

Abstract
Folding and maturation of most protein kinases require chaperone assistance. In higher eukaryotes, CDC37 is the predominant cochaperone that facilitates the transfer of kinase clients to HSP90. Kinase recognition is thought to occur through the N-terminal domain, which has, thus far, eluded structure determination. Client processing also requires the phosphorylation of the N-terminal tail at Ser13 by protein kinase CK2 (casein kinase 2). How phosphorylation alters the molecular properties of CDC37 is not understood. We show that the phosphorylation at Ser13 induces a large shift toward a more compact structure, based on ANS fluorescence, while modestly increasing secondary structure. Moreover, this transition requires interactions of the N-terminal domain and the remainder of CDC37. The stabilizing property of the phosphorylation event can be recreated in trans by a (phospho-Ser13) peptide derived from the N-terminal tail. However, the phosphorylation-induced transition is not dependent on the transferred phosphate group but rather the loss of serine-like properties at position 13. The complete absence of the N-terminal tail results in reduced secondary structure and unresponsiveness to subsequent addition of peptides. The N-terminal tail may therefore serve as an intramolecular chaperone that ensures that CDC37 assumes one of two readily interconvertible states in a manner that impacts the interaction of the client binding N-domain and the MC-domains, involved in dimerization and HSP90 binding.
Graphical abstract
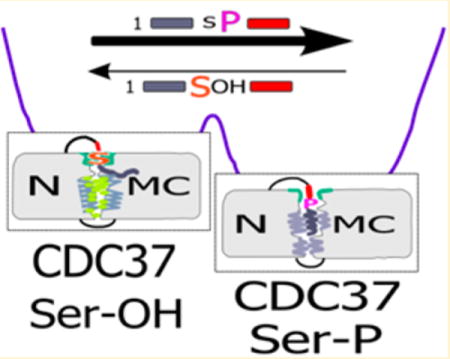
For many cellular proteins, initial folding and the subsequent maturation require complex cellular machinery. This machinery, consisting of an array of chaperones and cochaperones, engages its substrates, also referred to as clients, in a sequential manner. Chaperones that act in early client folding (e.g., HSP70) are distinct from those that assist at later stages of maturation. These later stages may involve broad specificity chaperones, such as HSP90, as well as protein class specific cochaperones, and transitions occur by client handover in large chaperone complexes (reviewed in ref 1). In the case of the majority of kinases or select ATP binding domains, the critical cochaperone is CDC37 (cell division cycle protein 37). Since CDC37–client interaction involves the select recognition of the client class, this interaction may be regarded not only as folding or maturation assistance but also as an early stage in quality control. Failure of a client to successfully pass through the successive rearrangements of the various client–chaperone complexes may result in its degradation. As demonstrated for mutations in CFTR (cystic fibrosis transmembrane conductance regulator), severe functional deficiencies may arise due to relatively minor alterations in client structure and highly stringent quality control.2,3
Functionally, CDC37 is thought to act as an adaptor or scaffold molecule facilitating the interaction of clients with HSP90.4 Since its first identification in 1980, the library of CDC37 clients has quickly expanded to cover over 350 proteins that involve almost exclusively ATP binding folds (mainly, but not exclusively, kinases).5 At least for a subset of kinase clients, CDC37 is capable of facilitating client processing independent of HSP90.6,7 Its kinase binding domain is sufficient to carry out this task, but this interaction has been proposed to reflect a less supervised mode of client maturation that is physiologically superseded by HSP90-dependent processing due to the low concentrations of CDC37 and abundance of HSP90.6 CDC37 does interact with select targets outside the kinase family, such as androgen receptor,8 reverse transcriptase,9 or the guanine nucleotide exchange factor Vav3. However, at least for the latter, the functional region of CDC37 is distinct from its kinase binding domain.10 Many oncogenic kinases heavily rely on CDC37 activity. Silencing of CDC37 expression by siRNA or shRNA depletes clients such as ERBB2, cRAF, CDK4, CDK6, and AKT in human colon, breast, and prostate cancer lines.11–13 Overexpression of CDC37 occurs in various cancers but at relatively modest levels,14 possibly reflecting its transitory involvement with a small intermittent pool for most of its clients. Targeting the interactions between CDC37 and its kinase clients is, therefore, a potential entry point for therapeutics aimed at individual overactive kinases or kinase pathways. However, the molecular events underlying molecular recognition of such a broad range of clients as well as the assembly and disassembly of HSP90 complexes remain poorly understood.
In-solution studies identified an inherently weak ability of CDC37 to dimerize with a micromolar dissociation constant.15 Current models of CDC37 action (reviewed in ref 16) involve the recognition of its client by a CDC37 dimer, binding of dimeric HSP90, and the subsequent dissociation of one CDC37 molecule to generate a relatively stable 1:1:2 complex of client, CDC37, and the HSP90 dimer.17,18 Of the three major domains of CDC37 (Figure 1), the N-terminal domain engages kinase clients19,20 and the central M-domain is responsible for the recruitment of HSP90 and homodimerization of CDC37.21–23 CDC37 function is regulated through phosphorylation at Ser13 by protein kinase CK2 (casein kinase 2). In studies of yeast CDC37, mutations of the CK2 target site result in severe growth deficiency and depletion of CDC37 clients.24,25 This suggests that CDC37 function as a class-specific cochaperone is largely not redundant. Available structural data on the M-domain and part of the C-terminal segment21,26 provide insight into the nature of the CDC37–HSP90 complex, but the N-domain is not resolved in this structure. Figure 1 shows a simplified version of the model that has been derived from several pieces of crystallographic data.21 It features a symmetric complex containing two molecules of CDC37. This model is complemented by an EM reconstruction of a 1:1:2 complex.17 However, the latter also failed to assign the location and mode of interaction of the N-domain relative to client. Biochemical data suggest that the dephosphorylation of the N-terminus of CDC37 is important for the maturation complex to proceed. This dephosphorylation within the HSP90 multiprotein complex is highly context-dependent and requires PP5 phosphatase.18 However, the mechanism by which phosphorylation achieves this objective is not understood, and it is not clear how CDC37 is capable of recognizing or identifying such a broad array of clients while rejecting some closely related kinases as clients.
Figure 1.
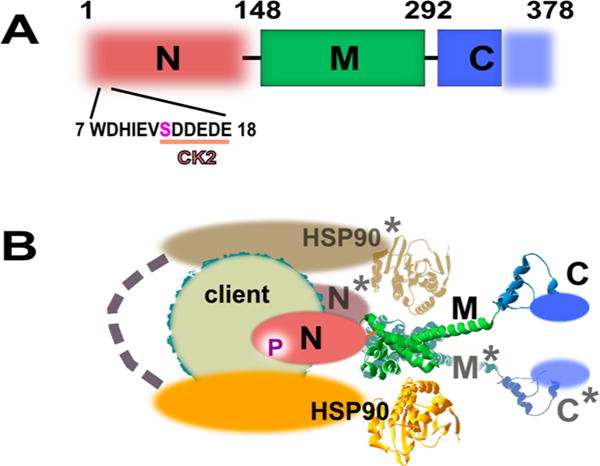
Domain organization of CDC37. (A) Only the middle domain (M) and part of the C-terminal domain (C) are structurally resolved by X-ray crystallography and depicted as solid blocks. The N-domain is responsible for kinase client recognition but is not structurally resolved. The N-terminal peptide contains an acidic substrate sequence for CK2 and the phosphorylation site at serine 13. (B) Model of CDC37/CDC37* dimer bound to HSP90 dimer and client. The representation is a simplified version of a model proposed by Vaughan et al.17 Modeled components are shown as spheres structured around the crystallographically confirmed backbone of the M and partial C-domain. Each M-domain interacts with an N-terminal ATP binding domain of HSP90.
The ability of CDC37 to establish defined interactions with its clients, yet retain a high degree of structural flexibility, represents a feature also found in intrinsically disordered proteins (IDP) or protein domains. For the most part, IDPs maintain mainly random coil or molten globule structure and manifest extraordinary structural flexibility and plasticity by sampling a variety of conformations that are in equilibrium.27–29 Alternatively, the ensembles represented in IDPs may also involve various alternative structured states that are in rapid equilibrium unless a specific state is selected out through modifications or interactions.30 Computational methods predict that over 30% of proteins in higher eukaryotes are IDPs or harbor long, intrinsically disordered segments defined as more than 50 consecutive residues.31,32 IDPs have been identified in key cellular processes such as transcription,33–38 signaling transduction,39,40 cell cycle control,41 and programmed cell death.42 There are already multiple cases in which either chaperone molecules function through their ID segment or IDPs are found to possess chaperone activity.43–45
Computational assessments indicate a strong correlation between intrinsic disorder and the occurrence of post-translational modifications, including phosphorylation, acetylation, and O-glycosylation.46–49 Post-translational modifications can induce transitions between order and disorder and thus regulate protein or domain interactions.34,50–52 In the before-mentioned example of CFTR, channel gating is regulated by its normally structured R-domain, which competes against the heterodimerization of two nucleotide binding domains, NBD1 and NBD2. The structural compactness of the R-domain is lost upon phosphorylation, thus favoring NBD heterodimerization and altering the state of the channel.50 The tasks being performed by CDC37, the proposed structural properties of the N-domain, and the mode of regulation by CK2 are consistent with those of a regulated IDP. In this study, we provide experimental evidence that phosphorylation causes significant changes in the level of compactness of CDC37, that this transition involves the tail region and the combined N and MC-domains, and that the resulting state is characterized by a high degree of stability. The transition depends on the loss of serine-like side chain properties, not phosphorylation itself. The ability of the phosphorylated N-terminal tail to induce structural changes can be recapitulated qualitatively in trans, using an exogenous phosphopeptide derived from its N-terminus. However, the overall change in secondary structure is very small, indicating a rearrangement of pre-existing structural elements rather than a transition from a largely disordered to a globular state.
