterochromatin in flies (Chiolo et al. 2011) were shown to shift away from the domain of repetitive sequences in a manner dependent on the SMC5/6 complex and its associated SUMO ligase, Mms21. Although Rad52 SUMOylation influenced damage relocation from the nucleolus to the nucleoplasm (Torres-Rosell et al. 2007), Rad52 was not needed for the relocation of a euchromatic DSB to pores (Horigome et al. 2014). In flies, the STUbL subunit Dgrn, along with the SMC5/6 complex, was necessary for DSB relocation away from heterochromatin (Chiolo et al. 2011; Ryu et al. 2015). This shift required the STUbL interactor Rad60 (ScEsc2/HsNIP45) and the Nup84 pore complex (Ryu et al. 2015).
The molecular link between pores and STUbL enzymes is less clear. Earlier work showed that the yeast STUbL Slx5/Slx8 can be precipitated with Nup84 from cell extracts (Nagai et al. 2008), yet it was unclear whether Slx5/Slx8 mediates damage relocation or instead processes breaks once they reach the periphery. Moreover, the damage-associated signal that triggers relocation remained unknown. Because STUbLs contain SUMO-interacting motifs (SIMs) (for review, see Sarangi and Zhao 2015) and because repair proteins of many different pathways are SUMOylated (Cremona et al. 2012; Psakhye and Jentsch 2012), it became important to test whether SUMO ligases contribute to the relocation of damage to the NE.
Here we dissect the role of SUMO-mediated events mediated by the E3 ligases Siz2 (PIAS in humans) and the SMC5/6-associated Mms21 in damage recognition by the yeast STUbL Slx5/Slx8 and examine how these components affect DSB relocation to the NE. We used rigorous assays that distinguish pore-binding from Mps3-binding sites and G1-phase from S-phase cells. Both loss-of-function and gain-of-function assays allowed us to correlate different sites of damage sequestration with the binding of Slx5, whose recruitment reflects recognition of a polySUMO modification. We found that SUMO chain length helps determine the perinuclear site for damage sequestration. These data likely reflect conserved rules governing damage relocation and appropriate repair pathway choice.
Results
SUMO E3 ligases affect DSB relocation
In addition to phosphorylation and ubiquitination (Smeenk and van Attikum 2013), many proteins become SUMOylated at sites of DNA damage (Cremona et al. 2012; Psakhye and Jentsch 2012). Targets include proteins implicated in basically every pathway of repair, including Rad52, Rad59, Srs2, RPA, Sae2, yKu, Sgs1, and Mre11. Indeed, the loss of Mre11 actually alters SUMOylation of other repair factors (Cremona et al. 2012). SUMO E3 ligases such as Siz2 and Mms21 themselves are recruited to damage (Zhao et al. 2004; Zhao and Blobel 2005; De Piccoli et al. 2006; Chung and Zhao 2015) and were shown to cooperate with Slx5/8 to suppress duplication-mediated genome rearrangements (Albuquerque et al. 2013). We therefore examined the role of SUMO E3 ligases in DSB relocation to the NE.
In budding yeast, there are four SUMO E3 ligases—namely, Siz1 and Siz2, the yeast PIAS homologs; Mms21, a highly conserved ligase component of the SMC5/6 complex; and Cst9, the meiosis-specific putative E3. Siz1 and Siz2 mediate the vast majority of SUMOylation in vegetatively growing yeast, with Siz1 primarily targeting cytoplasmic proteins, and Siz2 modifying a large number of factors involved in HR and other pathways of repair (Ferreira et al. 2011; Cremona et al. 2012; Psakhye and Jentsch 2012; Chung and Zhao 2015). Because Siz1 tends to compensate for survival of DNA damage in the absence of Siz2 (Cremona et al. 2012), we first monitored DSB relocation in cells lacking both PIAS homologs.
To do this, we used a high-resolution microscopic approach that monitors the subnuclear position of an HO-induced DSB at a GFP-LacI-tagged MAT locus in cells lacking both HM donor sites (Fig. 1A,B). A focal stack of images through a population of intact cells allowed us to determine the position of the break relative to the Nup49-CFP-tagged NE and score for the cell cycle phase based on bud size and nuclear position. PCR analysis monitored the efficiency DSB induction. This assay allowed us to differentiate G1-phase from S/G2-phase requirements for DSB relocation (Fig. 1B; Nagai et al. 2008; Horigome et al. 2014).
Figure 1.

SUMO E3 ligases are required for the DSB anchoring to the nuclear periphery. (A) Shown is chromosome III (Chr III) in GA-6844 bearing deleted homologous donor loci (hmlΔ/hmrΔ) and a lacO array inserted 4.4 kb from the HO cut site at MAT. GFP-LacI and CFP-Nup49 label DSB and pores, respectively. Relocation to Mps3 was previously shown to occur in S or G2 phase, coincident with extensive resection. (B) Locus position was scored relative to the nuclear diameter in the locus’ plane of focus using a spinning disc confocal image stack. Distance over diameter ratios are binned into three equal zones. (C) Position of cleaved MAT position relative to CFP-Nup49 in siz1Δ siz2Δ (GA-7968) and mms21ΔC (JC3654) after 120 min on galactose. The mutants compromise relocation in both G1- and S-phase cells. (#) Significantly nonrandom based on cell number and confidence values from a proportional test comparing random and experimental distributions; (*) significantly different distribution between wild type and the mutant; (red dotted line) 33% or random distribution. Cleavage efficiency, nuclei counted, and statistical significance for all imaging experiments are summarized in Supplemental Table S1. (D) Scoring of MAT colocalization with the pore cluster in nup133ΔN (GA-7314) after cut induction. Scoring criteria are shown at the left, and results comparing wild type (GA-7314) and siz2Δ (GA-7970) are at the right. A gray-shaded zone between dotted lines represents empirically (top) and computationally (bottom) determined limits of stochastic colocalization (Horigome et al. 2014, 2015). (E) Pore-ChIP (chromatin immunoprecipitation) was performed with Mab414 monoclonal (Abcam) with wild-type and siz2Δ isogenic derivatives of JKM179 (Lee et al. 1998). HO cleavage was induced for the indicated times on galactose. Quantitative PCR (qPCR) at 1.6 kb from the HO cut site was performed in triplicate on two biological replicates. See the Supplemental Material for normalization techniques. (F) Chromosome III in the MATa strain was used for ChIP, with the positions of primer/probe sets shown. ChIP of MAT colocalization with HA-tagged Mps3 is shown for wild-type (GA-8306) and siz2Δ (GA-8541) cells at the indicated times after cut induction. Data from two independent experiments quantified in triplicate are represented as mean ± SEM. (G) The position of the cleaved MAT relative to CFP-Nup49 in smt3-3KR (GA-9072) after 120 min on galactose, as in C. (n.s.) Not significantly different from random.
At 120 min after induction of the HO endonuclease, we note that the peripheral relocation of the induced DSB observed in wild-type cells was lost in the siz1Δ siz2Δ double mutant in both G1- and S-phase cells (Fig. 1C; Supplemental Table S1). We tested the deletion alleles individually and found that siz2Δ compromises perinuclear relocation more efficiently than siz1Δ alone, especially at early time points (Supplemental Fig. S1B). This is consistent with recent work showing a dominant role of Siz2 in damage-associated SUMOylation (Chung and Zhao 2015; for review, see Sarangi and Zhao 2015), although Siz1 may compensate over time (Supplemental Fig. S1B).
The SUMO E3 ligase Mms21, like all components of its associated SMC5/6 complex, is essential for yeast viability, although its SUMO ligase activity is not. Mms21 SUMOylation activity, on the other hand, is critical for survival of genotoxic stress and becomes essential in the absence of Siz1 and Siz2 activities (Cremona et al. 2012). We therefore tested the effects of a SUMO ligase-deficient Mms21, mms21ΔC, on DSB relocation. Similar to the siz1Δ siz2Δ double mutant, cells harboring the mms21ΔC allele were completely deficient in break relocation to the NE in both G1- and S-phase cells (Fig. 1C).
