

Abstract
Failure in energy metabolism and oxidative damage are associated with Huntington’s disease (HD). Ascorbic acid released during synaptic activity inhibits use of neuronal glucose, favouring lactate uptake to sustain brain activity. Here, we observe a decreased expression of GLUT3 in STHdhQ111 cells (HD cells) and R6/2 mice (HD mice). Localisation of GLUT3 is decreased at the plasma membrane in HD cells affecting the modulation of glucose uptake by ascorbic acid. An ascorbic acid analogue without antioxidant activity is able to inhibit glucose uptake in HD cells. The impaired modulation of glucose uptake by ascorbic acid is directly related to ROS levels indicating that oxidative stress sequesters the ability of ascorbic acid to modulate glucose utilisation. Therefore, in HD, a decrease in GLUT3 localisation at the plasma membrane would contribute to an altered neuronal glucose uptake during resting periods while redox imbalance should contribute to metabolic failure during synaptic activity.
Keywords: Ascorbic acid, vitamin C, astrocyte-neuron lactate shuttle (ANLS), brain energy metabolism
Introduction
The brain is an energy-demanding organ that relies on glucose as its main energy source. Glutamate uptake stimulates glucose transport, glycolysis and lactate release in astrocytes (astrocyte neuron lactate shuttle, ANLS). Neurons take up the released lactate to sustain synaptic-activity-related energetic demands (Pellerin & Magistretti, 1994). Moreover, glutamatergic synaptic activity stimulates the release of ascorbic acid from glial reservoirs (Wilson, 2000). Neuronal sodium vitamin C co-transporters (SVCT2) allow the uptake of ascorbic acid, which inhibits glucose transport and stimulates lactate uptake in neurons through a GLUT3-dependent mechanism (ascorbic acid metabolic switch: Beltrán et al., 2011, Castro et al., 2009).
Huntington’s disease (HD) is caused by an expansion of the CAG trinucleotide (coding for glutamine) in the huntingtin (HTT) gene (The Huntington’s Disease Collaborative Research Group, 1993). HD and other neurodegenerative diseases are characterised by oxidative stress (Przedborski and Ischiropoulos, 2005; Ischiropoulos and Beckman, 2003; Wang et al., 2013; Mochel et al., 2011) and a failure in brain energy metabolism (Beltrán et al., 2013). Interestingly, a decreased expression of the glucose transporters GLUT1 and GLUT3 was observed in post-mortem HD brains (Gamberino & Brennan, 1994). Moreover, a recent study found a correlation between higher GLUT3 gene dosage and a delayed onset of HD in a cohort of 987 individuals (Vittori et al., 2014), showing that GLUT3 should play an important role in the progression of the disease and more than likely, in energy metabolism and neuronal cell death.
We have described an early failure in energy metabolism and an alteration in ascorbic acid flux from astrocytes to neurons in HD models (Acuña et al., 2013). We also found that replenishing extracellular ascorbic acid during electrophysiological recordings completely restored the wild-type phenotype in striatal neurons from presymptomatic R6/2 mice. However, we did not elucidate yet whether this effect was due to the recovery of the ascorbic acid metabolic switch or to a decreased level of reactive oxidant species in our HD model. Our present aim is to study ascorbic acid inhibition of GLUT3 and glucose transport in HD neuronal models.
We describe here a decreased GLUT3 expression in HD models that is accompanied by a lower cell surface localisation of GLUT3 transporters in biotinylation assays. Ascorbic acid was unable to inhibit glucose transport in HD cells. Ability to inhibit was completely restored by overexpressing GLUT3, suggesting that low levels of this transporter may account for impaired metabolic modulation in HD. To our surprise, an ascorbic acid analogue with no antioxidant activity, 2-Phospho-L-ascorbate (phospho-ascorbate), inhibited glucose transport in wild-type (WT) and HD cells. Phospho-ascorbate also exhibit the ability to modulate neuronal metabolism in HD mice as seen from electrophysiological measurements. Thus, an intrinsic property of ascorbic acid is key to the modulatory behaviour of this molecule, rather than its antioxidant properties. The observed elevated oxidative stress in HD would make vast use of ascorbic acid, thus reducing the availability of this molecule for metabolic modulation. We see our results as a promising step forward in the future development of possible therapies that aim to ameliorate symptoms and neurodegeneration in HD.
Methods
Animals
All R6/2 mice (male and female, 3–12 weeks old) used were obtained from the breeding colony maintained at the University of California, Los Angeles (UCLA). Offspring were obtained from mouse pairings consisting of either male R6/2 hemizygote x female WT (strain B6CBAF1) or male WT x female WT with an ovarian transplant from a R6/2 hemizygote (obtained from the Jackson Laboratory, Bar Harbor, ME). All experimental procedures were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee (IACUC) at UCLA. Animals were maintained in a 12:12 light:dark cycle environment and supplied with commercial food pellets and water ad libitum. Studies were performed on two age groups of R6/2 mice and on age-matched WT control mice based on development of the overt motor phenotype. The first group of animals was tested before the appearance of overt behavioural symptoms at 3–4 weeks (Carter et al., 1999). The second animal group was tested after the appearance of the full behavioural phenotype at 8–12 weeks (Carter et al., 1999). Efforts were made to minimise the number of animals used for experimental purposes.
Cell cultures
Cell lines: Mouse striatal STHdhQ7 and STHdhQ111 cells (Q7 and Q111 cells; Trettel et al., 2000) were grown using DMEM (United States Biological) containing 10% foetal bovine serum (FBS: HyClone, Logan, UT, USA), 50 U/ml penicillin, 50 mg/ml streptomycin, 50 ng/ml amphotericin B and 2 mM L–glutamine (Nalgene, Rochester, NY, USA). Differentiation of Q7 and Q111 cells was induced by serum-free DMEM containing 10 ng/ml alpha-FGF (PROMEGA), 240 uM IBMX, 20 uM TPA, 48.6 uM forskolin and 5 uM dopamine (SIGMA) during 24 h at 37 °C (Trettel et al., 2000). Cells were treated with cholesterol (SyntheChol), MβCD (methyl-β-cyclodextrin), L-ascorbic acid, Phospho-L- ascorbic acid and glutathione reduced ethyl ester (Sigma-Aldrich, USA). The glioma cell lines (C6, ATCC® number CCL 107™) were grown using DMEM-F12 (United States Biological, Swampscott, MA) containing 10% foetal bovine serum (FBS; HyClone, Logan, UT), 50 U/ml penicillin, 50mg/ml streptomycin, 50 ng/ml amphoreticin B, and 2 mM L-glutamine (Gibco Invitrogen Corporation, Grand Island, NY). Primary cultures: Striatal neurons were obtained from 17-day-old embryos of Wistar rats as described previously (Acuña et al., 2013, Beltrán et al., 2011). Adult female rats were anaesthetised using isoflurane (1-chloro-2,2,2-trifluoroethyl difluoromethyl ether), embryo forebrains were removed and the striatum dissected. Tissue was digested with 0.12% trypsin (wt/vol, Gibco Co., Rockville, MD, USA) in 0.1 M phosphate buffer (PBS: pH 7.4, osmolarity 320 mOsm) and mechanically disrupted with a fire-polished Pasteur pipette. Cells were plated at 0.3×106 cells/cm2 in plates onto coverslips coated with poly-L-lysine (mol. wt > 350 kDa, Sigma-Aldrich Corp. St. Louis, MO, USA). After 20 min, floating cells were removed and attached cells were cultured for 5–7 days in Neurobasal Medium (Gibco) supplemented with B27 (Gibco), 50 U/ml penicillin, 50 mg/ml streptomycin, 50 ng/ml amphotericin B and 2 mM L-glutamine (Nalgene).