MATERIALS AND METHODS
Constructs and Proteins
The human ERBB2 and ERBB3 cDNA were expressed in a pFlag-myc-CMV-19 vector together with a C-terminal biotinylation acceptor peptide sequence (GLNDIFEAQKIEWHE). The Escherichia coli biotinyl transferase BirA was cloned into the pLXSN vector for cotransfection with biotinylation tagged constructs. The mammalian expression construct of HA-tagged CDC37 in the pKH3 vector was generously provided by Dr. Kerry Burnstein (University of Miami). From this construct, full length (1–378), Δ1–19 (20–378), N-terminal domain (1–148), and MC-domains (149–378) were derived and cloned into the bacterial expression vector pQE40. Point mutants including S13A, S13D, S13E, and S13C were created in pQE40 using the Agilent Technologies QuikChange mutagenesis system. A CK2 construct for bacterial expression was directly purchased from Addgene (plasmid 27083: pDB1 (CK2alpha)). For all in vitro studies, both CDC37 and CK2 constructs were expressed in the DH12S strain of E. coli (Invitrogen) and purified by FPLC (Biorad) on a Ni-NTA column (CDC37) or glutathione affinity column (CK2), respectively. Proteins were finally dialyzed and stored in PBS. The isolated N-domain of CDC37 shows a high tendency to aggregate during dialysis and storage. To overcome this issue, the N-domain was eluted from the Ni-NTA column with PBS containing 2 mM EDTA and processed immediately for CD spectroscopy and ANS assays by spin column desalting (Princeton Separations, CS-900).
In Vitro Phosphorylation/Dephosphorylation
In vitro phosphorylation of CDC37 by CK2 was carried out at 37 °C or room temperature for the indicated time periods. Reactions were close to complete after 3 h. The in vitro phosphorylation buffer contained 50 mM Tris-HCl, 10 mM MgCl2, 0.1 mM EDTA, 0.1 mM DTT, and 2.5 mM ATP. CDC37 concentrations were 1–6.5 μM with a 10:1 molar ratio of CDC37 to CK2. For studies on dephosphorylation efficiency, phosphorylation by CK2 was carried out for 3 h and blocked by the addition of 4,5,6,7-tetrabromobenzotriazole (TBB). For samples that were denatured prior to dephosphorylation, the indicated detergents were added and samples were boiled for 5 min. All samples were diluted 50-fold into phosphate buffer containing a high concentration of calf intestine alkaline phosphatase (0.1 U/μL final concentration). Samples were incubated for 1 h at the indicated temperatures.
Immunodepletion and Immunoprecipitation
To estimate the phosphorylation efficiency in vitro and in cell culture, three sequential immunodepletions of a phosphorylation reaction or cell lysate, respectively, were carried out. CDC37-containing solutions were diluted into 500 μL of PBS and incubated with 4 μL of CDC37 pS13 antibody (Epitomics, 3600-1) and 15 μL of protein A/G agarose beads (Santa Cruz, sc-2003). After incubation on a rotator for 30 min and centrifugation, the supernatant was subjected to a second and third round of immunodepletion. Detection of the remaining soluble CDC37 was done by CDC37 antibody (Santa Cruz, sc-13129) on western blots.
For the study of cellular interactions of recombinant CDC37 with ERBB clients in cell lysate, 5 × 105 BT474 cells were lysed in 500 μL of mild lysis buffer (20 mM Tris, 137 mM NaCl, 1% Triton X-100, 10% glycerol, 5 mM EDTA) containing 1 mM sodium orthovanadate and 1 mM PMSF and underwent predepletion with anti-His antibody. The cleared lysate was incubated with 15 pmol of in vitro phosphorylated recombinant CDC37, 5 μL of anti-His antibody (Genscript, A00186), and 20 μL of protein A/G beads. After 1 h of incubation, the beads were subjected to three washes with mild lysis buffer and analyzed by western blot using ERBB2 (Abcam, ab8054), ERBB3 (Santa Cruz, sc-285), or HSP90 (Stressgen, SPA-840) antibodies.
For coimmunoprecipitation experiments that were selective for cell surface receptors, N-terminally Flag epitope-tagged ERBB2 and ERBB3 were transiently expressed in the MCF7 breast cancer cell line in 6–well plates. Anti-Flag antibody (Sigma, F1804) was added to the culture medium and incubated on ice for 30 min to prevent internalization. Free antibody was removed by three washes with cold PBS, and cells were lysed in mild lysis buffer. The lysate was incubated with protein A/G beads for 1 h on a slow speed rotor. Beads were washed three times with lysis buffer, and associated proteins were analyzed by SDS-PAGE and western blots.
For the coimmunoprecipitation using biotinylation tags, MCF7 cells were transfected with ERBB2-bio or ERBB3-bio, BirA, and, when indicated, HA-CDC37. The biotag at the C-terminus of ERBBs encodes a biotinylation substrate peptide (GLNDIFEAQKIEWHE) for the BirA biotinyl transferase. The lysates were incubated with neutravidin agarose beads (Pierce, 29202) for 1 h. Were indicated, in vitro phosphorylated, E. coli expressed CDC37 was added to the incubation system. After the incubation, beads were washed three times with lysis buffer, and associated proteins were analyzed by western blots.
CD Spectrum
CD spectra were collected either directly, using the phosphorylation reaction, or following removal of CK2. For CK2 removal, in vitro phosphorylation mixtures containing both CDC37 and CK2 or CK2 alone were incubated with glutathione agarose resin, and the supernatant was loaded onto gel filtration spin columns for rebuffering just prior to CD analysis. The reconstitution buffer for CD analysis contained only 10 mM potassium phosphate (pH 7.5) with or without 1 M urea. The spectra were collected on a Jasco CD spectrometer (model J-815). The spectra obtained this way matched those obtained by measurements after desalting alone followed by subtraction of the CK2 control spectrum. Due to the substantially lower loss during handling, the latter approach was adopted for routine measurements of point mutants. Peptides representing the 19 N-terminal residues of CDC37, including regular or phosphorylated serine in position 13, were synthesized by Biomatik. The peptide sequences were MVDYSVWDHIEVSDDEDETC or MVDYSVWDHIEVpSD-DEDETC, including an additional cysteine in position 20 for optional immobilization. For CD measurements, the peptides were reconstituted in 10 mM potassium phosphate buffer (Ph 7.5) at a concentration of 300 μg/mL. For the N-terminal domain, the protein was eluted from the Ni-NTA column in PBS with 2 mM EDTA and subjected to spin column buffer exchange into the respective buffers for CD spectroscopy or ANS assays.
ANS Assay
The fluorescent spectrum of 1-anilinonaph-thalene-8-sulfonic acid (ANS) was determined in PBS in a Jasco fluorometer (model J-6500). As a reference, 10 μM ANS was incubated with 6 μM BSA, yielding maximal emission with an excitation wavelength of 380 nm. For CDC37 measurements, 20 μM ANS was incubated with 1 μM CDC37 in either phosphorylation buffer or PBS. For temperature control studies with DHFR, 0.64 μM DHFR and 4.62 μM CDC37 were incubated with and without a 10-fold molar excess of methotrexate. Synthetic peptides derived from the N-terminus of CDC37 were added at the ratio of 1:1 to the indicated CDC37 constructs.
Secondary Structure and Disorder Predictions
The secondary structure prediction for CDC37 was obtained using the PSIPRED algorithm53 using default settings at the Protein Sequence Analysis Workbench server.54 The propensity of segments of CDC37 for structural disorder was predicted using the PONDR suite of algorithms55 using default settings. The analysis presented included four algorithms: VL3,56 VSL2, VL-XT,57–59 and XL1-XT.
RESULTS
Assessment of E. coli Expressed CDC37
For most of our studies, we relied on human CDC37, expressed as recombinant protein in E. coli. Bacterial expressed CDC37 has been used in prior studies.60,61 However, we wanted to ascertain that the recombinant protein we generated was a reasonable substitute for mammalian expressed CDC37 for our objectives. We therefore evaluated the ability of CDC37 to be phosphorylated by protein kinase CK2 (casein kinase 2) at Ser13 and the ability of recombinant CDC37 to recognize mammalian clients with a selectivity profile that matches that of endogenous mammalian CDC37. The ability of CK2 to recognize and selectively phosphorylate Ser13 of CDC37 in vitro was confirmed by western blot analysis using a CDC37/pSer13-specific antibody for detection (Figure 2A). In different studies using either 3- or 2-fold sequential immunodepletions, we found 78% (± 7) of endogenous CDC37 to be constitutively phosphorylated in mammalian cells (Figure 2B). For recombinant human CDC37, expressed in MCF7 cells, approximately 90% of CDC37 is phosphorylated or resides in complexes carrying at least one phosphorylated Ser13 (Figure S1). Phosphorylation by CK2 is, therefore, reproducible in vitro and recreates a state that is representative of most of the CDC37 in a cellular setting.