To determine which of the two perinuclear anchorage sites, pores or Mps3, was affected by loss of SUMOylation, we used two independent assays. The most rigorous means to score pore association is with a microscopy-based assay that exploits a strain with a small N-terminal deletion in Nup133 that allows Nup49-CFP-tagged pores to form a single cluster in the NE without compromising the pore's transport function (Fig. 1D). Uncut controls and computer simulations of random distributions let us distinguish colocalization with the pore cluster above the level of stochastic coincidence (stochastic colocalization zone in Fig. 1D, shaded gray; see Horigome et al. 2014, 2015). As above, we can also determine cell cycle stage cell by cell. Colocalization with Mps3 is not possible to do by microscopy because the perinuclear rim staining of Mps3 in mitotic cells is weak (Horigome et al. 2014). Mps3 association is instead monitored by chromatin immunoprecipitation (ChIP). Previous studies showed that the association of resected breaks with Mps3 is S-phase-specific even though Mps3 is expressed throughout the cell cycle (Oza et al. 2009; Horigome et al. 2014).
We found that DSBs are enriched at the nuclear pore cluster for 2 h in G1-phase cells and for at least 40 min in S-phase cells (Fig. 1D). While resection through the lacO repeats attenuates the DSB signal in S-phase cells, nuclear pore-ChIP (using a monoclonal antibody against FG repeats) confirmed that DSBs persist at nuclear pores up to 4 h after the cut is induced. In the absence of Siz2, however, the DSB association with nuclear pores is significantly reduced both in G1- and S-phase cells, when monitored by either microscopy or pore ChIP (Fig. 1D,E). Intriguingly, siz2Δ did not alter DSB association with Mps3, suggesting that another signal addresses damage to Mps3 (Fig. 1F).
As shown in Figure 1C, we observed that loss of either the Siz2 or Mms21 SUMO ligase led to a loss of peripheral sequestration. One way to explain this double dependence is that the two ligases act sequentially to generate a polySUMOylation signal at damage, which in turn triggers relocation to the pore. Consistent with this model, it was observed that a number of Siz2 targets retain monoSUMOylation but lose polySUMO chains in the absence of Siz2, suggesting that another enzyme, such as Mms21, might deposit the initial SUMO conjugate (Mullen and Brill 2008; Psakhye and Jentsch 2012; D'Ambrosio and Lavoie 2014; Chung and Zhao 2015). Analogously, sequential action of Mms21 and Siz2 has been proposed to regulate sister chromatid segregation in yeast (Mullen and Brill 2008; D'Ambrosio and Lavoie 2014).
To see whether polySUMOylation is important for relocation, we evaluated DSB position in a mutant strain that cannot form SUMO chains due to mutations in three lysine residues of the N-terminal domain of the SUMO-1 homolog Smt3 (K11, K15, and K19) (Tatham et al. 2001; Bylebyl et al. 2003), compromising almost all polySUMOylation. In this smt3-3KR allele, DSB relocation is compromised in both G1- and S-phase cells (Fig. 1G), consistent with a requirement for SUMO chain formation. Given that this mutant alters all SUMO chain formation in the cell, affecting transcription as well as chromosome segregation, we cannot exclude that there are indirect effects of smt3-3KR. Nonetheless, the observed results are consistent with our hypothesis that Mms21 and Siz2 cooperate to deposit a polySUMOylation signal that might trigger DSB relocation. Intriguingly, smt3-3KR is synthetic lethal with both sgs1Δ and slx5Δ (Mullen et al. 2011).
PolySUMOylation is required for Slx5 recruitment and DSB relocation to pores in G1
Earlier work indicated that Slx5/Slx8 binds the Nup84 complex and showed that its recruitment to DSBs correlates with DSB relocation to pores, although positioning was not shown to depend on Slx5/Slx8 in the case of persistent DSBs (Nagai et al. 2008). Slx5 contains multiple SIMs, which are required for the formation of damage-induced foci of Slx5 in a SUMO-dependent manner (Cook et al. 2009). Moreover, the human homolog RNF4 preferentially binds polySUMO chains. We therefore asked whether Slx5 recruitment to the DSB at MAT is affected by the presence or absence of Siz2 or by the non-chain-forming SUMO mutant (smt3-3KR). Control ChIP for a fully functional HA-tagged Slx5 at MAT after 2 or 4 h of cut induction showed a strong enrichment for Slx5, which dropped to half the wild-type level in a siz2Δ strain, consistent with partial compensation by Siz1 (Fig. 2A). In the smt3-3KR mutant, on the other hand, Slx5 recruitment was completely eliminated (Fig. 2A; Bylebyl et al. 2003). We note that cleavage efficiency ranged from 75% to 90% in all samples, and ChIP data were normalized to cut efficiency at each time point.
Figure 2.

The STUbL Slx5/8 promotes DSB relocation to the nuclear pore. (A) ChIP for HA-tagged Slx5 monitored MAT locus binding of Slx5 at the indicated time after cut induction on galactose in wild-type (JC3020), siz2Δ (JC3668), smt3-3KR (JC3214), and a nontagged strain (JC727). Data from three independent experiments are represented as mean ± SEM. (B) MAT position relative to CFP-Nup49 in wild-type (GA-6844), slx5Δ (GA-7097), and slx8Δ (GA-7098) after 120 min on galactose. Symbols and scoring are as in Figure 1, C and G. (C) Scoring of MAT colocalization with the pore cluster in nup133ΔN (GA-7314) after cut induction as in Figure 1D. Strains scored at the indicated times on galactose were wild type (GA-7314) and slx5Δ (GA-7969). (Pink/red shaded region) Colocalization. (D) ChIP for HA-tagged Mps3 monitored MAT locus association with Mps3 at the indicated times after cut induction in wild-type (GA-8306) and slx5Δ (GA-8539) cells. Data from three independent experiments are represented as mean ± SEM.
We next asked whether Slx5 or Slx8 binding was necessary for DSB relocation to the NE. We monitored the position of the induced DSB at the GFP-lacI-tagged MAT locus in slx5Δ and slx8Δ strains, distinguishing G1-phase from S-phase cells (Fig. 2B). Loss of either subunit completely compromised DSB relocation to the nuclear periphery in G1-phase cells, but the effect was attenuated in S phase. Indeed, in S-phase cells, zone 1 enrichment of the DSB was still significant in slx5Δ and slx8Δ S-phase cells (Fig. 2B). From this, we conclude that the Slx5/Slx8 STUbL is essential for DSB–pore association in G1, while it accounts for only part of the shift in S.
We surmised that the partial effect of the STUbL mutants in S phase could reflect the fact that DSBs can bind Mps3 as well as pores in S phase, while Mps3 is not an option in G1. We therefore examined whether the loss of Slx5/Slx8 selectively compromises pore binding. Indeed, in the quantitative pore colocalization assay, the slx5 deletion strain lost DSB–pore association in both G1- and S-phase cells, while the interaction of the DSB with Mps3, as monitored by ChIP, was unchanged or even slightly increased (Fig. 2C,D). A similar loss of DSB–pore association was scored in slx5Δ and slx8Δ cells by pore-ChIP, with the effect being strongest very close to the DSB (Supplemental Fig. S2a). We conclude that the yeast STUbL is required for DSB recruitment selectively to nuclear pores, which can occur in either G1- or S-phase cells, while the S-phase relocation to Mps3 is Slx5/Slx8-independent. This does not mean that Mps3 interaction is SUMO-independent; indeed, the results shown in Figure 1 argue that it is also largely dependent on some form of SUMOylation.
LexA-polySUMO and LexA-Slx5 can trigger pore binding in the absence of damage and Siz2
While STUbL recruitment and damage movement to pores both correlate with polySUMOylation, it remained unresolved whether polySUMOylation is a sufficient signal for break relocation and whether it acts specifically by recruiting Slx5. Given the pleiotropic effects of the smt3-3KR mutant, we turned to a gain-of-function assay in which we targeted LexA fusion proteins to a GFP-tagged reporter. This allowed us to monitor the sufficiency of Smt3 moieties (SUMO) or specific ligands like Slx5 for relocation activity. We created a LexA-polySUMO fusion protein by stringing four Smt3s lacking the internal diglycine motifs together with LexA (see the Supplemental Material). This fusion was able to bind an undamaged genomic locus (PES4) thanks to a cluster of four LexA-binding sites inserted next to the lacO array (Fig. 3A). If we expressed LexA alone, the PES4 locus distributed randomly in the nucleoplasm (Fig. 3B), as did the locus in the absence of LexA (Taddei et al. 2004). We confirmed by microscopy that there was no change in the abundance of budded versus unbudded cells in cultures expressing LexA or the polySUMO fusion, arguing against any damage or checkpoint induction arising from expression of these constructs. However, the expression of LexA-polySUMO (LexA-4×Smt3) triggered a highly significant relocation of PES4 to the NE in G1-phase but not S-phase cells (Fig. 3B). Importantly, the LexA-polySUMO chain fusion lost its ability to shift to the NE in cells lacking Slx5 (Fig. 3B), arguing that Slx5 acts downstream from SUMO chain formation for DSB relocation (Fig. 3B). In siz2Δ cells, the LexA-polySUMO construct retained at least partial relocation activity, although it was significantly reduced (P = 1.8 × 10−4 in G1 phase) (Fig. 3B). This reduction in the absence of Siz2 may argue that Siz2 itself is part of the relocation machinery or might simply reflect pleiotropic effects arising from loss of the Siz2 E3 ligase (Fig. 3B).