Slice preparation
Mice were anaesthetised with isoflurane (1-chloro-2,2,2-trifluoroethyl difluoromethyl ether) for decapitation. Brains were removed into ice-cold low-Ca2+ oxygenated artificial cerebrospinal fluid (ACSF, in mM: 130 NaCl, 5 MgCl2, 1 CaCl2, 3 KC, 1.25 NaH2PO4, 26 NaHCO3 and 10 glucose) and coronal slices (350 mm) were obtained with a microslicer. Slices containing striatum and overlying cortex were maintained at room temperature (RT) in an incubation chamber filled with ACSF that was bubbled continuously with 95% O2 −5% CO2 for 1 h before being transferred to a laminar flow, thin-layer submersion recording chamber (Cepeda et al., 2003; Acuña et al., 2013).
Uptake assay using radiolabelled substances
Uptake assays were performed in 500 µl of incubation buffer (IB contains, in mM: 15 Hepes (pH 7.4), 135 NaCl, 5 KCl, 1.8 CaCl2, 0.8 MgCl2) containing 1–1.2 µCi of 2-deoxy-D-[3H]glucose [26.2 Ci/mmol; deoxyglucose (DOG), DuPont/NEN, Boston, MA]. Uptake was halted by washing cells with ice-cold IB containing 0.2mM HgCl2. Cells were dissolved in 200µl of lysis buffer (10mM Tris-HCl, pH 8.0, 0.2% SDS) and the incorporated radioactivity was measured by liquid scintillation spectrometry. For ascorbic acid effect assays (intracellular ascorbic acid), cells were preincubated in an IB containing ascorbic acid and 0.1mM DTT (IB-DTT) for 60 min at 37°C. Finally, 0.5mM DOG uptake was measured over a 10 sec period at RT.
Quantitative reverse transcription real-time PCR
The total RNA of R6/2 striatum and cell cultures was extracted with an E.Z.N.A.® Total RNA Kit II (Omega Biotek, Norcross, GA,USA) according to the manufacturer’s protocol. RNA quantification and purity were analysed with a spectrophotometer. cDNA was synthesised using the Maxima Universal First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Rockford, IL, USA) and PCR was performed using the Maxima SYBR Green qPCR Master Mix (Thermo Fisher Scientific). The following primers were used to analyse (i) GLUT3 expression, sense: 5’-TCATCTTCGCTGCCTTCCTCA-3’, antisense: 5’-CAGCACTCAGAAGCAGTCCTGGT-3’ (expected product size: 284 bp), (ii) β-actin expression, sense: 5’-TACCACCATGTACCCAGGCA-3’, antisense: 5’-CTCAGGAGGAGCAATGATCTTGAT-3’ (expected product size: 97 bp). A relative quantification of gene expression was performed, data represent the gene of interest in comparison to an internal control gene (actin) for each sample (2-ΔCt).
Western blot analysis
Total protein extracts were obtained from cell cultures. Cells were homogenised in buffer RIPA (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.1% sodium deoxycholate, 0.1% SDS, 5 mM EDTA) with 0.3% Triton X-100 and proteaseinhibitors: 2 µg/ml pepstatin A, 2 µg/ml leupeptin and 2 µg/ml aprotinin) and sonicated three to five times for 10 s at 4 °C. Proteins were resolved by SDS-PAGE (70 µg per lane for total protein extract) in a 10% (w/v) polyacrylamide gel, transferred to polyvinylidene difluoride membranes (0.45 mm pore; Amersham Pharmacia Biotech., Piscataway, NJ, USA) and probed with anti-GLUT3 (1:1,000, ab15311, Abcam, Cambridge, UK) and anti-β-actin antibodies (1:1,000, SC-81178, Santa Cruz Biotechnology, Santa Cruz, CA, USA). The reaction was developed using anti-rabbit and anti-mouse HRP-conjugated antibodies (1:10,000, 31460 and 31430, Thermo Fisher Scientific, Rockford, IL, USA) and the enhanced chemiluminescence Western blot method (Zambrano et al., 2010: Amersham Biosciences, Pittsburgh, PA, USA).
Immunofluorescence analyses
Cells grown on poly-L-lysine coated coverslips were fixed with HistoChoice® Tissue Fixative (Sigma-Aldrich, Chemical Co., St. Louis, MO). Samples were incubated overnight with antibodies anti-GLUT3 (1:100, SC-7582, Santa Cruz Biotechnology, CA, USA), anti-GM130 (1:100, BD 610823, BD Biosciences, CA, USA), anti-Calnexin (1:100, BD 610523, BD Biosciences, CA, USA), anti-EEA1 (1:100, SC-33585, Santa Cruz Biotechnology, CA, USA), anti-TfR (1:100, 136800, Invitrogen, Carlsbad, CA, USA), anti-MAP2 (1:100, ab36447, Abcam, MA, USA) and anti-Tau (1:100, ab64193, Abcam, MA, USA). Cells were washed and incubated with anti-rabbit, anti-mouse or anti-goat IgG-Alexa 568, IgG-Alexa 647 or IgG-Alexa 488 (1:300; A-10042, A-31571, A-11055, Invitrogen, Eugene, OR) and Hoechst stain solution (10 mg/ml; Sigma–Aldrich), and subsequently washed and mounted using fluorescence mounting medium (Dako, Carpinteria, CA). Cells were examined with an inverted Olympus FluoView 1000 confocal microscope (Castro et al., 2007, 2008; Acuña et al., 2013).
Biotinylation of surface proteins
Biotinylation assays were performed as previously described, with modifications by Acuña et al., 2013. Briefly, cells (1 × 108) were labelled with Sulfo-NHS-LC-Biotin (Thermo Fisher Scientific, Rockford, IL, USA). Cells were washed three times in ice-cold PBS 1X Ca++/Mg++ (pH 7.4) to remove any contaminating proteins and 1mM of Sulfo-NHS-LC-Biotin diluted in ice-cold PBS 1X Ca++/Mg++ (pH 7.4) was added per total reaction volume. Cells labelled with biotin were incubated at 4 °C for 30 min, washed three times with ice-cold PBS 1X Ca++/Mg ++ (pH 7.4) and the free biotin was blocked with a solution of 50 mM Tris-HCl and again washed three times with ice-cold PBS 1X Ca++/Mg++ (pH 7.4). The labelled cells were homogenised in 200 µl of buffer RIPA with Triton X-100 and protease inhibitors and then precipitated by continuous mixing with 40 µl of immobilised NeutrAvidin Protein (Thermo Fisher Scientific, Rockford, IL, USA) for 4 hours at 4 °C. The precipitates were washed four times in ice-cold PBS 1X Ca++/Mg++ (pH 7.4). The resin-bound complex was boiled in SDS-PAGE sample buffer. Samples were resolved by 10% (w/v) SDS– polyacrylamide gels and Western blot analysis was performed.