Figure 2.
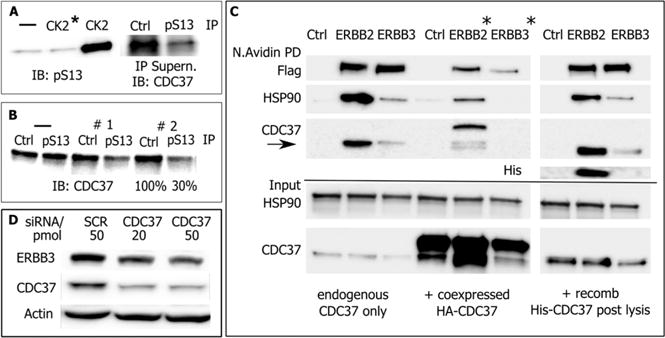
Evaluation of E. coli expressed CDC37. (A) (Left) In vitro phosphorylation of E. coli expressed His-CDC37/Ser13 probed by phosphorylation-site-specific antibody (IB: pSer). The control reaction with heat-deactivated CK2 is marked (*). (Right) Estimate of phosphorylation efficiency for in vitro phosphorylated E. coli expressed CDC37. pSer13/CDC37 was removed by three consecutive immunoprecipitations using immobilized pSer13 antibody or resin control. Approximately 85% of CDC37 was removed from the supernatant, and the remainder was detected by CDC37 antibody. (B) Estimate of phosphorylation efficiency of endogenous CDC37 in MCF7 cells. Two sequential immunoprecipitations with pSer13/CDC37 antibody remove approximately 70% from cell lysate. (C) Client binding preference of endogenous mammalian and in vitro phosphorylated, E. coli expressed CDC37. Recombinant, in vivo biotin tagged ERBB2 and ERBB3 receptors were analyzed for coimmunoprecipitated endogenous CDC37 (left). Cotransfected HA-tagged CDC37 and smaller size endogenous CDC37 (center) or E. coli expressed and in vitro phosphorylated CDC37 (right). E. coli expressed and endogenous CDC37 show the same client binding preferences. Note that ERBB2/3 levels in cotransfection experiments (*) are differentially impacted by the addition of endogenous CDC37. (D) Despite low binding in pull-down studies, the dependence of ERBB3 on CDC37 during early maturation is reflected in its sensitivity to CDC37 knockdown by siRNA.
As an additional functional test we evaluated the ability of recombinant CDC37 to bind HSP90 and client. For HSP90, clients fall into two categories: those requiring HSP90 only during maturation and those associating with it also in the mature state. We previously showed that ERBB3 (HER3), a receptor tyrosine kinase of the EGFR family, falls into the first category, with only a very small portion of ERBB3 at any time being transiently associated with HSP90 during maturation.62 By contrast, the closely related ERBB2 (HER2) receptor is a well-established representative of proteins requiring HSP90 throughout their life cycle.63 Before we could utilize this knowledge as a functional test for recombinant CDC37, we needed to establish whether this HSP90-derived interaction pattern also extended to CDC37. To this end, we expressed recombinant, biotinylation tagged ERBB2 or ERBB3 in MCF7 cells and measured the recovery of CDC37 or HSP90 that bound to neutraavidin resin purified receptors from cell lysate (Figure 2C). Endogenous CDC37 was recovered at significantly higher yield bound to ERBB2 than that to ERBB3, suggesting that the previously established binding preference of HSP90 carries over to CDC37. This preference is also evident for recombinant HA-tagged CDC37 expressed in MCF7 cells. In this case, the quantitative analysis is limited by the strong differential impact of overexpressed CDC37 on the levels of cotransfected ERBB receptors. We validated this binding preference for the binding of E. coli expressed and in vitro phosphorylated CDC37 to biotin tagged receptors in cellular lysate. Therefore, E. coli derived CDC37 demonstrated a binding preference that matches that of endogenous CDC37. The same results were obtained when we compared recombinant CDC37 expressed in mammalian cells and its selective interaction with cell surface, and hence mature, receptors (Figure S2) with the interaction of phosphorylated E. coli derived CDC37 and endogenous ERBB receptors in BT474 cells64,65 overexpressing both receptors in response to ERBB2 gene amplification (Figure S3). The failure of ERBB3 and CDC37 to coimmunoprecipitate reflects a CDC37 dependence that is limited to transient interactions in the nascent state. This dependency is, however, apparent in ERBB3 levels when CDC37 is knocked down by siRNA (Figure 2D). We therefore concluded that E. coli generated CDC37 is a suitable substitute for mammalian expressed CDC37 in our studies based on its own phosphorylation and differential client binding properties.
Phosphorylation at Serine 13 Increases Structural Compactness of the N-Terminus
The high yield of in vitro phosphorylation by CK2 facilitates the analysis of its impact on the structural dynamics of CDC37. An analysis of CDC37 by circular dichroism spectroscopy indicates a high prevalence of α helices (Figure 3A). This finding fits secondary structure predictions and partial structural information available for CDC37.21 On the basis of a deconvolution of the spectrum using the CDpro package of algorithms66 and a reference data set of soluble proteins, α helices constitute approximately 80% of CDC37, with the remainder representing coiled coil segments. CK2-mediated phosphorylation of CDC37 induced modest increases in helical content. For Figure 3A, samples had been depleted of CK2 by glutathione agarose prior to desalting and CD analysis. However, we observed the same shift when the contribution of CK2 was removed by subtraction of its CD spectrum (used for Figure 3B). While small in magnitude, the increases in helical content were highly reproducible between studies at 2.1 ± 0.3% (calculated from triplicate spectra of duplicate experimental sets). This increase occurred at the expense of random coil segments and translates into an estimated helix formation by approximately 10 additional amino acids. Interestingly, the increase is reversed when challenged with 1 M urea while the initially observed α-helical content remains intact (Figure 3B). This would suggest that phosphorylation induces structural changes that involve a well-defined region and is readily reversible without compromising the remainder of the protein structure. By contrast, CDC37 lacking the extreme N-terminal tail (CDC37 Δ1–19) shows a loss in helical content (Figure 3A) despite high stability and solubility of the purified protein.
Figure 3.
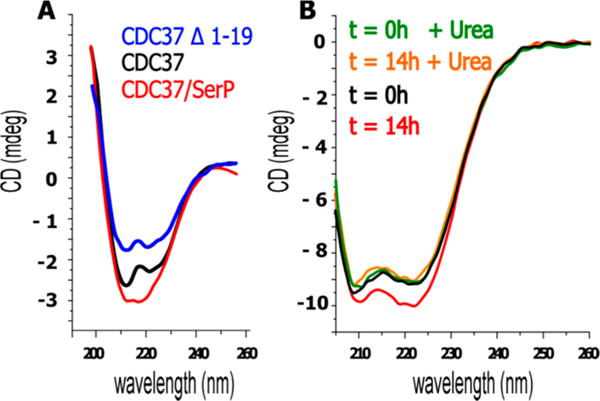
Phosphorylation on Ser13 monitored by CD spectroscopy. (A) CDC37 is largely composed of α-helices (black). After overnight phosphorylation by CK2 at room temperature, α-helical content of CDC37 increases (red). Deletion of the first 19 amino acids causes a significant loss of α-helical content (blue). Spectra were obtained after CK2 removal on GSH columns and desalting. (B) The CD spectrum after overnight phosphorylation, desalting, and subtraction of CK2 spectrum gives matching results but higher recovery. Upon phosphorylation the α-helical content of CDC37 increases by 2 to 3%. (data representative of duplicate studies and triplicate spectral measurements). The gain in α-helical content but not the remaining secondary structure is destabilized when 1 M urea is added after overnight phosphorylation. For experiments involving urea, data below 205 nm are not suitable for analysis and were omitted.
To further explore the phosphorylation-induced transition in CDC37, we applied the fluorescent dye, 1-anilinonaphthalene-8-sulfonic acid (ANS). This amphiphilic dye shows a strong increase in fluorescence when it obtains access to hydrophobic pockets of a protein. It is, therefore, widely used to obtain a measure of protein compactness. However, the intensity of the ANS signal depends on at least two major parameters. The first is the access to interior binding sites for ANS, which is being utilized for measurements of compactness. However, in order to bind, ANS does need structural elements with which to interact, and an all-out loss in secondary structure can, therefore, also result in reduced signal. Prior studies on the molten globule state of several proteins, including recombinant human growth hormone,67 show maximum fluorescence in the molten globule state and minimal fluorescent in the fully folded native and unfolded states.