Figure 3.

Targeted polySUMO and Slx5 promote chromatin relocation to the nuclear pore, bypassing Siz2 and Slx8 activities. (A) Scheme of the LexA fusion protein targeting to PES4::lacO-LexA for the zoning assay (GA-1461) and to LYS2::lacO-LexA in a strain bearing nup133ΔN (GA-8194) for nuclear pore colocalization of the intact targeted locus. (B,C) The position of lacO/LexA-tagged PES4 was visualized by GFP-LacI and scored in cells classified as G1 or S phase. Strains carried GFP-Nup49 (GA-1461) and the indicated LexA fusion proteins (polySUMO = 4×Smt3) (see the Supplemental Material) or LexA alone expressed from pAT4 derivatives. LexA fusion was expressed in wild-type (wt) (GA-1461), siz2Δ (GA-4447), slx5Δ (GA-4448), or slx8Δ (GA-4449) strains. Position was scored as in Figure 1C. (D) Pore cluster colocalization for LexA-tagged LYS2 in a strain bearing nup133ΔN (GA-8194) transformed with the indicated LexA fusion. Colocalization (pink to red) is as described in Figure 1D.
To determine whether the binding of Slx5 itself is sufficient to shift PES4 to pores, we expressed a LexA-Slx5 fusion in the same reporter strain. Like LexA-polySUMO, LexA-Slx5 was sufficient to shift its target locus to the NE in G1-phase cells, while LexA alone could not (Fig. 3C). Intriguingly, LexA-Slx5 did not relocate PES4 in S-phase cells, arguing that there may be cell cycle modifications of Slx5 or the Nup84 pore complex to which it binds (Fig. 3C). To see whether Slx5 is the key factor recruited by polySUMO with respect to the relocation event, we examined whether the artificial recruitment of Slx5 is sufficient to trigger relocation in the absence of Siz2. Indeed, LexA-Slx5 is able to shift PES4 to the periphery in the absence of Siz2 (Fig. 3C), arguing that, with respect to subnuclear targeting, an artificial recruitment of Slx5 (Fig. 3B) bypasses the requirement for Siz2. Furthermore, relocation by LexA-Slx5 was independent of an intact Slx8 (Fig. 3C) even though Slx8 is recruited to breaks with efficiency equal to that of Slx5 (Supplemental Fig. S2b). Consistently, a targeted LexA-Slx8 does not shift PES4 position to the NE (Fig. 3C). This result was not due to misfolding of the fusion protein, since the LexA-Slx8 fusion complements the impaired growth of a slx8Δ strain on HU (Supplemental Fig. S3a). We conclude that Slx8-mediated ubiquitination is not the signal for relocation to nuclear pores, although, in the living cell, when persistent DSBs are scored for their positioning, Slx8 contributes to pore association (Fig. 2, Supplemental Fig. S2a). This may reflect a role for Slx8 in promoting the stable binding of Slx5 at DSBs.
This partial divergence in function between Slx5 and Slx8 has precedents. Although Slx5 and Slx8 form a complex and the null alleles share many phenotypes, epistatic miniarray profiling (EMAP) analysis suggests that loss of Slx5 is more deleterious than loss of Slx8 on damaging agents (Nagai et al. 2008; Hustedt et al. 2015). Indeed, slx5Δ populations tend to generate polyploid cells, which is not the case in slx8 mutants, and Slx5 can form repair foci in the absence of Slx8 (Cook et al. 2009).
To determine whether the Slx5/polySUMO chain-mediated positioning at the NE reflected binding at pores or at Mps3, we coupled a LacI-LexA-tagged internal locus (LYS2, chromosome 2) with the nup133ΔN mutant in which pores cluster on one side of the nucleus. This allowed us to monitor the position of the reporter relative to a pore cluster. Upon the targeting of LexA-polySUMO to LYS2, we observed a strong colocalization with the nuclear pore cluster (Fig. 3D). This result is remarkably consistent with our finding that DSB relocation to pores is Slx5-dependent in G1-phase cells and that relocation to Mps3 is not affected by loss of Slx5 (Fig. 2D). Similarly, LexA-Slx5 was sufficient to shift the tagged LYS2 locus to the nuclear pore cluster in G1 only, like LexA-polySUMO (Fig. 3B–D). These sufficiency studies lack the complex signals that arise from checkpoint activation and the recruitment of irrelevant DNA repair factors. They allow us to establish a dependency between Slx5- and Siz2-mediated polySUMOylation for locus recruitment to nuclear pores. Indeed, we bypassed the requirement for Siz2-mediated polySUMOylation by targeting Slx5 to a tagged locus (Fig. 3C). These gain-of-function studies argue that Slx5 binding may be sufficient to shift the break to pores, at least in G1 phase. We hypothesize that S-phase cells incur other modifications that block Slx5's relocation activity or else that additional S-phase constraints impair relocation, provoking a need for factors other than Slx5.
In flies, it was shown that dRad60 (the homolog of Rad60 in fission yeast, Esc2 in budding yeast, and NIP45 in humans) contributes to the shift of DSBs away from heterochromatin in irradiated Drosophila cells (Ryu et al. 2015). This class of factors, called RENi, is thought to stimulate the ubiquitination activity of the Slx5/8 STUbL in yeast (Prudden et al. 2007). Therefore, we tested a complete deletion of the S. cerevisiae RENi homolog Esc2 to see whether it would phenocopy loss of Slx5/Slx8, which blocks relocation primarily in G1. Indeed, we found that esc2Δ cells show a significant drop in relocation of the DSB to the NE only in G1 phase and not in S phase (cf. Fig. 3B and Supplemental Fig. S2b). Unlike Slx5, however, the targeting of a LexA-Esc2 fusion did not trigger a shift to the NE (Supplemental Fig. S3c), arguing that it is not directly involved in pore association. Rather, like Slx8, Esc2 may help stabilize Slx5 binding to polySUMO motifs at damage.
MonoSUMOylation relocates exclusively to Mps3-binding sites in S-phase cells
In Figure 1, we showed that the loss of SUMO E3 ligases impaired break relocation in S phase as well as in G1, unlike slx5Δ, and found strong effects of mms21ΔC, which is thought to mediate primarily monoSUMOylation (D'Ambrosio and Lavoie 2014). Given the inability of LexA-polySUMO to relocate damage in S-phase cells, we next examined whether the fusion of a single Smt3 moiety to LexA might promote the positioning of an undamaged site in S phase. We expressed two monoSUMO LexA fusions (i.e., to either a single Smt3 moiety or a smt3-3KRmoiety) and monitored the position of PES4 locus tagged with lacO-LexA sites. In both cases, the randomly distributed undamaged locus shifted to zone 1 upon expression of the LexA-monoSUMO construct only in S-phase and not in G1-phase cells (Fig. 4A). The LexA-smt3-3KR fusion was also expressed in a siz2Δ strain, and there was a slightly reduced but still significant Siz2-independent relocation activity (Fig. 4A). We next determined whether the relocated locus showed enrichment at the nuclear pore cluster using the LYS2::lacO-LexA strain bearing nup133ΔN and Nup49-CFP. In contrast to the LexA-polySUMO construct, the LexA-smt3-3KR (monoSUMO) fusion did not colocalize with the pore cluster (Fig. 4B), suggesting that the relocated locus binds Mps3. To address this hypothesis, we overexpressed the soluble Mps3 N-terminal domain, which can displace other nucleoplasmic ligands from interaction with membrane-bound Mps3-binding sites (Ferreira et al. 2011). The soluble Mps3N′ domain indeed successfully competed for LexA-smt3-3KR's relocation activity (Fig. 4C). Consistent with the fact that Mps3 functions as a tethering site only in S phase, the monoSUMO fusion can only mediate relocation in S-phase cells. Taken together, our data argue that, in S-phase cells, monoSUMOylation events mediate DSB relocation to Mps3, whereas, in G1, relocation to the nuclear pore requires a polySUMO chain that acts through the recruitment of Slx5. Given that LexA-Slx5 can bypass the requirement for Siz2, we suggest that Slx5 does not need to be SUMOylated to interact with Nup84 (see the model in Fig. 4D), although, in S-phase, there may be other factors at play. Nonetheless, we note that not all Slx5 foci that arise from ionizing radiation are associated with nuclear pores (Supplemental Fig. S4); thus, there may be ways to impair Slx5–pore association in S phase. Alternatively, Slx5–pore binding may be transient in nature, allowing some foci to shift away from the NE after processing of the damage.