Live cell imaging of fluorescent glucose analogue uptake
Time course uptake assays were performed in 400 µl of IB containing 0.5 mM 2-NBDG (2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxyglucose; Invitrogen), a fluorescent analogue of glucose. Fluorescent incorporation into cells was examined using a Fluoview 1000 confocal microscope. The fluorescence signal was normalised according to:
| (1) |
where F0 correspond to the mean of fluorescence intensities measured in the first 10 frames. For experiments in the presence of intracellular ascorbic acid, cells were preloaded using a solution of 1 mM ascorbic acid, for 60 min at 37 °C (Beltrán et al., 2011).
TIRF microscopy analysis
Cells expressing GLUT3-EGFP were visualised with an objective-based total internal reflection fluorescence (TIRF) microscope. A 473-nm solid-state diode laser was focused onto a single-mode optical fibre and transmitted via the rear illumination port of an Olympus IX71 inverted microscope. Laser light was reflected on a dichroic mirror (Chroma Technology), passed through a high numerical aperture objective (x 60, NA 1.49, oil immersion, Olympus) and was totally internally reflected at the glass-water interface. Under these experimental conditions, fluorescent objects were observed at the evanescent field (180 nm of the glass-water interface under our experimental conditions). Treatments were performed with 10 mM MβCD for 15 minutes and with 30 µg/ml SyntheChol for 20 minutes.
ROS determination in living cells
Production of reactive oxygen species (ROS) was measured by oxidation of DCFH-DA (2′,7′-dichlorofluoroscein-diacetate) to fluorescent DCF as previously described with modifications by He et al., 2003. Cells were seeded onto 96 well plates at a density of 1.5 × 104. They were preincubated in the presence of 0 or 1 mM ascorbic acid for 30 minutes. After this, 5 µg/ml DCFH-DA was added. Fluorescence intensity was measured using a Synergy 2 Multi-Mode microplate reader with filters at 485 nm excitation and 520 nm emission. Units of relative fluorescence (RFU) were recorded at 40 s intervals for 10 min periods.
Plasmids and transfection
Plasmid constructs, GLUT3-EGFP, encoding full-length rat GLUT3 fused at their C termini to EGFP were kindly provided by Dr. J.P. Bolaños (Departamento de Bioquìmica y Biologìa Molecular, Universidad de Salamanca, Spain). GLUT3-mCherry, encoding full-length GLUT3 fused at their C termini to mCherry (red fluorescent protein) was subcloned from GLUT3-EGFP to pcDNA 3.1-mCherry with the restriction enzymes Xba and XhoI. Plasmid constructs, mHtt-dsRed, encoding amino acids 1–588 of human Htt containing 138 glutamine repeats fused to dsRed were kindly provided by Dr C. Hetz (Instituto de Ciencias Biomédicas, Universidad de Chile, Santiago, Chile). Cells maintained in Optimem defined media (Gibco Invitrogen Corporation, Grand Island, NY) were transfected using Lipofectamine 2000 (Gibco Invitrogen Corporation, Grand Island, NY) according to the manufacturer’s instructions.
Electrophysiological assays
Recordings were made from medium-sized spiny neurons from the striatum using the whole-cell patch-clamp configuration in voltage-clamp. Medium-sized spiny neurons were visualized in the dorsolateral striatum using infrared illumination with differential interference contrast optics (IR-DIC microscopy) and identified by their somatic size and basic membrane properties. A −70mV holding potential was maintained during the experiment. Electrophysiological data were acquired at 10 Khz. Recording electrodes consisting of borosilicate glass had an impedance of 5–7MO. The pipette internal solution was: 131 mmol/L potassium gluconate, 1 mmol/L EGTA, 1 mmol/L MgCl2, 2 mmol/L ATP-K2, 0.3 mmol/L NMDA GTP-Na, 6 mmol/L KCl, 1 mmol/L NaCl and 5 mmol/L HEPES (pH 7.3) osmolarity 290 mOsm/L. Postsynaptic currents were evoked by a tungsten electrode placed into the corpus callosum to stimulate the corticostriatal pathway. Stimulation pulses were 0.1–0.2 ms in duration of varying amplitude and were applied every 200 ms. pCLAMP 8 software (Axon Instruments) was used to generate stimuli and for data display, acquisition and storage (Castro et al. 2007, Acuña et al., 2013). Drugs were superfused with gassed ACSF (4-CIN) or were included within the patch pipette (ascorbic acid, 2-phospho-ascorbate). During ascorbic acid (Asc) and Phospho-L-ascorbic acid (P-Asc) recordings anti-SVCT2 antibody (SC-9926, Santa Cruz Biotechnology, CA, USA) was included within the patch pipette.
Statistical analyses
Statistical comparison between two or more groups of data was respectively performed using a Student`s t-test or analysis of variance (ANOVA) plus Bonferroni post-test. Regressions were calculated using SigmaPlot v9.0 software (correlations >0.9 were accepted).
Results
Ascorbic acid modulation of glucose uptake is impaired in HD
As was mentioned above, we showed an impaired ascorbic acid metabolic switch in HD (Acuña et al., 2013). But we did not study glucose use in HD cells. We have now investigated this in detail using an HD cell model and studying GLUT3 expression in STHdhQ7 and STHdhQ111 cell lines. These cell lines correspond to immortalised striatal neurons obtained from HD mouse knock-in embryos, expressing endogenous levels of normal (Q7, WT phenotype) and mutant (Q111, HD phenotype) huntingtin (Htt) respectively. To determine whether intracellular ascorbic acid was able to inhibit glucose uptake in HD cells, we measured radio-labelled deoxyglucose (DOG) in both Q cell types (Fig. 1b). DOG is a glucose analogue that is translocated through the plasma membrane by GLUTs (Maher et al., 1996). Then, Q cells were incubated with ascorbic acid for 60 min at 37°C, which was then rapidly replaced with a DOG containing solution. Ascorbic acid rapidly inhibited DOG uptake (10 s) in Q7 cells that had been preloaded with 1 mM ascorbic acid. However, no effect was seen in similar experiments with Q111 cells (Fig. 1b). Thus, we conclude that intracellular ascorbic acid is unable to efficiently modulate glucose consumption in HD cells. DOG uptake in absence of acorbic acid (basal uptake of DOG) was decreased in Q111 cells (Fig. 1a). We have previously demonstrated that ascorbic acid turns neuronal glucose consumption off through inhibition of GLUT3 (Beltrán et al., 2011). Thus, we decide to investigate whether the lack of ascorbic acid effect on DOG uptake and the decrease in basal DOG uptake may be related to a deficiency in GLUT3 expression in Q111 cells.
Figure 1. Glucose uptake inhibition by intracellular ascorbic acid is impaired in HD cells.