The fluorescence of full-length CDC37 (NMC, representing its individual domains) drops significantly following phosphorylation by CK2 (NMC, t = 14 h). Most of the decrease occurs within 3 h and is complete after 14 h at room temperature. The CDC37 Δ1–19 truncation mutant displays a fluorescence similar to that of phosphorylated full-length CDC37, whereas the middle and C-terminal segments of CDC37 (MC) show very low ANS fluorescence. Together with the very favorable protein properties during synthesis and storage, this suggests a stable and compact structure for MC and would suggest that the large variation in ANS signal is derived from phosphorylation-induced changes of the N-terminal domain (N). However, in isolation, the N-terminal segment does not display a high ANS signal. For direct comparison, Figure 4A also shows the calculated sum of both signals, which is comparable in concentration and composition to that for wild-type protein. The sum of the N and MC segments amounts to approximately 65% of the signal obtained from phosphorylated CDC37, or less than 30% of the unphosphorylated reference. Yet, in sharp contrast to the MC-domains, the N-domain exhibits a high tendency to aggregate postpurification (Figure 4B). In fact, all N-domain-related studies were carried out immediately after purification with spin column desalting replacing dialysis in order to obtain reproducible results. To address this apparent contradiction between the obtained ANS signal and protein properties, we determined the CD spectrum of the N-domain. Consistent with the low ANS signal, this indicated α-helical structure (Figure 4C). The intensity of the concentration-adjusted CD spectrum is proportional to the size of the N-domain compared to full-length CDC37, suggesting that the freshly isolated N-domain does not display a lack of secondary structure.
Figure 4.
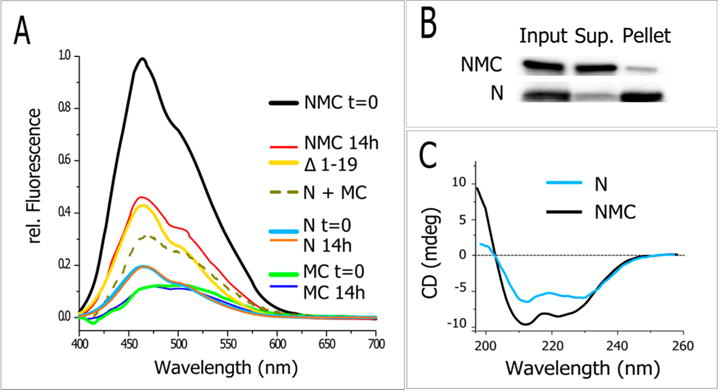
On the basis of ANS fluorescence, CDC37 becomes more compact upon phosphorylation. Legend is arranged in order of spectral height. (A) Overnight phosphorylation markedly decreases the ANS fluorescence of full-length CDC37 (NMC 14h), whereas isolated N and MC-domains show a low and phosphorylation-insensitive fluorescence. The sum of N and MC-domains fluorescence is added for comparison and molar equivalent but is reduced in intensity. Low ANS fluorescence was observed for CDC37 lacking the first 19 amino acids (Δ1–19). Data are normalized to the fluorescence of untreated NMC. For all ANS assays, excitation occurred at 380 nm and CDC37 concentrations were normalized based on the OD280 estimated protein concentration. (B) The N-domain in isolation is prone to rapid aggregation in PBS. N and NMC constructs were compared by high-speed centrifugation and western blot analysis for their solubility after dialysis and comparable storage. (C) Comparative characterization of the purified N-domain by CD. The concentration-corrected spectrum was obtained immediately after purification and spin column desalting.
The Chemical Characteristics of Serine Are Needed To Retain CDC37 in Its Less Compact State
Phosphorylation at serine or threonine can, in many cases, be mimicked by substitutions with glutamic or aspartic acid. We therefore investigated the impact of these substitutions on the ANS signal (Figure 5). Indeed, both aspartic and glutamic acid substitutions result in ANS binding that resembles the phosphorylated state. However, the same is true for substitution with alanine. These data suggest that the observed transition is not driven by the introduction of the phosphate group but by the loss of serine.
Figure 5.
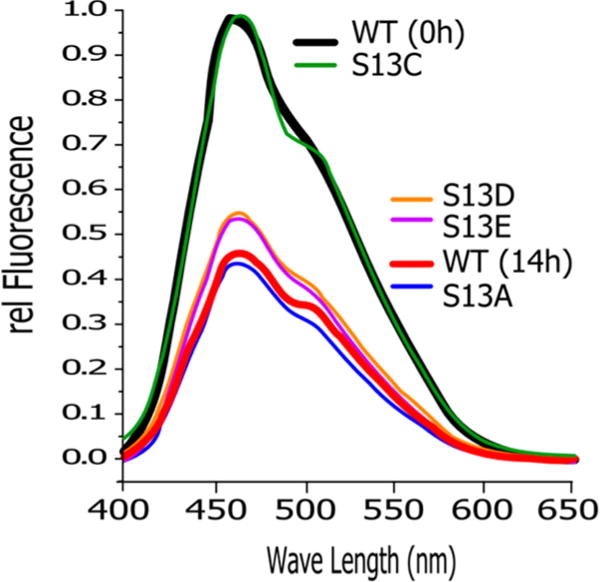
Increase in compactness requires the removal of Ser-like properties. The ANS assay shows a marked decrease in fluorescence that is comparable to the phosphorylation of wild-type CDC37 (WT (14 h)) when serine 13 is mutated to alanine, aspartic acid, or glutamic acid. The biochemically more conserved mutant S13C retains the fluorescence pattern of wild-type CDC37.
By comparison, the substitution of serine with cysteine creates a wild-type-like signal. The same data were obtained for substitution with threonine (Figure S4). However, the substitution by threonine is less straightforward in its interpretation. Its properties resemble those of the unphosphorylated state, but the potential presence of phosphorylation by CK2 cannot be ruled out definitively with the tools available. Overall, substitutions studies indicate that the loss of the side chain properties of Ser13, not the introduction of a phosphate group, causes the observed transitions in the structure of CDC37.
The N-Terminal Segment of CDC37 May Be Structurally Adaptable
The above studies show substantial structural rearrangements upon phosphorylation, resulting in a more compact end state. Furthermore, this appears to involve relatively small net changes in actual secondary structure components. Similar phosphorylation-driven transitions between states of increased compactness have been described for intrinsically disordered proteins (IDP).34,50–52 We therefore analyzed the CDC37 primary sequence with the PONDR suite of algorithms (Predictor of Naturally Disordered Regions).53 Figure 6A shows the superimposition of the four individual algorithms that were combined in this analysis. The consensus of the four predictions assigns a high probability of disorder to two regions of CDC37: the M to C domain transition (approximately residues 250–325) and most of the N-domain (approximately residues 25–170). Yet, both regions are assigned a high α-helical content in secondary structure prediction (Figure 6B). The secondary structure prediction matches available crystallographic data, where available, including the exceptionally long α helix found in the M section.21 With the exception of the VL3 algorithm (which evaluates a 30 amino acid window), the remaining algorithms show a high agreement in predicting a tightly defined area of disorder at the extreme N-terminus. This small segment of predicted disorder is centered on Ser13.
Figure 6.
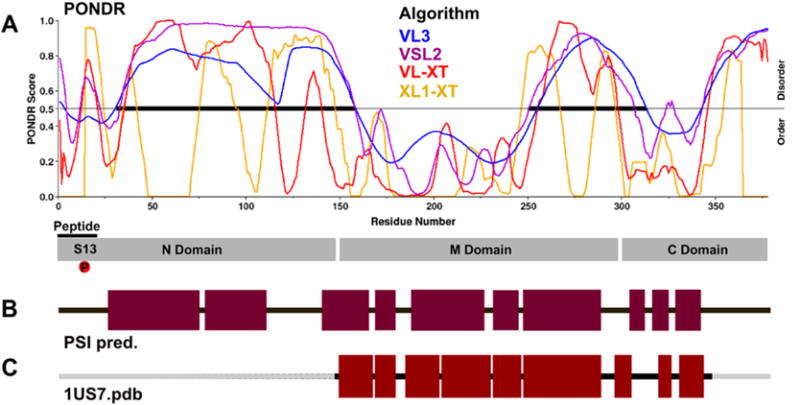
N-domain of CDC37 is predicted to have properties of intrinsically disordered proteins and a high α-helical content. (A) Prediction of disorder using the PONDR suite of algorithms. The disorder (PONDR) score is shown for four different algorithms. Note that VL3 uses a larger sequence window for its prediction. (B) Despite the predicted disorder, the N-domain is also predicted to contain large segments of α-helical structure based on a PSI-pred prediction of secondary structure elements. (C) For structurally resolved segments of CDC37, the predicted secondary structure correlates well with experimental observations (1US7.pdb).