Figure 4.

The targeted monoSUMO construct shifts chromatin to the nuclear periphery in S phase but not to nuclear pores. (A) The indicated LexA fusion proteins are expressed in the wild-type (wt) strain (GA-1461) and siz2Δ (GA-4447). The position of lacO/LexA-tagged ARS607 was visualized by GFP-LacI and scored as in Figure 1C. (B) Pore cluster colocalization for LexA-tagged LYS2 in a strain bearing nup133ΔN (GA-8194) transformed with the indicated LexA fusions. Colocalization (pink to red) was scored as in Figure 1D. (C) In a wild-type strain (GA-1461) expressing LexA-smt3-3KR and either an empty vector or the Mps3N′ construct, PES4 position was scored as in A. (D) Summary of the chromatin positioning roles of monoSUMO and polySUMO chains based on the targeting assays.
Slx5 binding to Nse5, part of the SMC5/6 complex, is compromised by the L247A mutation
In addition to being a target of SUMO E3 ligases, and binding SUMO chains, Slx5 and its orthologs have been shown to interact with a component of the SMC5/6 complex (i.e., Nse5), with factors involved in kinetochore function (Ndc10), and with the repair factor family RENi (Rad60, Esc2, and Nip45) (summarized in Cook et al. 2009). The SMC5/6 complex contains the Nse2/Mms21 E3 SUMO ligase as well as Nse1, an E3 ubiquitin ligase, and the Nse3–6 subunits (Supplemental Fig. S5a; Jeppsson et al. 2014). High-throughput yeast two-hybrid (Y2H) screens suggested that budding yeast Nse5 might bind Slx5 (Hazbun et al. 2003), like its Schizosaccharomyces pombe homolog (Prudden et al. 2007). Nse5 contributes to SMC5/6 complex stability and its recruitment to sites of replication stress by binding Nse6 (Cook et al. 2009; Bustard et al. 2012), but Nse5 also binds directly to Smt3 and indirectly to E3 ligases Siz1, Siz2, and Mms21 and the upstream E2 ligase Ubc9 (DE Bustard and JA Cobb, pers. comm.). To see whether Nse5 contributes to the recruitment of Slx5/Slx8, we first characterized the proposed protein–protein interaction between Nse5 and Slx5 by Y2H assays.
With galactose-inducible expression of LexA-Nse5 as the bait and of a Slx5 activation domain (AD) fusion as prey, we scored a >103-fold increase in β-galactosidase activity in cells exposed to galactose. This reflects an efficient interaction between Slx5 and Nse5 (Supplemental Fig. S5b). Given that both Slx5 and Nse5 are reported to interact with SUMO (Hazbun et al. 2003), we asked whether the Y2H interaction might be compromised in strains that fail to efficiently deposit SUMO chains (i.e., either smt3-3KR, which blocks SUMO chain formation, or smt3-331, which reduces SUMOylation efficiency). The Slx5–Nse5 binding was reduced, yet there was still interaction that was 350-fold above background (Supplemental Fig. S5b), suggesting that Nse5 and Slx5 may interact independently of SUMO. Consistently, a mutant form of Slx5 in which the two demonstrated SIM elements A and B were mutated (alanine substitutions at amino acids 25–27 and amino acids 93–96) (Xie et al. 2007) did not significantly reduce Nse5–Slx5 interaction (Supplemental Fig. S5b). In all cases, both constructs were efficiently expressed on galactose (Supplemental Fig. S6).
In earlier work, SMC5/6 was shown to facilitate the relocation of DSBs away from the nucleolus (Torres-Rosell et al. 2007) and away from heterochromatin in Drosophila cells (Chiolo et al. 2011), yet it was unclear whether it acts solely by delivering the SUMO E3 ligase Mms21 or whether it stabilizes the recruitment or binding of the STUbL complex. In order to be able to test the relevance of the Nse5–Slx5 interaction in relocation assays, we screened previously isolated point mutations in Nse5 for one that would lose interaction with Slx5 without disrupting the stability of SMC5/6 and the SUMO ligase activity of Mms21. The nse5 L247A mutant protein completely abrogated the Nse5–Smt3 interaction (Supplemental Fig. S5c) but retained wild-type levels of binding to Nse6 (Supplemental Fig. S5c) and, unlike the mms21-11 mutant, did not induce MMS sensitivity (DE Bustard and JA Cobb, pers. comm.). It did, however, abolish Nse5's interaction with Slx5 as monitored by Y2H (Supplemental Fig. S5c). While this may be partly through a loss of SUMO recognition, the L247A allele appeared to be a useful tool to perturb the Slx5–SMC5/6 complex interaction without interfering with SMC5/6 complex integrity or its SUMOylation activity. In contrast, the temperature-sensitive smc6-9 allele both disrupts the SMC5/6 complex (Torres-Rosell et al. 2005; De Piccoli et al. 2006; Sollier et al. 2009) and compromises its E3 ligase function (Bermudez-Lopez et al. 2015). A summary of the relevant interactions disrupted by Nse5 L247A is shown in Figure 5A.
Figure 5.

The SMC5/6 complex, the recruiter Rtt107, and the efficient interaction of Nse5 with Slx5/Slx8 facilitate DSB anchoring to Mps3. (A) Depiction of Nse5 interactions with SUMO (Smt3), Slx5, and Nse6 and the effects of the nse5L247A mutation (Supplemental Fig. S5; see the text). (B) The position of the cleaved MAT locus relative to CFP-Nup49 in wild-type (GA-6844), nse5-L247A (JC3161), smc6-9 (JC3131), and rtt107Δ (GA-7092) cells after 120 min on galactose at 30°C is shown. Binning into G1 and S as well as the symbols are as in Figure 1C. (C) ChIP for Nup84-13MYC monitored the MAT locus after 240 min on galactose in wild-type (GA-4133), nse5-L247A (JC3154), and smc6-9 (JC3150) cells grown at 30°C. Data from four experiments are represented as mean ± SEM. PCR probes were at the indicated distances from the HO cut site. (D) ChIP against 3HA-Mps3 (anti-HA) at the indicated times on galactose. Enrichment of MAT (0.6 kb from the cut site) over uncut SMC2 was quantified by qPCR in wild type (JC3167) and nse5-L247A (JC3114). For the smc6-9 temperature-sensitive alleles (JC3115), strains were grown at 25°C and then transferred for 1 h to 37°C to inactivate the smc6-9 allele before HO induction at 35°C. Data from three independent experiments are represented as mean ± SEM. (E) ChIP for HA-tagged Slx5 monitored MAT locus association at the indicated time after cut induction in wild-type (JC3020), nse5-L247A (JC3621), smc6-9 (JC3198), and nontagged (JC727) cells at 30°C. Data from three independent experiments are represented as mean ± SEM.