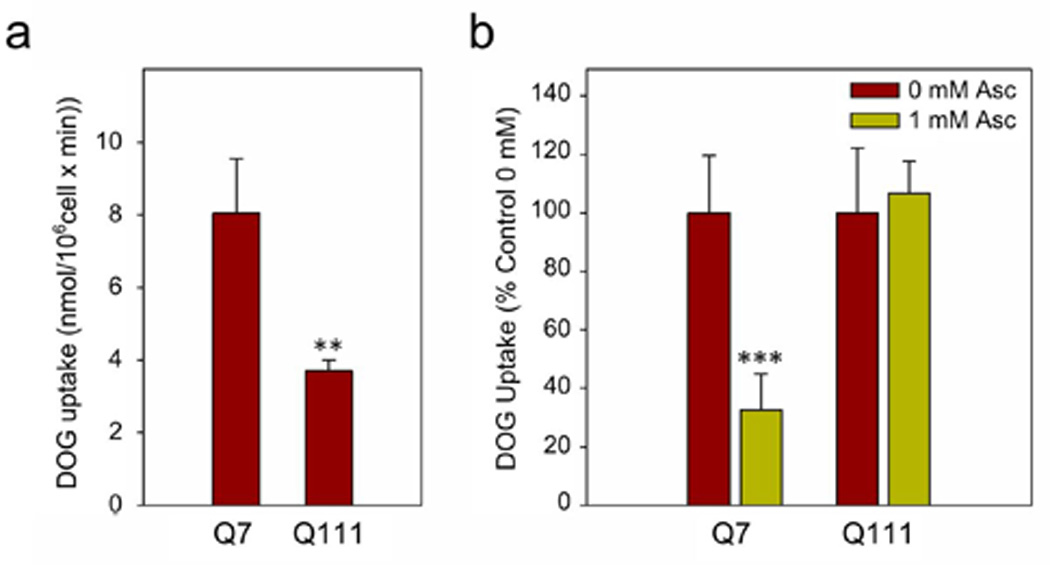
(a) 3H-deoxyglucose (DOG) transport analysis using a 10-sec uptake assay in Q7 and Q111 cells. Bars plot for absolute values of DOG uptake. Student’s t-test, n=3, ** p<0.01. (b) 3H-deoxyglucose (DOG) transport analysis using a 10 s uptake assay in the presence of intracellular ascorbic acid. Q7 and Q111 cells were preloaded in a 1 mM ascorbic acid (Asc) solution for 60 min at 37 °C. Student’s t-test, n=3, *** p<0.001.
GLUT3 expression at the plasma membrane is impaired in HD
We analysed mRNA and protein levels in samples obtained from Q cells and R6/2 mice striatum (Fig. 2). R6/2 is a transgenic mouse model that expresses a mutation on exon 1 of human Htt protein (containing approximately 150 CAG repeats), and it is characterized by an early abnormal brain metabolism (Cepeda-Prado et al., 2012). RT-qPCR analyses showed decreased levels of mRNA coding for GLUT3 in presymptomatic R6/2 mice (Fig. 2a). Thus, in the first stages of HD, GLUT3 expression is impaired. We also measured mRNA levels coding for GLUT3 in Q cells. A decrease in GLUT3 expression was seen in Q111 cells (Fig. 2b). Also Q111 cells showed a decrease in relative GLUT3 protein levels (Fig. 2C). We did not observe a decrease in the intensity of fluorescence corresponding to the immunoreactivity to GLUT3 in Q111 cells (Fig. 2d). This could be explained by the different shape and size observed between Q7 and Q111 cells. While Q7 cells exhibit a widespread cytoplasm, Q111 are more compact (Trettel et al., 2000). No apparent changes in subcellular distribution were evidenced by immunofluorescence (Fig. 2d).
Figure 2. Low expression of GLUT3 in HD mice and HD cells.
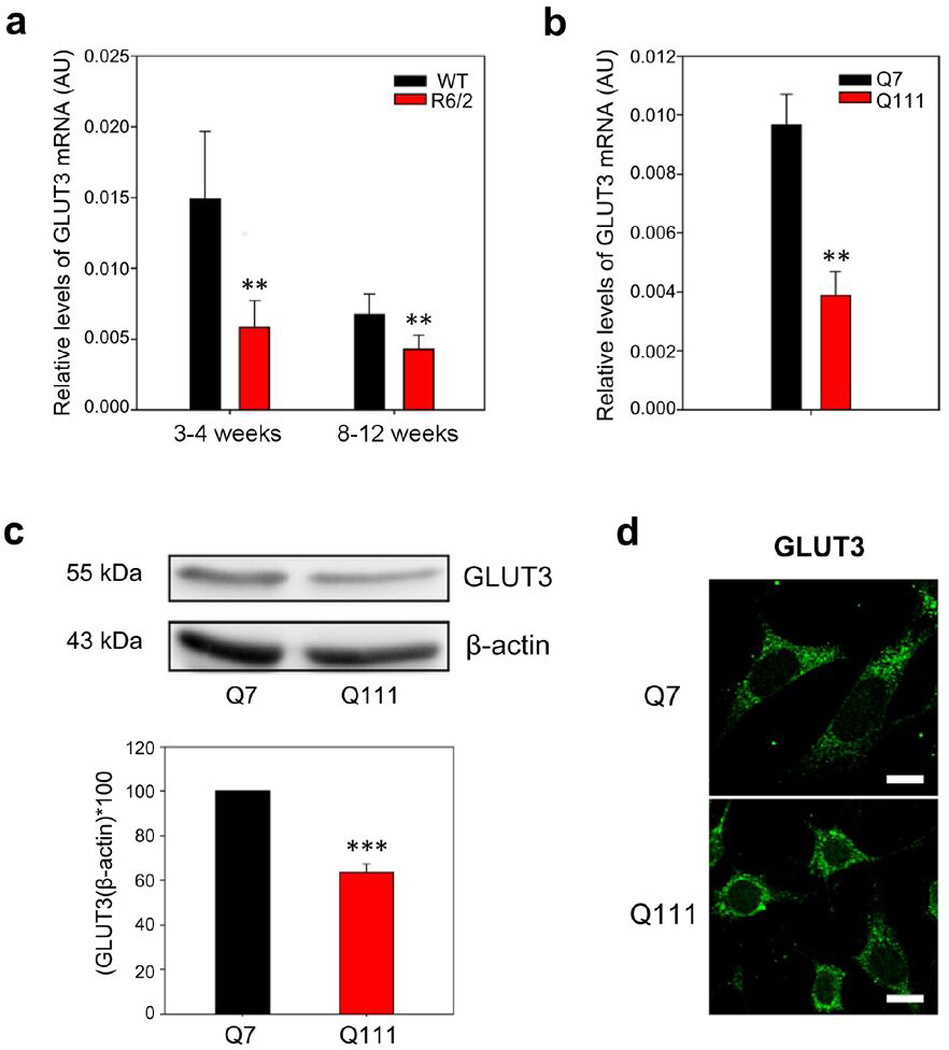
(a) qPCR analyses for GLUT3 from mRNA extracts of striatum from presymptomatic (3–4 weeks old) and symptomatic (8–12 weeks old) R6/2 mice. Striatum samples from littermates (WT) were used as controls. Results were normalised using specific primers to amplify mRNAs coding for β-actin. Analysis of variance (ANOVA) followed by the Bonferroni post-test,, n=4, ** p<0.01 and * p<0.05. (b) qPCR analyses for mRNA coding for GLUT3 in Q7 and Q111 cells. Student’s t-test, n=4, ** p<0.01. (c) Western blot assay for GLUT3 of total protein extracts from Q7 and Q111 cells. Bar plots show quantification of GLUT3 Western blot by densitometric scanning analysis, n=3, *** p<0.001, ** p<0.01. (d) Immunofluorescence analyses for GLUT3 (green) in Q7 and Q111 cells. Representative images for four independent experiments. Scale bar is 10 µm.