A Phosphopeptide Derived from the N-Terminal Tail of CDC37 Induces the Structural Changes Observed after Phosphorylation
Since the phosphorylation of the N-terminus correlates with the observed decreases in ANS binding and since this segment is set apart as a distinct entity in the prediction of intrinsic disorder, we generated two peptides that represent residues 1–19 of the N-terminus. The peptides differed by carrying either serine or phosphoserine at position 13. At 25 °C, the addition of both peptides induces a moderate decrease in the ANS signal (Figure 7A, panel II). Although this decrease appeared to be more pronounced for the (phospho-Ser13) peptide, differences were not large enough to be conclusive. We therefore tested whether the addition of peptides in trans influences the compactness of CDC37 under thermal stress. To this end, we measured ANS fluorescence at an elevated temperature of 50 °C. As a proof of principle for this assay, we first used dihydrofolate reductase (DHFR) and its stabilization by methotrexate (MTX) (Figure 7A, panel I). The binding of methotrexate to DHFR is well-known to exert a strong stabilizing force, strong enough to block the unfolding of DHFR fusion constructs in the context of post-translational mitochondrial import.68 In the absence of a destabilizing temperature challenge, DHFR receives a measurable, but relatively small, additional stabilization from MTX. The stabilizing effect of MTX is significantly enhanced at 50 °C. While by itself nonphysiological, this stabilization at elevated temperature provides a much better representation of the strong barrier to unfolding that methotrexate binding introduces under physiological conditions. CDC37-derived peptide in isolation or when added to DHFR did not alter the ANS signal. When the same temperature challenge is applied to CDC37, a stabilization by the phophopeptide becomes readily apparent, whereas the stabilization by the unphosphorylated peptide remains small. These observations suggest that the impact of phosphorylation at Ser13 induces interactions of the peptide with the remainder of the protein that convey a more compact structure that is more resistant to structural changes which expose ANS binding sites. This stabilization is readily evident in the phosphorylated species of CDC37 (Figure 7A, panel III), which is effectively undisturbed by the increase in temperature. The deletion of the N-terminal tail (Figure 7A, panel IV) does generate an intermittent state that is only partially responsive to peptide addition. More importantly, it does not discriminate with regard to the nature of the peptide.
Figure 7.
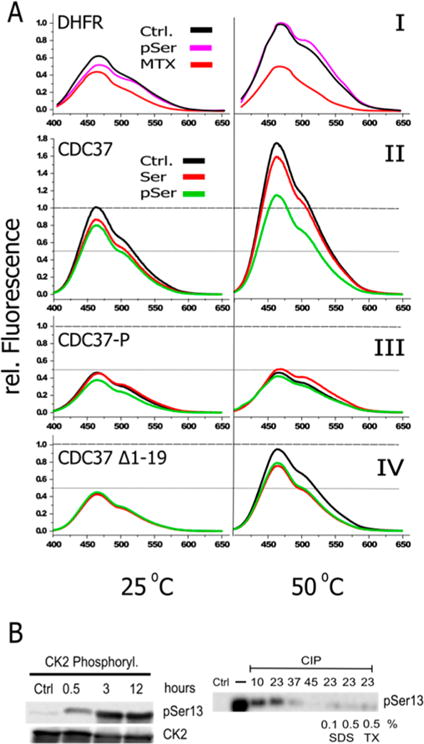
Phosphorylation at Ser13 or the addition of a (phospho-Ser13) peptide in trans stabilizes CDC37. (A) Panel I: Validation of ANS assay at elevated temperatures using DHFR and its stabilization by methotrexate at room temperature and 50 °C. Methotrexate, but not the pSer13 peptide (pSer), stabilizes DHFR upon temperature elevation. Panel II: ANS binding to unphosphorylated CDC37 rises at elevated temperature. A clearly discernible stabilization by the (phospho-Ser13) peptide (pSer) occurs at 50 °C. All peptides were added at a 1:1 molar ratio. Panel III: Compared to that of unphosphorylated CDC37, phosphorylated CDC37 has greatly reduced ANS binding at room temperature and is resistant to temperature increases. Quantitatively, the impact of the unphosphorylated peptide is small, but it is the only scenario in which peptide addition caused an increase in ANS fluorescence. Panel IV: The Δ1–19 truncation construct shows comparable starting fluorescence to that of pCDC37 but lacks its thermal stability. Modest stabilization occurs for both peptides, regardless of their phosphorylation state. (B) Phosphorylated serine 13 is partially shielded from phosphatases in a temperature-dependent manner. (Left) Phosphorylation at Ser13 by CK2 is close to maximal after 3 h. (Right) The access of pSer13 to a high concentration (0.1 U/μL) of alkaline phosphatase for 1 h is incomplete despite a large access of phosphatase. Dephosphorylation efficiency increases sharply above room temperature. Room-temperature dephosphorylation is complete when CDC37 is heat-denatured by SDS or Triton X-100 (TX), at the indicated concentrations, prior to a 10-fold dilution into phosphatase reaction buffer.
A remarkable feature of the phosphorylation at Ser13 is the physiological requirement for a specific phosphatase to carry out dephosphorylation in the context of the client and HSP90 containing complex. Outside of this structural context, in vitro dephosphorylation of pCDC37 can be achieved with aggressive alkaline phosphatase treatment. We therefore wanted to evaluate whether the structural transitions that we observed alter the ability of a generic phosphatase to gain access to Ser13 (Figure 7B). A time course of phosphorylation by CK2 shows that close to the maximum attainable phosphorylation can be achieved within 3 h at room temperature. Following inhibition with 4,5,6,7-tetrabromobenzotriazole (TBB, a CK2-specific inhibitor), treatment with a high concentration of alkaline phosphatase (CIP) ensures that enzyme levels are not a limiting factor. Under those aggressive dephosphorylation conditions, phosphorylation at Ser13 can be reversed, but it exhibits a very pronounced temperature dependence. At low temperatures, dephosphorylation remains partial despite large phosphatase access. Dephosphorylation efficiency increases rapidly above room temperature. Enzyme levels or the enzyme’s temperature dependence are not the limiting factors at and below room temperature, as CDC37 that was subjected to structure-disrupting treatments prior to dilution into dephosphorylation buffer is efficiently dephosphorylated, even at room temperature.
DISCUSSION
As a cochaperone that delivers a broad range of kinase clients to HSP90, CDC37 is postulated to undergo a complex series of transitions in protein interactions. Phosphorylation at the N-terminal tail by CK2 is critical in the overall function of CDC37 and its interaction with the HSP90 machinery, but it is not clear what the structure of the N-terminal tail, and indeed the entire N-terminal domain, is and how phosphorylation alters function. In this study, we show that upon phosphorylation CDC37 shows a small but reproducible increase in secondary structure that is accompanied by a much more pronounced change in ANS binding. This suggests a more compact end state that is experimentally characterized by a remarkably high resistance to opening up at elevated temperatures. Indeed, on the basis of this readout, the stabilization that occurs as a result of phosphorylation is comparable to the stabilization that DHFR receives from binding methotrexate. For CDC37 molecules that initially folded with an intact N-terminal tail, this two-state transition can be induced in trans by a synthetic (phospho-Ser13) peptide representing the first 19 amino acids. The ability of the phospho- and nonphosphopeptide to compete in trans against existing N-terminal tails in the opposite phosphorylation state suggests a hierarchy in interactions. The higher stability of the phosphorylated state is evident from the ability of the (phospho-Ser13) peptide to achieve thermal stabilization of CDC37 in trans, even in the presence of a competing unphosphorylated tail in cis. The opposite scenario of the addition of unphosphorylated peptide to phosphorylated CDC37 is quantitatively too small to be statistically significant. However, it is qualitatively the only instance in our studies in which the addition of peptide ever increased the ANS signal.
Figure 8 presents a conceptual model for the different interconvertible states of CDC37 in terms of the energy landscape and rationale for the observed changes in ANS signal and secondary structure. The more compact mode observed for the phosphorylated state is also the default for the isolated N and MC-domains, and the generation of a more ANS accessible ground state requires both the interaction of the N and MC-domains as well as the regulating presence of the unphosphorylated N-terminal tail. This model of interaction between the N-terminal tail and the MC domain is also consistent with reports that tyrosine phosphorylation at positions 4 and 298 disrupts the interaction between CDC37 and client.69 The incorporation of the tail segment in Figure 8 reflects this concept of a dependency on N–MC interactions, but the specific placement of the tail interaction site is arbitrary, and the tail could alternatively influence the N–MC interaction allosterically.
Figure 8.
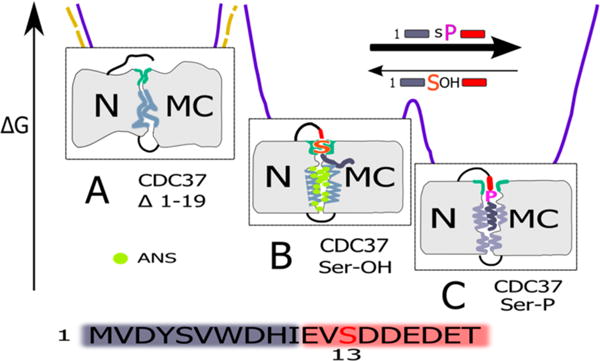
Visual presentation of the hypothesis for CDC37 structural transitions, derived from current data. The model is not intended to describe specific structural features but, instead, to provide a conceptual framework that best reconciles the current data. Detailed discussion is provided in the text. Note that current data cannot distinguish between a direct tail interaction in the N–MC interface (depicted here) and potential allosteric regulation across domains. Gray areas represent amphiphilic to hydrophobic segments in the interface and tail sequence as well as their packing and degree of secondary structure. To maintain a state in which secondary structure elements remain accessible to ANS requires stabilizing interactions, specifically interactions that rely on the biochemical properties of serine 13. The loss of these properties by phosphorylation (or biochemically divergent mutation) allows for additional, strong interactions of the tail peptide that are accompanied by ANS (green) exclusion and increase in secondary structure. This equilibrium is reflected in the relative competition strength of the respective peptides in trans (indicated by arrows). Complete absence of the tail during folding (yellow dashed energy landscape) results in irreversible and incorrect interactions when the N and MC-domains are held in close proximity. The presence of the tail segment (purple landscape) steers the folding process away from this trapped and largely irreversible state. Maximal ANS binding (green) occurs in state B when binding-suitable secondary structure elements are both present (absent in A) and accessible (reduced in C). The peptide coloring scheme (amplified below) reflects the very pronounced separation into an amphiphilic to hydrophobic N-terminus followed by an exceptionally acidic segment.