Using the temperature-sensitive smc6-9 allele, we first asked whether the disruption of SMC5/6 would affect DSB relocation to the NE. Not unexpectedly, like the E3 ligase-deficient allele (mms21ΔC) (Fig. 1C), the smc6-9 mutant lost perinuclear anchorage in both G1- and S-phase cells (Fig. 5B). SMC5/6 is known to be recruited to DSBs by Rtt107 when phosphorylated by the DNA damage checkpoint kinase Mec1 (De Piccoli et al. 2006; Ohouo et al. 2010; Leung et al. 2011; Ullal et al. 2011). Consistently, the rtt107Δ strain also compromised relocation of DSBs to the NE in both stages of the cell cycle (Fig. 5B). In contrast to the effects of smc6-9 and mms21ΔC mutants, break relocation to the NE in the nse5L247A allele has a reduced but still significant perinuclear association in G1 (Fig. 5B). This may reflect its ability to sustain functional Mms21 activity in G1-phase cells, which neither smc6-9 nor rtt107Δ does.
This model predicts that the smc6-9 mutation might affect recruitment to both pores and Mps3, phenocopying the loss of Mms21's SUMOylation activity, while the nse5L247A allele might only impair S-phase recruitment to Mps3, since the Mms21 SUMOylation activity, necessary for the polySUMO signal in G1, would be intact. We tested this with both pore-ChIP and Mps3-ChIP assays and, whenever possible, elevated the temperature to 37°C to induce the smc6-9 defect (Fig. 5C,D). Indeed, the nse5 L247A protein allowed DSB association with Nup84 (see the 1.6-kb probe in Fig. 5C), while smc6-9 did not (both probes in Fig. 5C). Thus, the Nse5 interaction with Slx5 may not be essential for G1-phase relocation to pores. In the case of Mps3, both mutants showed a similar reduction to levels less than twofold above background (Fig. 5D). We asked whether the nse5L247A allele actually reduces Slx5 binding at the break by performing HA-Slx5 ChIP in the nse5L247A and smc6-9 strains (Fig. 5E). Slx5-HA recruitment was indeed compromised by nse5L247A at the DSB, suggesting that Nse5 at least partially stabilizes Slx5 binding, probably acting in S-phase and not in G1-phase cells, since relocation to the pore was intact in the nse5L247A mutant. We were unable to test the smc6-9 allele combined with Slx5-HA at 37°C due to poor growth of this strain on galactose at this temperature. However, at a semipermissive 30°C, we could score a partial reduction in Slx5 recruitment in the smc6-9 strain, consistent with a loss of Mms21-mediated SUMOylation.
In addition to its effect on Slx5/Slx8 recruitment, we propose that the SMC5/6 complex plays a Slx5/8-independent role in relocation of breaks to Mps3, acting at least in part through Mms21. The nse5L247A mutant protein may fail to bind an S-phase ligand necessary for efficient relocation to Mps3. This ligand most likely is not Slx5, since LexA-Slx5 was unable to shift PES4 to either nuclear pores or Mps3 in S-phase cells (Fig. 3C).
Our results establish a hierarchy of molecular interactions that mediate the relocation of DSBs to distinct perinuclear sites where the relative importance of these pathways varies with the cell cycle. We found that polySUMOylation has a specific role in G1, acting at least in part through Slx5, while monoSUMOylation functions in S phase, acting through an unknown ligand. It is not clear what the relocation-relevant targets of SUMOylation are, although it seems likely that multiple targets could function in this case given that LexA-polySUMO had a fairly efficient relocation activity. Given the conservation of SUMOylation across species, the conservation of STUbL enzymes, and the fact that SMC5/6 and the recruitment factors responsible (Rtt107/Esc4 and Esc2/Rad60) are important for break relocation in flies and yeast, we suggest that SUMO conjugation and STUbL recruitment will be relevant positioning signals in other species.
Slx8 and pore proteins facilitate BIR and imprecise end-joining
To determine whether the functional hierarchy of factors for break relocation translates into a repair function, we scored for cell survival after 1, 2, or 4 h of growth on galactose, during which the HO endonuclease would be induced. After this incubation, cells were plated on glucose-containing plates to score for the efficiency of repair through the number of colony-forming units (CFUs) recovered (viable cells in Fig. 6A). We first asked whether slx5Δ and nse5L247A would be epistatic, and, indeed, while the loss of Slx5 compromises cell survival far more than the other single mutants, there was no additive loss of viability when combined with nse5L247A (Fig. 6A, light gray). This was not the case when siz2Δ, which reduced survival only moderately at 2 h, was combined with slx5Δ; in this case, the double mutant was strongly additive (Fig. 6A, dark gray). This is consistent with the notion that Siz2 has additional roles at damage, promoting survival by factors other than those that recruit and activate Slx5/Slx8. Nonetheless, Slx5 has a profound effect on survival following induction of a persistent DSB.
Figure 6.

Slx8 and pore proteins favor repair by BIR and imprecise end-joining. (A) Cell survival after HO cut induction. Cleavage at the MAT locus by the HO endonuclease was induced by galactose for the indicated times. Cells were washed and plated on YPAD plates, and CFUs were scored after 2 d at 30°C. The rates of the viable cells in wild-type (GA-6844), siz2Δ (GA-6858), nse5-L247A (JC3161), slx5Δ (GA-7097), siz2Δ slx5Δ (GA-9206), and nse5-L247A slx5Δ (GA-9355) strains were normalized to cell count before cut induction. (B) BIR assay. A recipient cassette composed of a 3′ truncated LYS2 gene (ly), a 36-base-pair cut site for the HO endonuclease, and a kanR marker was incorporated into chromosome V (ChrV). A donor cassette composed of a TRP1 marker and a 5′ truncation of LYS2 (ys2) was inserted 60 kb from the telomere of chromosome I (ChrI). The two mutant lys2 fragments share 2.1 kb of homology. The HO endonuclease was expressed under the control of a galactose promoter. Lys+ cells lacking kanamycin resistance were scored for wild-type (GA-8994), nup60Δ (GA-9185), slx8Δ (GA-9186), and pol32Δ (GA-9090) cells. Error bars indicate standard deviation. Significance was determined by Student's t-test. (C) Precise NHEJ and imprecise NHEJ were performed and analyzed as described in the Materials and Methods on GA-8860 (wild type) and GA-8471 (nup84Δ).
There were multiple pathways of repair that led to CFUs in the survival assay scored above. To get insight into which repair pathways Slx5 and pore binding affects, we performed an assay designed to monitor repair of a DSB by strand invasion into an ectopic donor (BIR). We scored for the loss of the distal arm of the invaded chromosome to ensure that we monitored BIR rather than another reciprocal recombination event. A recent study observed an ∼50% drop in efficiency of survival in both the nup84Δ and slx5Δ strains after induction of a DSB in a subtelomeric region (Chung et al. 2015). Survival was correlated with a recombination-based pathway that required Rad52 and Rad32, although it was not a specific assay for ectopic BIR, in which a one-ended DSB is repaired by an error-free mechanism involving recombination-mediated replication fork restart. Since Nup84 had already been tested in a BIR-like assay, we tested another pore component that is associated with the Nup84 complex, Nup60. Nup60 is recruited to breaks and shows similar EMAP sensitivities (Nagai et al. 2008). Monitoring a strand invasion event from chromosome V to chromosome I (Fig. 6B), we observed that slx8Δ and nup60Δ strains have a 60% and 40% drop in BIR efficiency, respectively (Fig. 6B). We propose that an irreparable or persistent DSB resembles a collapsed fork in that it has a damaged end with no immediate donor for repair (no intact sister chromatid).
The other potential pathway for repair of such lesions would be either precise NHEJ or imprecise NHEJ. Compromising the Nup84 complex did not compromise precise NHEJ at an HO-induced break to any detectable extent (Fig. 6C), but imprecise end-joining (also called alternative or microhomology-mediated end-joining) was reduced by 50%, like BIR (Fig. 6C). Both BIR and imprecise end-joining involve resection at the cut site; thus, one scenario that explains our results is that recruitment of DSBs without intact sisters to nuclear pores by Slx5/Slx8 triggers the degradation of a complex that might be blocking end resection. Resection would then allow alternative strand invasion or annealing pathways to proceed in a last-ditch attempt by the cell to force repair and survive. Intriguingly, sequestration at Mps3 appears to have the opposite effect: Mps3 suppresses ectopic recombination and thus may counteract the repair pathway promoted at nuclear pores (Ferreira et al. 2011; Horigome et al. 2014; Chung et al. 2015).