Htt is an essential integrator of intracellular transport of vesicles, organelles and trafficking of proteins to the cell surface (Caviston et al., 2009; Twelvetrees et al., 2010). With that in mind, we decided to study the subcellular localisation of GLUT3 in Q cells. By means of colocalisation assays and immunofluorescence analysis in Q7 cells, we observed a partial colocalisation of GLUT3 (in green, Fig 3a, 3b and 3c) with at least three different intracellular compartments: the Golgi apparatus (GM130, in red, Fig. 3a), endoplasmic reticulum (Calnexin, in red, Fig. 3b), and early endosomes (EEA1, in red, Fig. 3c). Using a confined displacement algorithm (Ramírez et al., 2010) we observed no changes in colocalisation in Q111 cells (Fig 3a, 3b and 3c). No apparent polarisation of GLUT3 was observed (Fig. 3d). GLUT3 was equally distributed in axons and dendrites (Tau, in red and MAP2, in cyan, respectively: Fig. 3d). However, a decrease in GLUT3 expression at the plasma membrane was determined in Q111 cells by means of biotinylation assays (Fig 3e, 3f).
Figure 3. GLUT3 expression at the cell surface is decreased in HD cells.
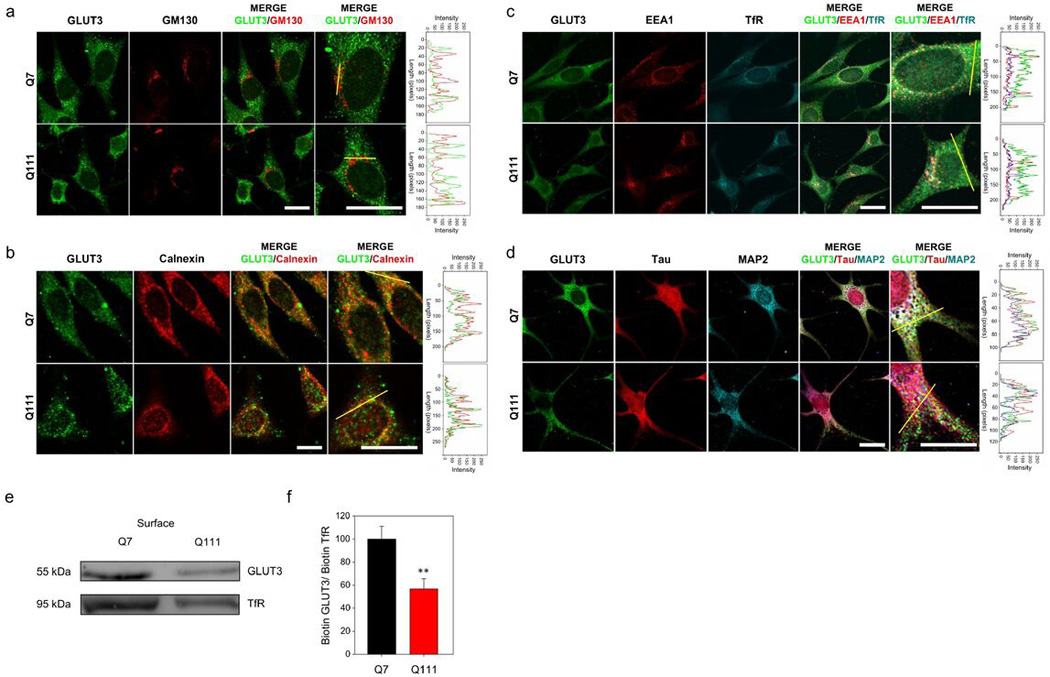
(a, b, c and d) Immunofluorescence analyses in Q7 and Q111 cells for GLUT3 (green) and GM130 (red) in (a), for Calnexin (red) in (b), for EEA1 (red) and TfR (cyan) in (c), and for TAU (red) and MAP2 (cyan) in (d). Merged images and magnification of the merged images is shown. Line-scan data for yellow lines drawn across cells in the merged panels is shown at right of each set. Representative images for three independent experiments. Scale bars are 10 µm. (e) Western blot analysis of biotinylated proteins from Q7 and Q111 cells. (f) Bar plots for densitometric scanning analysis of the GLUT3 Western blot. Student’s t-test, n=4, ** p<0.01.
GLUT3 overexpression recovers the modulation of glucose uptake otherwise impaired in HD cells
We have already shown that a decrease in both GLUT3 expression and GLUT3 localisation occurs at the cell surface in Q111 cells. We have also observed that ascorbic acid mediated inhibition of glucose uptake is impaired in these cells. Because the effect of ascorbic acid on glucose uptake occurs only in cells expressing functional GLUT3 (Beltrán et al., 2011), we reasoned that GLUT3 overexpression in Q111 cells may increase the number of transporters at the cell surface and perhaps correct the impaired inhibition of glucose uptake. To test this idea we performed kinetics studies using the fluorescent glucose analogue 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxyglucose (2-NBDG) in Q cells and in primary cultures of striatal neurons (Fig. 4). Uptake of 2-NBDG was evidenced by an increase in cytoplasmic fluorescence (Fig. 4b and 4d). In Q7 cells, 2-NBDG uptake was nearly 5 fold larger than the rate of uptake seen in Q111 cells (Fig. 4a and 4b, closed circles. The initial velocity for Q7 cells in the presence of 0 mM ascorbic acid was 1.5×10−3 +/− 4.8×10−5 arbitrary units/sec, in contrast, the initial velocity for Q111 cells in the presence of 0 mM ascorbic acid was 3.2×10−4 +/− 3.0×10−5 arbitrary units/sec. Likewise, 2-NBDG uptake was 4.6 times higher in WT neurons than the rate of uptake observed in cultured neurons expressing mHtt-dsRed (Fig. 4d, 4e and 4f, closed circles). The initial velocity for WT neurons in the presence of 0 mM ascorbic acid was 1.4×10−2 +/−3.7×10−5 arbitrary units/sec, while the initial velocity for neurons expressing mHtt in the presence of 0 mM ascorbic acid was increased up to 3.2×10−3 +/− 5.2×10−5 arbitrary units/sec. As for the previous experiments using DOG (Fig 1 and Castro et al., 2007), 2-NBDG uptake was inhibited by intracellular ascorbic acid in Q7 cells (Fig. 4a, open circles). The initial velocity for Q7 cells in the presence of 1 mM ascorbic acid was 3.5×10−4 +/− 2.9×10−5 arbitrary units/sec and cultured neurons (Fig. 4d, open circles), and the initial velocity for cultured neurons in the presence of 1 mM ascorbic acid was 7.2×10−6 +/− 3.9×10−7 arbitrary units/sec. As expected, 2-NBDG uptake was not inhibited in cells expressing mHtt (Fig. 4b and e, open circles). The initial velocity