The properties of the N-terminal deletion construct (CDC37 Δ1–19) suggest that it has assumed a state that is outside of the framework of controlled transitions that we see for phosphorylated and unphosphorylated CDC37. On the basis of ANS assays, it appears very compact, and the overall protein properties are consistent with this assumption. However, this stands in contrast to the loss in secondary structure components based on CD measurements and its inability to discriminate between the addition of phosphorylated and unphosphorylated tail peptides added in trans (Figure 7, panel IV). The apparently contradictory ANS signal may reflect the fact that reduced ANS binding, in most cases, reflects an increase in compactness but can also be the consequence of a loss of secondary structure elements that serve as binding pockets. In the case of CDC37 Δ1–19, both may apply. This would suggest an additional function for the N-terminal tail, besides that of a reversible switch for CDC37 conformations. The more open state represented by the unphosphorylated state may be prone to collapse in a way that involves inappropriate N–MC interactions, unless this is actively being blocked by the tail segment. This assumption is consistent with our observation that both the phosphorylated and unphosphorylated states form distinct interactions with the remainder of the protein. The state that is obtained when CDC37 folds in the complete absence of the N-terminus would, therefore, represent a soluble and stable, but nevertheless unproductive, dead end in the folding landscape of CDC37. In this context, the N-terminal tail may serve as an intramolecular chaperone.
The observed changes in ANS binding are far more extensive than those seen by CD spectroscopy. The latter only involve the transition of a few amino acids (estimated 10) from a random-coil to an α-helical conformation. Furthermore, a PSI-pred secondary structure prediction is in good agreement with experimental data for the M and C-domains as well as CD data for the isolated N-domain in predicting a large proportion of α-helical content, including for most of the N-domain. The above observations can be reconciled with the assumption that the observed structural transitions involve significant changes in the packing behavior of already formed α helices. Several predictors of disorder assign a high aggregate probability of IDP properties to portions of the MC-domains. Moreover, except for the extreme N-terminal tail, the entire N-domain receives high scores that are either contiguous or interrupted by short segments of predicted order, depending on the algorithm. With respect to alternative concepts on IDPs, our data for CDC37 are, therefore, more in line with preformed structural elements whose interactions are in a state of flux. This would also explain why the N-terminal domain shows a high degree of α-helical content yet is far more prone than any other CDC37 segment or mutant to aggregate in solution over a short period of time. Such a property to readily reorganize structural elements may contribute to the ability of CDC37, and specifically it is N-domain, to recognize a broad range of kinase clients. The N-terminal tail, in either phosphorylation state, would serve to maintain two interaction capable and interconvertible states instead of the state observed for CDC37 Δ1–19. This model is consistent with earlier studies that assigned functional contributions to the tail region in client binding and maturation.20
Protein phosphorylation has been observed before as a functional switch in IDPs.34,50–52 Furthermore, a preponderance of acidic residues has been identified as favoring a disordered state.70 However, studies on the impact of further phosphorylation, especially at the N-terminus of proteins, have come to divergent conclusions.71,72 A striking feature of the mechanism by which the N-terminal tail segment regulates CDC37 is that the introduction of the phosphate group is not causal for the observed transitions. Transitions occur due to the loss of serine-like properties at position 13. Cysteine, and most likely threonine, are functionally neutral replacements that retain the ability to keep CDC37 in the more open state. At the same time, the phosphorylated state is not characterized solely by the loss of serine-dependent interactions but establishes a different set of interactions. Figure 8 incorporates these observations by assigning serine-dependent interactions a lower degree of stability, whereas phosphorylation, or other non-serine-like replacements, remove this barrier and allow segments of the tail region, especially its very amphiphilic to hydrophobic first 10 amino acids, to engage the remainder of the protein. The number of amino acids involved and the experimental CD data would fit a model in which this extreme N-terminus assumes α-helical character. However, if the most N-terminal amino acids are, in fact, induced to form an α helix, then it would be strictly dependent on stabilizing interactions with its immediate surroundings. Neither peptide in isolation (even with considerable addition of stabilizers) forms an α helix in solution (Figure S5).
In most cases, phosphorylation is a trigger that in some form or another results in the transmission of a signal, with few but very prominent exceptions in metabolic regulation. Even in the case of IDP-based regulation, phosphorylation is generally regarded as being the causal modification. The resulting view of molecular processes is effectively kinase- and phosphorylation-centric. Although considered to be critical, phosphatases are regarded as a means to turn off a signal initiated by phosphorylation, even if this signal involves altering the structural state of an IDP-like domain. A hallmark of most of the above reactions is the involvement of highly specific kinases and phosphorylation events that can be reversed by either generic phosphatases, or more often, class specific phosphatases with a range of clients. In addition, the phosphorylated state usually comprises a small portion of the total protein substrate pool. For CDC37, the inverse is true in all cases. Dephosphorylation occurs in the CDC37–HSP90 complex and selectively requires PP5 phosphatase to join the complex. In the context of the HSP90 complex, pSer13 is not sensitive to the attack of generic phosphatases.18 By contrast, phosphorylation occurs by a protein kinase with broad substrate specificity and constitutively high activity,73 and the phosphorylated species comprises most of the cellular CDC37. Hence, phosphoserine serves as a structural building block in the ground state of wild-type CDC37, and selective dephosphorylation provides a stability switch that is based on the specific introduction of a nonphosphorylated side-chain with serine-like properties.
What are the possible implications of this stability switch? An estimate of the relative abundance of the phosphorylated state of endogenous CDC37 in MCF7 cells revealed that it is effectively the default state, with a minimum of 70–80% of CDC37 or CDC37 complexes being phosphorylated at Ser13. This may, in part, reflect the high activity of CK2. Assuming a dimeric state, the approach taken would potentially overestimate the degree of phosphorylation if a semiphosphorylated state is abundant. However, such an asymmetric state would also have significant functional consequences and, based on the micromolar estimates for dimer stability, is unlikely to be recovered at high yield under stringent immunoprecipitation conditions, let alone 3-fold sequential immunodepletion. Whether phosphorylation does modulate dimerization is not clear from existing and current data, although earlier studies have implicated dephosphorylation in the dissociation of CDC37 from the client HSP90 complex, which is preceded by the dissociation of the CDC37 dimer.17,18 Although our current data cannot distinguish direct interactions of the tail segment with the N–MC interface from allosteric interactions, the consequences of tail phosphorylation on the packing of CDC37 require the interaction of the N and MC-domains. This would couple changes in the client-binding N-domain to the dimerization and HSP90 binding MC-domains. Our observation that the exact nature by which Ser13 is altered is not relevant for the internal transitions of CDC37 may no longer apply at later stages of complex formation. Instead, interactions outside the CDC37 monomer may make direct use of the properties of the phosphate group. The phosphorylation state of CDC37 as it is released from these complexes may well impact its stability. Together with the abundance of CK2, this may contribute to the low abundance of the unphosphorylated state in a cellular setting. Previous studies had already highlighted the fact that potential phosphomimetics and noncharged replacements of serine alike generate similar phenotypes, both of which are deficient compared to the actual phosphorylation of Ser13.18,74 In a cellular setting with low CDC37 levels and high levels of HSP90, isolated CDC37–client complexes are very short-lived compared to the more stable ternary complex involving HSP90. It is, therefore, difficult to differentiate deficiencies in the processing of the ternary complex from a reduced ability of CDC37 to initially engage client. This may explain some of the divergent data on the impact of mutations in the N-terminal tail of CDC37 on its function. The specific contributions of the phosphorylated tail segment of CDC37 are likely to change as the nature of protein complexes is being rearranged during client maturation. While the current data are clearly limited in their ability to outline the nature of specific complexes, they provide new insight into dynamic transitions within the CDC37 cochaperone as it positions itself to engage client, and it provides an expanded and testable hypothesis of its mode of operation.
Supplementary Material
Acknowledgments
We thank Dr. Kerry Burnstein for CDC37 plasmid constructs, Dr. Sylvia Daunert for access to CD spectroscopic equipment, and Dr. Amjad Farooq (UM) and Dr. Ioannis Gelis (USF) for valuable discussions.
Funding
This work was supported by the NIH/NCI (R01-CA98881; R.L.) and the Braman Family Breast Cancer Institute (R.L.) as well as collaborative funding by the Bankhead-Coley Biomedical Research Program, 3BN08 (R.L., James N. Wilson).