Discussion
We identified two distinct SUMO-dependent pathways that trigger relocation of a DSB to the NE (Fig. 7). In G1, SUMO chain formation leads to recruitment of Slx5 and subsequent break relocation to the nuclear pore. G1 polySUMOylation depends on Siz2 and Mms21. Loss of either SUMO E3 ligase compromises relocation of DSBs to pores. In S phase, on the other hand, monoSUMOylation by Mms21 leads to DSB movement to Mps3 but not to the nuclear pore. Interestingly, Slx5 contributes to, but is not sufficient for, S-phase movement of breaks to pores and is stabilized at damage in an Nse5-dependent manner. Thus, our data also suggest that the SMC5/6 complex contributes in several ways to break relocation; in both G1 and S phase, it recruits Mms21 to mediate monoSUMOylation, but its Nse5 subunit may have additional S-phase-specific roles.
Figure 7.
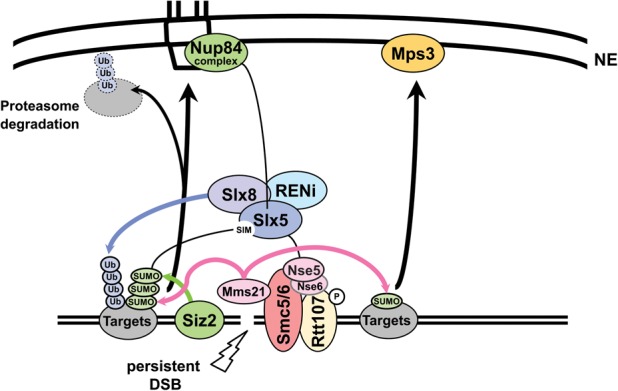
Model for the role of SUMO and the Slx5 STUbL subunit in break relocation. Siz2 is damage-associated and deposits SUMO on various repair substrates that are monoSUMOylated by Mms21. Slx5/Slx8 is itself SUMOylated and recruited to the DSB in a polySUMOylation-dependent manner. Siz2, polySUMOylation, and its recognition by Slx5/Slx8 are required for DSB relocation to pores in both G1 and S. Nup84 components are recognized by Slx5. In contrast, monoSUMOylation can shift chromatin to Mps3 in an SMC5/6-dependent but Slx5/8-independent manner. Rtt107 phosphorylation by Mec1 recruits the SMC5/6 complex to DSBs. A component of SMC5/6, Nse5, interacts with Slx5. The SUMO E3 ligase Mms21 is recruited with the SMC5/6 complex. Ubiquitination of SUMOylated target proteins by the Slx5/Slx8 STUbL most likely results in subsequent degradation by the proteasome at the NE to enable repair pathways such as BIR.
Our findings allow a generalization of the phenomena to damage relocation from heterochromatin in Drosophila cells (Chiolo et al. 2011). Chiolo et al. (2011) similarly found that ablation of the fly Slx5/Slx8 homologs SMC5/6 and impaired SUMOylation interfered with the relocation of DSBs from heterochromatin to perinuclear sites of repair (Ryu et al. 2015). It has also been reported elsewhere that the repair of intrastrand cross-links by the Fanconi anemia complex and PML body-mediated recovery from arsenic-induced damage (Tatham et al. 2008; Kim and D'Andrea 2012) are controlled by SUMOylation, STUbL enzymes, and SMC5/6. Finally, Mms21 in parallel with Slx5/8 and SMC5/6 was shown to suppress spontaneous gross chromosomal rearrangements, although the mechanism was not clarified (Hwang et al. 2008; Albuquerque et al. 2013). Our study suggests that damage can be directed to subcompartments at the NE by distinct signals, where distinct repair activities seem to be favored. From genetic data, one would argue that the proteasome acts on the same pathway as nuclear pores and Slx5/Slx8 (Nagai et al. 2008), possibly requiring Slx8-mediated ubiquitination. Above all, we establish here for the first time a strong correlation between the spatial sequestration of damage and the extent of SUMO chain formation.
We note that ubiquitination and SMC5/6 are also necessary for telomere maintenance and PML body formation in ALT cancer cells (Potts and Yu 2007), where BIR is the primary mechanism of telomere maintenance. Consistently, Slx8 promotes type II survivor formation in yeast mutants that lack functional telomerase (Azam et al. 2006). Also, in yeast, eroded telomeres relocate to nuclear pores (V Geli, pers. comm.). These repair events all entail ectopic strand invasion, as occurs in BIR. Thus, it is tempting to speculate that a processing event that enables nonsister chromatid strand invasion is favored by shifting breaks to nuclear pores.
With respect to relocation to pores in G1-phase cells, in a gain-of-function targeting assay, we found that LexA-Slx5 can bypass the requirement for polySUMOylation. Moreover, the relocation mediated by LexA-4×Smt3 (polySUMO) fusion is dependent on Slx5 (Fig. 3). This is consistent with the notion that any number of appropriately modified targets might suffice to recruit Slx5. Indeed, it may be the extent of modification rather than the specific substrate that is critical for site-specific recruitment. Still, the question remains: Which targets of SUMOylation are relevant in this context? Do they vary with cell cycle or types of damage?
There are a number of DSB-bound factors that are targets and ligands of either Mms21, Siz2, or both. Siz2 binding at sites of damage appears to be facilitated by RPA binding to ssDNA, mediated by the Rfa2 C-terminal winged helix domain (Chung and Zhao 2015). We did not find a requirement for Rad52 in DSB relocation to nuclear pores (Horigome et al. 2014), although Rad52-KR mutants that attenuate its SUMOylation affect damage relocation out of the yeast nucleolus (Torres-Rosell et al. 2007). Similarly, whereas yKu70-SUMOylation and yKu80-SUMOylation mediate the interaction of telomeres with Mps3, these constructs do not target DNA to Nup84 (Ferreira et al. 2011; Chung et al. 2015). Htz1 SUMOylation was implicated in DSB relocation to Mps3 (Kalocsay et al. 2009), but, given that Htz1 is a necessary chaperone for the localization of Mps3 in the inner nuclear membrane (Gardner et al. 2011), these data must be complemented with controls that show that Mps3 was properly localized in these mutants (Gardner et al. 2011). The fact that Htz1 deletion affects DSB relocation to both pores and Mps3 (Dion and Gasser 2013; Horigome et al. 2014) may argue for a role of this variant histone in break relocation. Future studies will have to address this by mapping and mutating specific interaction sites between Htz1 and Mps3. Whatever the target may be, our work illustrates the crucial importance of SUMOylation signals at DSBs for the targeting of damage to distinct nuclear subcompartments that mediate repair.
Materials and methods
Plasmids, yeast strains, and techniques
Standard yeast methods and media were used, and constructs, strains, growth conditions, and Y2H assays are described in detail in the Supplemental Material and Supplemental Table S2. Cut efficiencies are summarized in Supplemental Table S1.
The LexA fusions were created in the plasmid pAT4 (Taddei et al. 2004). The 4×Smt3 construct (LexA-polySUMO) was created by cloning four Smt3 genes tandemly into the vector pAT4 using a 4×Smt3 construct synthesized by Bio Basic, Inc., gene synthesis. To prevent processing of the internal SUMO proteins by Ulp1, the constructs were truncated at I96 to remove the diglycine motif. The fourth SUMO residue in the chain retained the full SUMO sequence, including the diglycine motif. Targeted binding is described in Neumann et al. (2012) and Horigome et al. (2014).
Microscopy and statistical analyses
Fluorescence microscopy and quantification were performed according to published methods (Meister et al. 2010; Horigome et al. 2015) using a Metamorph-driven spinning disk confocal system with a Yokogawa CSU-X1 scan head and an EM-CCD Cascade II (Photometrics) camera. LacI-GFP position was determined with a through-focus stack of 16–21 0.2-μm steps and was measured by ImageJ (National Institutes of Health) and the plug-in software PointPicker (Meister et al. 2010). The numbers of nuclei scored are in Supplemental Table S1.
To determine zone enrichment, we applied a χ2 test comparing zone 1 with a random distribution (degree of freedom, 2; confidence limit, 95%). P-values are listed in Supplemental Table S1. To compare the perinuclear enrichment of two different strains, we used a proportional analysis with a confidence limit of 95%. The error bars of ChIP experiments represent the SEM.