for Q111 cells in the presence of 1 mM ascorbic acid was 1.1×10−4 +/− 6.2×10−6 arbitrary untits/sec (Fig. 4e, open circles) and the initial velocity for neurons expressing mHtt-dsRed in the presence of 1 mM ascorbic acid was 7.2×10−6 +/− 5.3×10−7 arbitrary untits/sec. GLUT3 overexpression induced an increase in the rate of 2-NBDG uptake (6.8, 24.9, 1.3 and 1.7 fold for Q7 cells, Q111 cells, cultured neurons and neurons expressing mHtt-dsRed respectively: Fig. 4, red triangles). The initial velocity for Q cells overexpressing GLUT3-mCherry in the presence of 0 mM ascorbic acid was 1.1×10−2 +/− 5.3×10−4 arbitrary units/sec (Q7) and 8.0×10−3 +/− 5.6×10−4 arbitrary units/sec (Q111). The initial velocity for primary cultures of striatal neurons overexpressing GLUT3-EGFP in the presence of 0 mM ascorbic acid was 2.0×10−2 +/− 3.1×10−4 arbitrary units/sec (cultured neurons) and 1.9×10−3 +/− 3.6×10−4 arbitrary unts/sec (cultured neurons expressing mHtt-dsRed). All the data on transport kinetics suggest an increased expression of functional GLUT3 at the plasma membrane. In fact, a marked intensity at the plasma membrane is seen in transfected cells by direct fluorescence (in red, Fig. 4c and in green, Fig. 4f) and by inmunofluoresence (Fig. 5a). The effect of ascorbic acid on 2-NBDG uptake showed that HD cells (Q111 cells and cultured neurons expressing mHtt) overexpressing GLUT3 behaved in the same way as WT cells (Q7 and cultured neurons), that is, ascorbic acid inhibited 2-NBDG uptake. The initial velocity for Q7 cells overexpressing GLUT3-mCherry in the presence of 1 mM ascorbic acid was 4.5×10−5 +/− 2.1×10−6 arbitrary units/sec; the initial velocity for cultured neurons overexpressing GLUT3-mCherry in the presence of 1 mM ascorbic acid was 2.1×10−8 +/− 1.9×10−9 arbitrary units/sec; the initial velocity for Q111 cells overexpressing GLUT3-mCherry in the presence of 1 mM ascorbic acid was 1.7×10−4 +/− 1.9×10−5 arbitrary units/sec and the initial velocity for cultured neurons expressing mHtt-dsRed and overexpressing GLUT3-mCherry in the presence of 1 mM ascorbic acid was 3.7×10−4 +/−1.1×10−5 arbitrary units/sec. Thus, we conclude that GLUT3 overexpression is enough to rescue the impairment of ascorbic acid-induced glucose uptake modulation in Q111 cellss.
Figure 4. Overexpression of GLUT3 rescues the ability of ascorbic acid to inhibit glucose analogue uptake in HD cells.
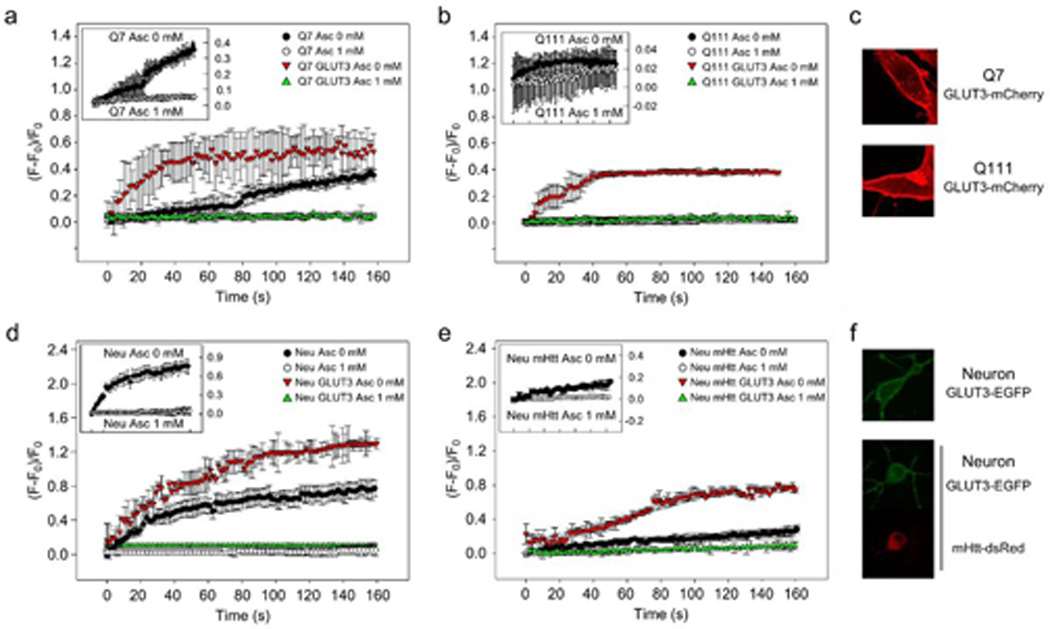
(a, b, c) 2-NBDG transport in Q7 (a) and Q111 (b) cells. 2-NBDG transport was performed in the absence (closed circles and red triangles) or presence (open circles and green triangles) of intracellular ascorbic acid and where indicated, in GLUT3-mCherry expressing cells (red and green triangles). For clarification purposes, the inset shows plots for Q7 and Q111 cells (open and closed circles). GLUT3-mCherry expression is shown in red (c). n=4. (d, e, f) 2-NBDG transport in primary cultures of striatal neurons (d) and primary cultures of striatal neurons expressing mHtt-dsRed (e). 2-NBDG transport was performed in the absence (closed circles and red triangles) or presence (open circles and green triangles) of intracellular ascorbic acid and where indicated, in GLUT3-EGFP expressing cells (red and green triangles). For clarification purposes, the inset shows plots for cultured neurons and cultured neurons expressing mHtt-dsRed (open and closed circles). GLUT3-EGFP and mHtt-dsRed expression is shown in green and red, respectively (f). n=4.
Figure 5. GLUT3 behaviour at the plasma membrane does not change in HD cells.

(a) Immnufluorescence analyses in Q7 and Q111 cells for GLUT3 (green). Direct fluorescence corresponding to GLUT3-mCherry is shown in red. Line-scan data for yellow lines drawn across cells in the merged panels is shown. (b) TIRF microscopy images for representative Q7 and Q111 cells expressing GLUT3-EGFP. Cells were treated with cholesterol (30 µg/ml) or methyl-β-cyclodextrin (MβCD: 10 mM) for the time indicated. Bar plots for normalised fluorescence intensity obtained from TIRF microscopy images in Q cells expressing GLUT3-EGFP. Analysis of variance (ANOVA) followed by the Bonferroni post-test, n=3.