Footnotes
Supporting Information
Figure S1 shows an estimate of the relative percent of Ser13 phosphorylated protein for recombinant CDC37 expressed in MCF7 cells. Figure S2 further evaluates client specificity of recombinant CDC37 (ERBB2 versus ERBB3 client) shown in Figure 2c. However, in this case, receptor immunoprecipitation was limited to cell surface localized and therefore mature receptor. Figure S3 further validates the client specificity of recombinant CDC37 but in reverse. In vitro phosphorylated, recombinant CDC37 was used as bait for binding studies using lysate of the naturally ERBB2 overexpressing BT474 breast cancer cell line. Figure S4 shows that both S13C and S13T mutations display ANS binding and fluorescence that mirrors that of wild-type CDC37 (complementing Figure 5). Figure S5 shows the CD spectrum of peptide representing residues 1–19 of CDC37 in isolation. Both the phosphorylated and unphosphorylated peptide display a CD spectrum characteristic of a random coil. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Pearl LH, Prodromou C. Structure, function, and mechanism of the Hsp90 molecular chaperone. Adv Protein Chem. 2001;59:157–186. doi: 10.1016/s0065-3233(01)59005-1. [DOI] [PubMed] [Google Scholar]
- 2.Loo MA, Jensen TJ, Cui L, Hou Y, Chang XB, Riordan JR. Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 1998;17:6879–6887. doi: 10.1093/emboj/17.23.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, Gurkan C, Kellner W, Matteson J, Plutner H, Riordan JR, Kelly JW, Yates JR, III, Balch WE. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 2006;127:803–815. doi: 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 4.Miyata Y. Protein kinase CK2 in health and disease: CK2: the kinase controlling the Hsp90 chaperone machinery. Cell Mol Life Sci. 2009;66:1840–1849. doi: 10.1007/s00018-009-9152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S. Quantitative analysis of HSP90–client interactions reveals principles of substrate recognition. Cell. 2012;150:987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee P, Rao J, Fliss A, Yang E, Garrett S, Caplan AJ. The Cdc37 protein kinase-binding domain is sufficient for protein kinase activity and cell viability. J Cell Biol. 2002;159:1051–1059. doi: 10.1083/jcb.200210121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tatebe H, Shiozaki K. Identification of Cdc37 as a novel regulator of the stress-responsive mitogen-activated protein kinase. Mol Cell Biol. 2003;23:5132–5142. doi: 10.1128/MCB.23.15.5132-5142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao J, Lee P, Benzeno S, Cardozo C, Albertus J, Robins DM, Caplan AJ. Functional interaction of human Cdc37 with the androgen receptor but not with the glucocorticoid receptor. J Biol Chem. 2001;276:5814–5820. doi: 10.1074/jbc.M007385200. [DOI] [PubMed] [Google Scholar]
- 9.Wang X, Grammatikakis N, Hu J. Role of p50/CDC37 in hepadnavirus assembly and replication. J Biol Chem. 2002;277:24361–24367. doi: 10.1074/jbc.M202198200. [DOI] [PubMed] [Google Scholar]
- 10.Wu F, Peacock SO, Rao S, Lemmon SK, Burnstein KL. Novel interaction between the co-chaperone Cdc37 and Rho GTPase exchange factor Vav3 promotes androgen receptor activity and prostate cancer growth. J Biol Chem. 2013;288:5463–5474. doi: 10.1074/jbc.M112.390963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith JR, Workman P. Targeting CDC37: an alternative, kinase-directed strategy for disruption of oncogenic chaperoning. Cell Cycle. 2009;8:362–372. doi: 10.4161/cc.8.3.7531. [DOI] [PubMed] [Google Scholar]
- 12.Smith JR, Clarke PA, de Billy E, Workman P. Silencing the cochaperone CDC37 destabilizes kinase clients and sensitizes cancer cells to HSP90 inhibitors. Oncogene. 2009;28:157–169. doi: 10.1038/onc.2008.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gray PJ, Jr, Stevenson MA, Calderwood SK. Targeting Cdc37 inhibits multiple signaling pathways and induces growth arrest in prostate cancer cells. Cancer Res. 2007;67:11942–11950. doi: 10.1158/0008-5472.CAN-07-3162. [DOI] [PubMed] [Google Scholar]
- 14.Stepanova L, Yang G, DeMayo F, Wheeler TM, Finegold M, Thompson TC, Harper JW. Induction of human Cdc37 in prostate cancer correlates with the ability of targeted Cdc37 expression to promote prostatic hyperplasia. Oncogene. 2000;19:2186–2193. doi: 10.1038/sj.onc.1203561. [DOI] [PubMed] [Google Scholar]
- 15.Siligardi G, Panaretou B, Meyer P, Singh S, Woolfson DN, Piper PW, Pearl LH, Prodromou C. Regulation of Hsp90 ATPase activity by the co-chaperone Cdc37p/p50cdc37. J Biol Chem. 2002;277:20151–20159. doi: 10.1074/jbc.M201287200. [DOI] [PubMed] [Google Scholar]
- 16.Mayer MP, Prodromou C, Frydman J. The Hsp90 mosaic: a picture emerges. Nat Struct Mol Biol. 2009;16:2–6. doi: 10.1038/nsmb0109-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaughan CK, Gohlke U, Sobott F, Good VM, Ali MM, Prodromou C, Robinson CV, Saibil HR, Pearl LH. Structure of an Hsp90–Cdc37–Cdk4 complex. Mol Cell. 2006;23:697–707. doi: 10.1016/j.molcel.2006.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaughan CK, Mollapour M, Smith JR, Truman A, Hu B, Good VM, Panaretou B, Neckers L, Clarke PA, Workman P, Piper PW, Prodromou C, Pearl LH. Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of Cdc37. Mol Cell. 2008;31:886–895. doi: 10.1016/j.molcel.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grammatikakis N, Lin JH, Grammatikakis A, Tsichlis PN, Cochran BH. p50(cdc37) acting in concert with Hsp90 is required for Raf-1 function. Mol Cell Biol. 1999;19:1661–1672. doi: 10.1128/mcb.19.3.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shao J, Irwin A, Hartson SD, Matts RL. Functional dissection of Cdc37: characterization of domain structure and amino acid residues critical for protein kinase binding. Biochemistry. 2003;42:12577–12588. doi: 10.1021/bi035138j. [DOI] [PubMed] [Google Scholar]
- 21.Roe SM, Ali MM, Meyer P, Vaughan CK, Panaretou B, Piper PW, Prodromou C, Pearl LH. The mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(Cdc37) Cell. 2004;116:87–98. doi: 10.1016/s0092-8674(03)01027-4. [DOI] [PubMed] [Google Scholar]
- 22.Shao J, Grammatikakis N, Scroggins BT, Uma S, Huang W, Chen JJ, Hartson SD, Matts RL. Hsp90 regulates p50(cdc37) function during the biogenesis of the activeconformation of the heme-regulated eIF2 alpha kinase. J Biol Chem. 2001;276:206–214. doi: 10.1074/jbc.M007583200. [DOI] [PubMed] [Google Scholar]
- 23.Zhang W, Hirshberg M, McLaughlin SH, Lazar GA, Grossmann JG, Nielsen PR, Sobott F, Robinson CV, Jackson SE, Laue ED. Biochemical and structural studies of the interaction of Cdc37 with Hsp90. J Mol Biol. 2004;340:891–907. doi: 10.1016/j.jmb.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 24.Bandhakavi S, McCann RO, Hanna DE, Glover CV. A positive feedback loop between protein kinase CKII and Cdc37 promotes the activity of multiple protein kinases. J Biol Chem. 2003;278:2829–2836. doi: 10.1074/jbc.M206662200. [DOI] [PubMed] [Google Scholar]
- 25.Mandal AK, Lee P, Chen JA, Nillegoda N, Heller A, DiStasio S, Oen H, Victor J, Nair DM, Brodsky JL, Caplan AJ. Cdc37 has distinct roles in protein kinase quality control that protect nascent chains from degradation and promote posttranslational maturation. J Cell Biol. 2007;176:319–328. doi: 10.1083/jcb.200604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sreeramulu S, Jonker HR, Langer T, Richter C, Lancaster CR, Schwalbe H. The human Cdc37·Hsp90 complex studied by heteronuclear NMR spectroscopy. J Biol Chem. 2009;284:3885–3896. doi: 10.1074/jbc.M806715200. [DOI] [PubMed] [Google Scholar]
- 27.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272:5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 28.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 29.Babu MM, Kriwacki RW, Pappu RV. Structural biology. Versatility from protein disorder. Science. 2012;337:1460–1461. doi: 10.1126/science.1228775. [DOI] [PubMed] [Google Scholar]
- 30.Espinoza-Fonseca LM. Reconciling binding mechanisms of intrinsically disordered proteins. Biochem Biophys Res Commun. 2009;382:479–482. doi: 10.1016/j.bbrc.2009.02.151. [DOI] [PubMed] [Google Scholar]
- 31.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 32.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Intrinsically disordered protein. J Mol Graphics Modell. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 33.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 34.Kar S, Sakaguchi K, Shimohigashi Y, Samaddar S, Banerjee R, Basu G, Swaminathan V, Kundu TK, Roy S. Effect of phosphorylation on the structure and fold of transactivation domain of p53. J Biol Chem. 2002;277:15579–15585. doi: 10.1074/jbc.M106915200. [DOI] [PubMed] [Google Scholar]