ChIP
ChIP with anti-HA or anti-MYC epitope-tagged strains was carried out as previously described (Yoshida et al. 2010) with modifications described in the Supplemental Material. Absolute enrichment was calculated as follows: For each time point, the signal from a site near the HO DSB at MAT was normalized to that from either the genomic SMC2 locus or the mitochondrial OLI1 locus in ChIP and input DNA samples. For each time point and site, normalized ChIP signals were normalized to input DNA signals.
Repair assays
Precise NHEJ and imprecise NHEJ were performed as described in van Attikum et al. (2007) using isogenic derivatives of GA-1081 (i.e., GA-8860 [wild type] and GA-8471 [nup84Δ]). The DSB survival assay is described in the Supplemental Material; the BIR assay was performed and the frequency of recombinants was calculated as previously described (Donnianni and Symington 2013).
Supplementary Material
Acknowledgments
We thank Dr. S. Nagai (Stanford University, CA) for data in Supplemental Figure S2, acquired while in the Gasser laboratory. We thank J.E. Haber for yeast strains, and the Friedrich Miescher Institute Facility for Advanced Imaging and Microscopy for technical help. C.H. was supported by a Marie Curie International Incoming Fellowship and a Japan Society for the Promotion of Science Post-doctoral Fellowship for Research Abroad. The Gasser laboratory thanks the Novartis Research Foundation, the Swiss National Science Foundation, and the Swiss Cancer League for grants awarded to S.M.G. and a student fellowship awarded to I.M. D.E.B. was supported by a graduate studentship from Alberta Innovates Health Solutions. The Cobb laboratory was supported by operating grants from Canadian Institutes of Health Research (MOP-82736) and Natural Sciences and Engineering Research Council (418122) awarded to J.A.C. C.H. performed experiments, interpreted results, and prepared figures; D.E.B., I.M., M.T.-P., and N.D. performed experiments, interpreted results, and prepared figures; J.A.C. performed experiments and supervised D.E.B.; S.M.G. interpreted results and supervised C.H., I.M., M.T.-P., and N.D.; and all contributed to writing the paper.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.277665.116.
References
- Agmon N, Liefshitz B, Zimmer C, Fabre E, Kupiec M. 2013. Effect of nuclear architecture on the efficiency of double-strand break repair. Nat Cell Biol 15: 694–699. [DOI] [PubMed] [Google Scholar]
- Albuquerque CP, Wang G, Lee NS, Kolodner RD, Putnam CD, Zhou H. 2013. Distinct SUMO ligases cooperate with Esc2 and Slx5 to suppress duplication-mediated genome rearrangements. PLoS Genet 9: e1003670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam M, Lee JY, Abraham V, Chanoux R, Schoenly KA, Johnson FB. 2006. Evidence that the S. cerevisiae Sgs1 protein facilitates recombinational repair of telomeres during senescence. Nucleic Acids Res 34: 506–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CB, Lewis LK, Karthikeyan G, Lobachev KS, Jin YH, Sterling JF, Snipe JR, Resnick MA. 2001. Genes required for ionizing radiation resistance in yeast. Nat Genet 29: 426–434. [DOI] [PubMed] [Google Scholar]
- Bermudez-Lopez M, Pocino-Merino I, Sánchez H, Bueno A, Guasch C, Almedawar S, Bru-Virgili S, Garí E, Wyman C, Reverter D, et al. 2015. ATPase-dependent control of the Mms21 SUMO ligase during DNA repair. PLoS Biol 13: e1002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukata L, Parker SL, D'Angelo MA. 2013. Nuclear pore complexes in the maintenance of genome integrity. Curr Opin Cell Biol 25: 378–386. [DOI] [PubMed] [Google Scholar]
- Bustard DE, Menolfi D, Jeppsson K, Ball LG, Dewey SC, Shirahige K, Sjogren C, Branzei D, Cobb JA. 2012. During replication stress, non-SMC element 5 (NSE5) is required for Smc5/6 protein complex functionality at stalled forks. J Biol Chem 287: 11374–11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bylebyl GR, Belichenko I, Johnson ES. 2003. The SUMO isopeptidase Ulp2 prevents accumulation of SUMO chains in yeast. J Biol Chem 278: 44113–44120. [DOI] [PubMed] [Google Scholar]
- Bystricky K, Van Attikum H, Montiel MD, Dion V, Gehlen L, Gasser SM. 2009. Regulation of nuclear positioning and dynamics of the silent mating type loci by the yeast Ku70/Ku80 complex. Mol Cell Biol 29: 835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Rondinelli B, D'Andrea AD. 2016. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol 26: 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, Bellaoui M, Boone C, Brown GW. 2002. A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc Natl Acad Sci 99: 16934–16939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiolo I, Minoda A, Colmenares SU, Polyzos A, Costes SV, Karpen GH. 2011. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 144: 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung I, Zhao X. 2015. DNA break-induced sumoylation is enabled by collaboration between a SUMO ligase and the ssDNA-binding complex RPA. Genes Dev 29: 1593–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung DK, Chan JN, Strecker J, Zhang W, Ebrahimi-Ardebili S, Lu T, Abraham KJ, Durocher D, Mekhail K. 2015. Perinuclear tethers license telomeric DSBs for a broad kinesin- and NPC-dependent DNA repair process. Nat Commun 6: 7742. [DOI] [PubMed] [Google Scholar]
- Cook CE, Hochstrasser M, Kerscher O. 2009. The SUMO-targeted ubiquitin ligase subunit Slx5 resides in nuclear foci and at sites of DNA breaks. Cell Cycle 8: 1080–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona CA, Sarangi P, Yang Y, Hang LE, Rahman S, Zhao X. 2012. Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the mec1 checkpoint. Mol Cell 45: 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrosio LM, Lavoie BD. 2014. Pds5 prevents the polySUMO-dependent separation of sister chromatids. Curr Biol 24: 361–371. [DOI] [PubMed] [Google Scholar]
- De Piccoli G, Cortes-Ledesma F, Ira G, Torres-Rosell J, Uhle S, Farmer S, Hwang JY, Machin F, Ceschia A, McAleenan A, et al. 2006. Smc5-Smc6 mediate DNA double-strand-break repair by promoting sister-chromatid recombination. Nat Cell Biol 8: 1032–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion V, Gasser SM. 2013. Chromatin movement in the maintenance of genome stability. Cell 152: 1355–1364. [DOI] [PubMed] [Google Scholar]
- Dion V, Kalck V, Seeber A, Schleker T, Gasser SM. 2013. Cohesin and the nucleolus constrain the mobility of spontaneous repair foci. EMBO Rep 14: 984–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnianni RA, Symington LS. 2013. Break-induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci 110: 13475–13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira HC, Luke B, Schober H, Kalck V, Lingner J, Gasser SM. 2011. The PIAS homologue Siz2 regulates perinuclear telomere position and telomerase activity in budding yeast. Nat Cell Biol 13: 867–874. [DOI] [PubMed] [Google Scholar]
- Gardner JM, Smoyer CJ, Stensrud ES, Alexander R, Gogol M, Wiegraebe W, Jaspersen SL. 2011. Targeting of the SUN protein Mps3 to the inner nuclear membrane by the histone variant H2A.Z. J Cell Biol 193: 489–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geli V, Lisby M. 2015. Recombinational DNA repair is regulated by compartmentalization of DNA lesions at the nuclear pore complex. Bioessays 37: 1287–1292. [DOI] [PubMed] [Google Scholar]
- Hazbun TR, Malmstrom L, Anderson S, Graczyk BJ, Fox B, Riffle M, Sundin BA, Aranda JD, McDonald WH, Chiu CH, et al. 2003. Assigning function to yeast proteins by integration of technologies. Mol Cell 12: 1353–1365. [DOI] [PubMed] [Google Scholar]
- Horigome C, Oma Y, Konishi T, Schmid R, Marcomini I, Hauer MH, Dion V, Harata M, Gasser SM. 2014. SWR1 and INO80 chromatin remodelers contribute to DNA double-strand break perinuclear anchorage site choice. Mol Cell 55: 626–639. [DOI] [PubMed] [Google Scholar]
- Horigome C, Dion V, Seeber A, Gehlen LR, Gasser SM. 2015. Visualizing the spatiotemporal dynamics of DNA damage in budding yeast. Methods Mol Biol 1292: 77–96. [DOI] [PubMed] [Google Scholar]