It has been described that cholesterol and fatty acid metabolism are altered in HD (Block et al., 2010), thus, it is possible that GLUT3 localisation at cellular surface could be associated to changes in cholesterol concentration and/or distribution at plasma membrane. To test this idea we measured GLUT3-mCherry using total internal reflection microscopy, TIRM. The pattern of GLUT3-mCherry at the cellular surface was seen to be similar in Q7 and Q111 cells and it was unaffected by changes in the content of plasma membrane cholesterol induced by incubations with Methyl-β–cyclodextrin (MβCD) and cholesterol(Fig. 5b).
Redox imbalance sequesters the ability of ascorbic acid to modulate glucose uptake
Maintaining a redox balance is essential for normal cellular functions. A redox imbalance has been described for neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease and HD (Przedborski and Ischiropoulos, 2005; Ischiropoulos and Beckman, 2003; Wang et al., 2013; Mochel et al., 2011). Ascorbic acid, a potent antioxidant (Padh, 1990) is used in the brain to maintain a redox balance. Thus, under conditions of oxidative stress, ascorbic acid may be consumed, leaving less-than-enough molecules available for inhibition of glucose uptake. Elevated ROS levels were observed in intact Q111 cells (Fig. 6a). Antioxidants such as ascorbic acid and glutathione (GSH) demonstrate their capacity to reduce ROS in Q7 and other cells (Fig. 6b). No significant reduction of ROS levels was observed in Q111 cells in presence of GSH (Fig. 6b). Knowing this, we tested whether other antioxidants might be able to inhibit glucose uptake in a mechanism that depends on the presence of GLUT3. To do this, we measured radiolabelled DOG uptake in the presence of intracellular GSH in Q cells and glioma C6 cells (Fig. 6c). In a previous work, we demonstrated that DOG uptake was inhibited by intracellular ascorbic acid, likely by a mechanism that depends on the expression of GLUT3 in C6 cells (Beltran et al., 2011). In that study, by using shRNA, we engineered a stable C6 cell line (C6 shGLUT3) in which the expression of GLUT3 was suppressed up to 50% (Beltran et al., 2011). Intracellular GSH was able to strongly inhibit DOG uptake in glioma C6 cells (Fig. 6c). A less significant effect was observed in C6 shGLUT3 (Fig. 6c). However, no significant changes were observed in Q7 and Q111 cells (Fig. 6c). Inhibition of DOG uptake by GSH was GLUT3 independent (Fi. 6c), suggesting that GSH may directly inhibit glucose uptake, by reducing GLUT3 residues, or it may revert oxidative stress making endogenous ascorbic acid available for modulation of glucose uptake.
Figure 6. HD cells show elevated ROS levels.
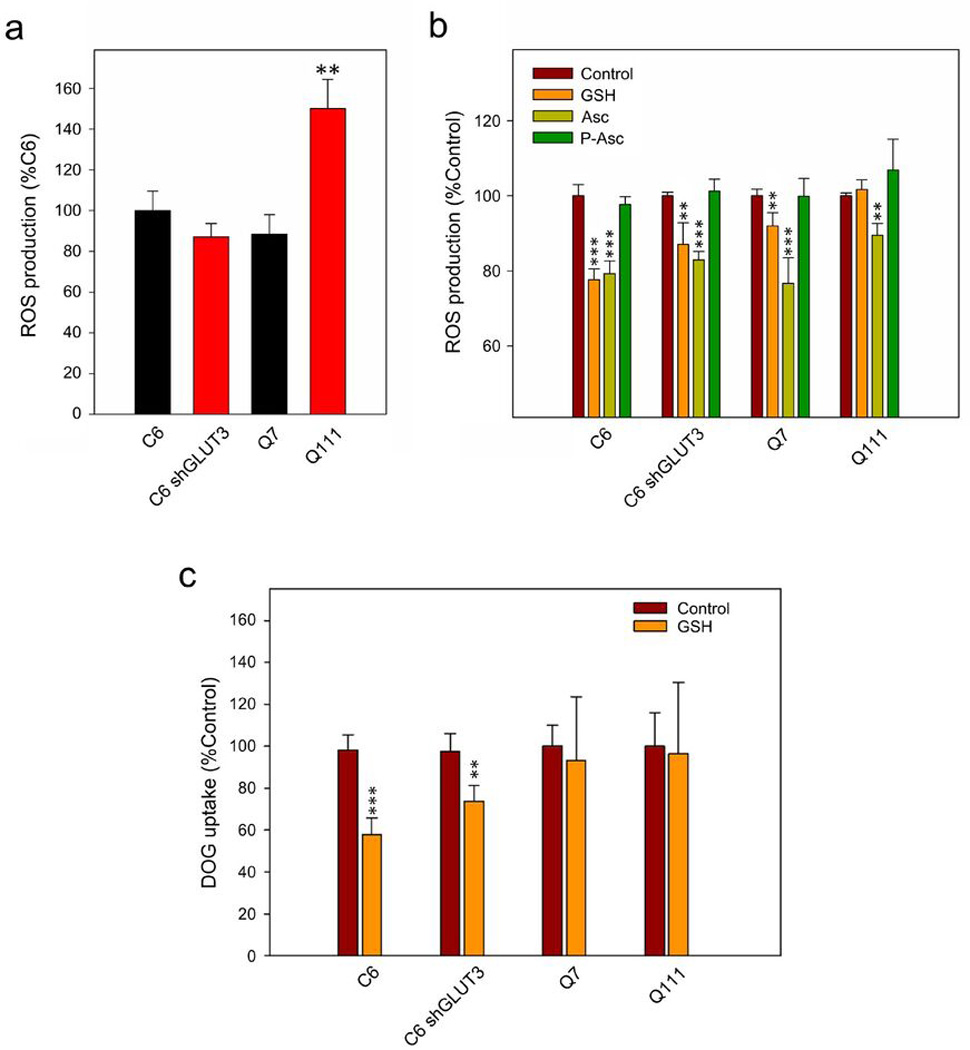
(a) ROS production determination in C6, C6-shGLUT3, Q7 and Q111 cells. Analysis of variance (ANOVA) followed by the Bonferroni post-test, n=3, ** p<0.01. (b) Decrease in ROS production by ascorbic acid (Asc), gluthathione (GSH) and phospho-ascorbate (P-Asc). Cells were preincubated with or without 1mM ascorbic acid and DCFH-DA (5 µg/ml) for 30 min at 37 °C. Fluorescence intensity of each well was measured in a plate reader at 485 nm (excitation) and 530 nm (emission) wavelengths. Relative fluorescence units (RFU) were recorded at 4 s intervals for a period of 10 min. Analysis of variance (ANOVA) followed by the Bonferroni post-test, n=3, *** p<0.001, ** p<0.01. (c) 3H-deoxyglucose (DOG) transport analysis using a 10-sec uptake assay (37 °C) in C6, C6-shGLUT3 and Q7 and Q111 cells. Cells were preincubated for 15 min with 0.01 mM GSH (GSH). Analysis of variance (ANOVA) followed by the Bonferroni post-test, n=3, *** p<0.001, ** p<0.01