- 35.Lum JK, Neuweiler H, Fersht AR. Long-range modulation of chain motions within the intrinsically disordered transactivation domain of tumor suppressor p53. J Am Chem Soc. 2012;134:1617–1622. doi: 10.1021/ja2078619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McDowell C, Chen J, Chen J. Potential conformational heterogeneity of p53 bound to S100B(betabeta) J Mol Biol. 2013;425:999–1010. doi: 10.1016/j.jmb.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 37.Rajagopalan S, Jaulent AM, Wells M, Veprintsev DB, Fersht AR. 14-3-3 activation of DNA binding of p53 by enhancing its association into tetramers. Nucleic Acids Res. 2008;36:5983–5991. doi: 10.1093/nar/gkn598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Dieck J, Teufel DP, Jaulent AM, Fernandez-Fernandez MR, Rutherford TJ, Wyslouch-Cieszynska A, Fersht AR. Posttranslational modifications affect the interaction of S100 proteins with tumor suppressor p53. J Mol Biol. 2009;394:922–930. doi: 10.1016/j.jmb.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 39.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–584. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 40.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Uversky VN, Obradovic Z. Functional anthology of intrinsic disorder. 1 Biological processes and functions of proteins with long disordered regions. J Proteome Res. 2007;6:1882–1898. doi: 10.1021/pr060392u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ou L, Waddell MB, Kriwacki RW. Mechanism of cell cycle entry mediated by the intrinsically disordered protein p27(Kip1) ACS Chem Biol. 2012;7:678–682. doi: 10.1021/cb200487h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng Z, Xue B, Kurgan L, Uversky VN. Resilience of death: intrinsic disorder in proteins involved in the programmed cell death. Cell Death Differ. 2013;20:1257–1267. doi: 10.1038/cdd.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kovacs D, Tompa P. Diverse functional manifestations of intrinsic structural disorder in molecular chaperones. Biochem Soc Trans. 2012;40:963–968. doi: 10.1042/BST20120108. [DOI] [PubMed] [Google Scholar]
- 44.Bardwell JC, Jakob U. Conditional disorder in chaperone action. Trends Biochem Sci. 2012;37:517–525. doi: 10.1016/j.tibs.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tompa P, Kovacs D. Intrinsically disordered chaperones in plants and animals. Biochem Cell Biol. 2010;88:167–174. doi: 10.1139/o09-163. [DOI] [PubMed] [Google Scholar]
- 46.Iakoucheva LM, Radivojac P, Brown CJ, O’Connor TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eisenhaber B, Eisenhaber F. Posttranslational modifications and subcellular localization signals: indicators of sequence regions without inherent 3D structure? Curr Protein Pept Sci. 2007;8:197–203. doi: 10.2174/138920307780363424. [DOI] [PubMed] [Google Scholar]
- 48.Fukuchi S, Hosoda K, Homma K, Gojobori T, Nishikawa K. Binary classification of protein molecules into intrinsically disordered and ordered segments. BMC Struct Biol. 2011;11:29. doi: 10.1186/1472-6807-11-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kurotani A, Tokmakov AA, Kuroda Y, Fukami Y, Shinozaki K, Sakurai T. Correlations between predicted protein disorder and post-translational modifications in plants. Bioinformatics. 2014;30:1095–1103. doi: 10.1093/bioinformatics/btt762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bozoky Z, Krzeminski M, Chong PA, Forman-Kay JD. Structural changes of CFTR R region upon phosphor-ylation: a plastic platform for intramolecular and intermolecular interactions. FEBS J. 2013;280:4407–4416. doi: 10.1111/febs.12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen L, Brown JW, Mok YF, Hatters DM, McKnight CJ. The allosteric mechanism induced by protein kinase A (PKA) phosphorylation of dematin (band 4.9) J Biol Chem. 2013;288:8313–8320. doi: 10.1074/jbc.M112.438861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen YX, Du JT, Zhou LX, Liu XH, Zhao YF, Nakanishi H, Li YM. Alternative O-GlcNAcylation/O-phosphorylation of Ser16 induce different conformational disturbances to the N terminus of murine estrogen receptor beta. Chem Biol. 2006;13:937–944. doi: 10.1016/j.chembiol.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 53.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 54.Buchan DW, Minneci F, Nugent TC, Bryson K, Jones DT. Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Res. 2013;41:W349–357. doi: 10.1093/nar/gkt381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta. 2010;1804:996–1010. doi: 10.1016/j.bbapap.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Radivojac P, Obradovic Z, Brown CJ, AK D. Prediction of boundaries between intrinsically ordered and disordered protein regions. Pac Symp Biocomput. 2003:216–227. [PubMed] [Google Scholar]
- 57.Romero O, Dunker K. Sequence data analysis for long disordered regions prediction in the calcineurin family. Genome Inf Ser. 1997;8:110–124. [PubMed] [Google Scholar]
- 58.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 59.Li X, Romero P, Rani M, Dunker AK, Obradovic Z. Predicting protein disorder for N-, C-, and internal regions. Genome Inf Ser. 1999;10:30–40. [PubMed] [Google Scholar]
- 60.Miyata Y, Nishida E. Analysis of the CK2-dependent phosphorylation of serine 13 in Cdc37 using a phospho-specific antibody and phospho-affinity gel electrophoresis. FEBS J. 2007;274:5690–5703. doi: 10.1111/j.1742-4658.2007.06090.x. [DOI] [PubMed] [Google Scholar]
- 61.Miyata Y, Nishida E. CK2 controls multiple protein kinases by phosphorylating a kinase-targeting molecular chaperone, Cdc37. Mol Cell Biol. 2004;24:4065–4074. doi: 10.1128/MCB.24.9.4065-4074.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gerbin CS, Landgraf R. Geldanamycin selectively targets the nascent form of ERBB3 for degradation. Cell Stress Chaperones. 2010;15:529–544. doi: 10.1007/s12192-009-0166-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu W, Mimnaugh E, Rosser MF, Nicchitta C, Marcu M, Yarden Y, Neckers L. Sensitivity of mature Erbb2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J Biol Chem. 2001;276:3702–3708. doi: 10.1074/jbc.M006864200. [DOI] [PubMed] [Google Scholar]
- 64.Alimandi M, Romano A, Curia MC, Muraro R, Fedi P, Aaronson SA, Di Fiore PP, Kraus MH. Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene. 1995;10:1813–1821. [PubMed] [Google Scholar]
- 65.Kraus MH, Popescu NC, Amsbaugh SC, King CR. Overexpression of the EGF receptor-related proto-oncogene erbB-2 in human mammary tumor cell lines by different molecular mechanisms. EMBO J. 1987;6:605–610. doi: 10.1002/j.1460-2075.1987.tb04797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sreerama N, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 67.Fink AL, Calciano LJ, Goto Y, Kurotsu T, Palleros DR. Classification of acid denaturation of proteins: intermediates and unfolded states. Biochemistry. 1994;33:12504–12511. doi: 10.1021/bi00207a018. [DOI] [PubMed] [Google Scholar]
- 68.Eilers M, Schatz G. Binding of a specific ligand inhibits import of a purified precursor protein into mitochondria. Nature. 1986;322:228–232. doi: 10.1038/322228a0. [DOI] [PubMed] [Google Scholar]
- 69.Xu W, Mollapour M, Prodromou C, Wang S, Scroggins BT, Palchick Z, Beebe K, Siderius M, Lee MJ, Couvillon A, Trepel JB, Miyata Y, Matts R, Neckers L. Dynamic tyrosine phosphorylation modulates cycling of the HSP90–P50-(CDC37)–AHA1 chaperone machine. Mol Cell. 2012;47:434–443. doi: 10.1016/j.molcel.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zetina CR. A conserved helix-unfolding motif in the naturally unfolded proteins. Proteins. 2001;44:479–483. doi: 10.1002/prot.1113. [DOI] [PubMed] [Google Scholar]
- 71.Smart JL, McCammon JA. Phosphorylation stabilizes the N-termini of alpha-helices. Biopolymers. 1999;49:225–233. doi: 10.1002/(SICI)1097-0282(199903)49:3<225::AID-BIP4>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 72.Szilak L, Moitra J, Krylov D, Vinson C. Phosphorylation destabilizes alpha-helices. Nat Struct Biol. 1997;4:112–114. doi: 10.1038/nsb0297-112. [DOI] [PubMed] [Google Scholar]
- 73.Pinna LA. Protein kinase CK2: a challenge to canons. J Cell Sci. 2002;115:3873–3878. doi: 10.1242/jcs.00074. [DOI] [PubMed] [Google Scholar]
- 74.Shao J, Prince T, Hartson SD, Matts RL. Phosphorylation of serine 13 is required for the proper function of the Hsp90 co-chaperone, Cdc37. J Biol Chem. 2003;278:38117–38120. doi: 10.1074/jbc.C300330200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.