- Hustedt N, Seeber A, Sack R, Tsai-Pflugfelder M, Bhullar B, Vlaming H, van Leeuwen F, Guenole A, van Attikum H, Srivas R, et al. 2015. Yeast PP4 interacts with ATR homolog Ddc2–Mec1 and regulates checkpoint signaling. Mol Cell 57: 273–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JY, Smith S, Ceschia A, Torres-Rosell J, Aragon L, Myung K. 2008. Smc5–Smc6 complex suppresses gross chromosomal rearrangements mediated by break-induced replications. DNA Repair 7: 1426–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppsson K, Kanno T, Shirahige K, Sjogren C. 2014. The maintenance of chromosome structure: positioning and functioning of SMC complexes. Nat Rev Mol Cell Biol 15: 601–614. [DOI] [PubMed] [Google Scholar]
- Kalocsay M, Hiller NJ, Jentsch S. 2009. Chromosome-wide Rad51 spreading and SUMO-H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol Cell 33: 335–343. [DOI] [PubMed] [Google Scholar]
- Khadaroo B, Teixeira MT, Luciano P, Eckert-Boulet N, Germann SM, Simon MN, Gallina I, Abdallah P, Gilson E, Geli V, et al. 2009. The DNA damage response at eroded telomeres and tethering to the nuclear pore complex. Nat Cell Biol 11: 980–987. [DOI] [PubMed] [Google Scholar]
- Kim H, D'Andrea AD. 2012. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev 26: 1393–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. 1998. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94: 399–409. [DOI] [PubMed] [Google Scholar]
- Lemaître C, Grabarz A, Tsouroula K, Andronov L, Furst A, Pankotai T, Heyer V, Rogier M, Attwood KM, Kessler P, et al. 2014. Nuclear position dictates DNA repair pathway choice. Genes Dev 28: 2450–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung GP, Lee L, Schmidt TI, Shirahige K, Kobor MS. 2011. Rtt107 is required for recruitment of the SMC5/6 complex to DNA double strand breaks. J Biolo Chem 286: 26250–26257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeillet S, Palancade B, Cartron M, Thierry A, Richard GF, Dujon B, Doye V, Nicolas A. 2005. Genetic network interactions among replication, repair and nuclear pore deficiencies in yeast. DNA Repair 4: 459–468. [DOI] [PubMed] [Google Scholar]
- Meister P, Gehlen LR, Varela E, Kalck V, Gasser SM. 2010. Visualizing yeast chromosomes and nuclear architecture. Methods Enzymol 470: 535–567. [DOI] [PubMed] [Google Scholar]
- Mullen JR, Brill SJ. 2008. Activation of the Slx5–Slx8 ubiquitin ligase by poly-small ubiquitin-like modifier conjugates. J Biol Chem 283: 19912–19921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen JR, Das M, Brill SJ. 2011. Genetic evidence that polysumoylation bypasses the need for a SUMO-targeted Ub ligase. Genetics 187: 73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai S, Dubrana K, Tsai-Pflugfelder M, Davidson MB, Roberts TM, Brown GW, Varela E, Hediger F, Gasser SM, Krogan NJ. 2008. Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science (New York, NY) 322: 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai S, Heun P, Gasser SM. 2010. Roles for nuclear organization in the maintenance of genome stability. Epigenomics 2: 289–305. [DOI] [PubMed] [Google Scholar]
- Neumann FR, Dion V, Gehlen LR, Tsai-Pflugfelder M, Schmid R, Taddei A, Gasser SM. 2012. Targeted INO80 enhances subnuclear chromatin movement and ectopic homologous recombination. Genes Dev 26: 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohouo PY, Bastos de Oliveira FM, Almeida BS, Smolka MB. 2010. DNA damage signaling recruits the Rtt107–Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol Cell 39: 300–306. [DOI] [PubMed] [Google Scholar]
- Oza P, Jaspersen SL, Miele A, Dekker J, Peterson CL. 2009. Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev 23: 912–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palancade B, Liu X, Garcia-Rubio M, Aguilera A, Zhao X, Doye V. 2007. Nucleoporins prevent DNA damage accumulation by modulating Ulp1-dependent sumoylation processes. Mol Biol Cell 18: 2912–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, et al. 2009. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 35: 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts PR, Yu H. 2007. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat Struct Mol Biol 14: 581–590. [DOI] [PubMed] [Google Scholar]
- Prudden J, Pebernard S, Raffa G, Slavin DA, Perry JJ, Tainer JA, McGowan CH, Boddy MN. 2007. SUMO-targeted ubiquitin ligases in genome stability. The EMBO J 26: 4089–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psakhye I, Jentsch S. 2012. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 151: 807–820. [DOI] [PubMed] [Google Scholar]
- Ryu T, Spatola B, Delabaere L, Bowlin K, Hopp H, Kunitake R, Karpen GH, Chiolo I. 2015. Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat Cell Biol 17: 1401–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarangi P, Zhao X. 2015. SUMO-mediated regulation of DNA damage repair and responses. Trends Biochem Sci 40: 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeenk G, van Attikum H. 2013. The chromatin response to DNA breaks: leaving a mark on genome integrity. Annu Rev Biochem 82: 55–80. [DOI] [PubMed] [Google Scholar]
- Sollier J, Driscoll R, Castellucci F, Foiani M, Jackson SP, Branzei D. 2009. The Saccharomyces cerevisiae Esc2 and Smc5–6 proteins promote sister chromatid junction-mediated intra-S repair. Mol Biol Cell 20: 1671–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su XA, Dion V, Gasser SM, Freudenreich CH. 2015. Regulation of recombination at yeast nuclear pores controls repair and triplet repeat stability. Genes Dev 29: 1006–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Hediger F, Neumann FR, Bauer C, Gasser SM. 2004. Separation of silencing from perinuclear anchoring functions in yeast Ku80, Sir4 and Esc1 proteins. EMBO J 23: 1301–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatham MH, Jaffray E, Vaughan OA, Desterro JM, Botting CH, Naismith JH, Hay RT. 2001. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J Biol Chem 276: 35368–35374. [DOI] [PubMed] [Google Scholar]
- Tatham MH, Geoffroy MC, Shen L, Plechanovova A, Hattersley N, Jaffray EG, Palvimo JJ, Hay RT. 2008. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat Cell Biol 10: 538–546. [DOI] [PubMed] [Google Scholar]
- Therizols P, Fairhead C, Cabal GG, Genovesio A, Olivo-Marin JC, Dujon B, Fabre E. 2006. Telomere tethering at the nuclear periphery is essential for efficient DNA double strand break repair in subtelomeric region. J Cell Biol 172: 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Rosell J, Machin F, Farmer S, Jarmuz A, Eydmann T, Dalgaard JZ, Aragon L. 2005. SMC5 and SMC6 genes are required for the segregation of repetitive chromosome regions. Nat Cell Biol 7: 412–419. [DOI] [PubMed] [Google Scholar]
- Torres-Rosell J, Sunjevaric I, De Piccoli G, Sacher M, Eckert-Boulet N, Reid R, Jentsch S, Rothstein R, Aragon L, Lisby M. 2007. The Smc5–Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat Cell Biol 9: 923–931. [DOI] [PubMed] [Google Scholar]
- Ullal P, Vilella-Mitjana F, Jarmuz A, Aragon L. 2011. Rtt107 phosphorylation promotes localisation to DNA double-stranded breaks (DSBs) and recombinational repair between sister chromatids. PLoS One 6: e20152. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- van Attikum H, Fritsch O, Gasser SM. 2007. Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J 26: 4113–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Kerscher O, Kroetz MB, McConchie HF, Sung P, Hochstrasser M. 2007. The yeast Hex3⋅Slx8 heterodimer is a ubiquitin ligase stimulated by substrate sumoylation. J Biol Chem 282: 34176–34184. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Shimada K, Oma Y, Kalck V, Akimura K, Taddei A, Iwahashi H, Kugou K, Ohta K, Gasser SM, et al. 2010. Actin-related protein Arp6 influences H2A.Z-dependent and -independent gene expression and links ribosomal protein genes to nuclear pores. PLoS Genet 6: e1000910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Blobel G. 2005. A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc Natl Acad Sci 102: 4777–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Wu CY, Blobel G. 2004. Mlp-dependent anchorage and stabilization of a desumoylating enzyme is required to prevent clonal lethality. J Cell Biol 167: 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.