To investigate this possibility, we used an ascorbic acid analogue, phospho-ascorbate, which lacks antioxidant properties (Wilson et al., 1996). Accordingly, phospho-ascorbate was unable to reduce ROS levels in Q111 cells (Fig. 6b). In previous works we have shown that intracellular ascorbic acid inhibits the ability of glucose to sustain excitatory postsynaptic currents (EPSCs) when neuronal lactate uptake is inhibited (Castro et al., 2007, Acuña et al., 2013). In the present study, we recorded EPSCs in medium-sized spiny neurons from WT mouse striatal slices by stimulating the corticostriatal pathway (Fig 7a and 7b). After glucose deprivation, glucose application produced a rapid recovery of synaptic responses (100 +/− 7.0 %). According to our data (Castro et al., 2007, Acuña et al., 2013), restoration of EPSCs by glucose was less efficient (32.9 +/− 11.6 %, Fig. 7b, Control, 4CIN) in the presence of external 100 µM alpha-cyano-4-hydroxycinnamic acid (4-CIN), an inhibitor of the neuronal monocarboxylate transporter MCT2. Therefore, the inhibition of neuronal monocarboxylate uptake produces a drop in the synaptic activity even in the presence of glucose. When ascorbic acid uptake through SVCT2 is inhibited by using an intracellular anti-SVCT2 antibody, glucose is now able to sustain EPSCs in the presence of 200 µM 4-CIN (Figure 7b). This antibody inhibits ascorbic acid uptake by binding to the endofacial side of the ascorbic acid transporter, SVCT2 (Castro et al., 2007; Acuña et al., 2013). To test the effect of intracellular ascorbic acid or phospho-ascorbate on the ability of glucose to sustain synaptic activity (which is a consequence of the modulation of the glucose uptake), we performed a set of experiments using either ascorbic acid or phospho-ascorbate together with the antibody anti-SVCT2 in the recording pipette. Both ascorbic acid and phospho-ascorbate were able to inhibit glucose ability to sustain synaptic responses (ascorbic acid and phospho-ascorbate were able to inhibit glucose consumption, 29.2 +/− 0.3 % for ascorbic acid and 30.7 +/− 5.6 % for phospho-ascorbate, Fig 7a and 7b, Asc and P-Asc). In good agreement with these results, phospho-ascorbate was able to inhibit DOG uptake at the same rate as ascorbic acid in Q7 cells (Fig. 7c). However, phospho-ascorbate inhibited DOG uptake in Q111 cells while ascorbic acid did not modulate uptake in these cells (Fig. 7c, Fig. 1). Phospho-ascorbate is able to modulate glucose consume while it is not able to act as antioxidant. On the other hand, ascorbic acid is an antioxidant able to modulate glucose consumption. Because Q111 cells exhibit high levels of oxidant species (Fig. 6a), we reasoned that in Q111 cells, oxidative stress might be “sequestering” ascorbic acid to maintain the redox balance. This can be understood as a displacement in the ascorbic acid redox equilibrium, making the interaction of the oxidised molecule with targets (i.e. proteins associated with the modulation of DOG uptake) less likely. In contraposition, phospho-ascorbate is not subject for being sequestered simply because this molecule has no antioxidant activity. Thus phospho-ascorbate will always be available to interact with putative targets responsible to the modulation of DOG uptake. In conclusion, oxidative stress seems to contribute to metabolic failure in HD by reducing the availability of biological molecules that normally modulate glucose uptake in neurons during periods of activity.
Figure 7. Ascorbic acid inhibits glucose transport via intrinsic molecular properties that are independent of the antioxidant effect.
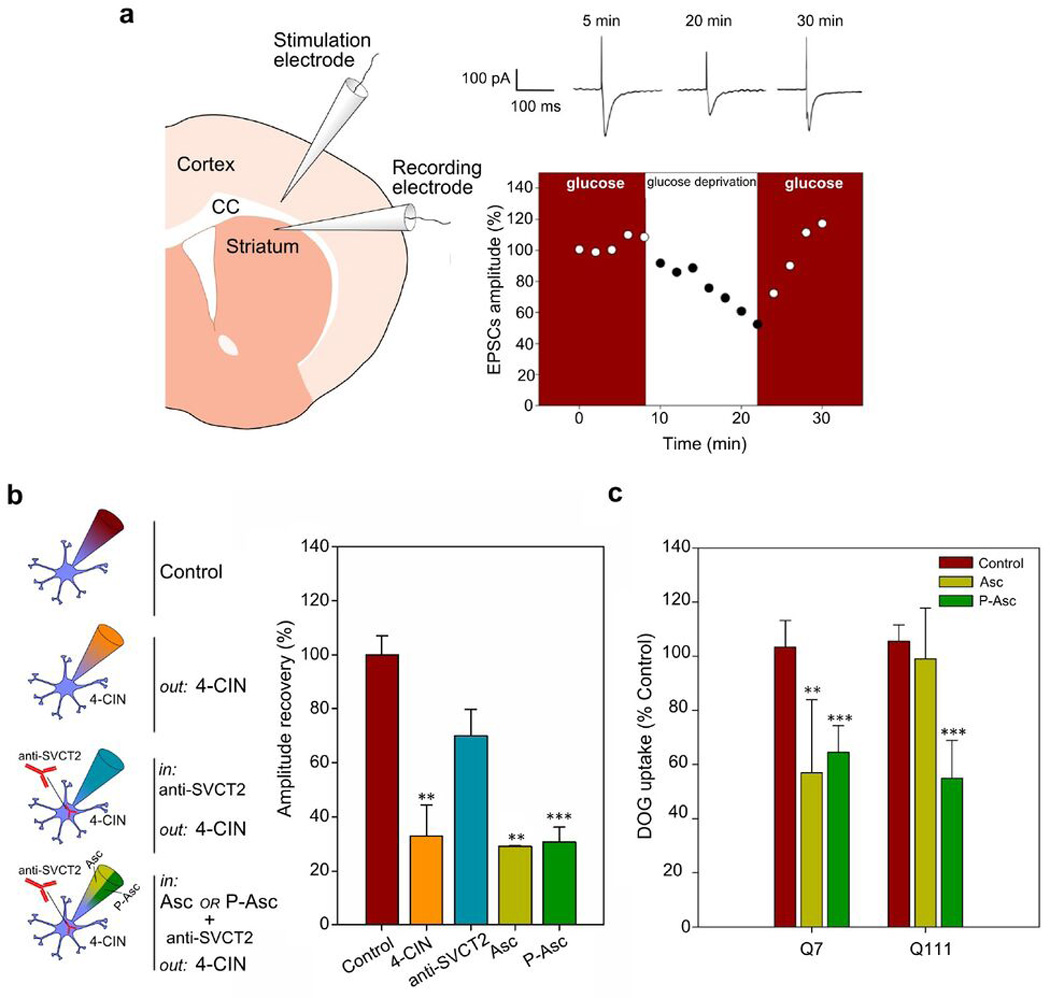
(a) Schematic drawing of the experimental set-up showing positions of the stimulating electrode at the corpus callosum (CC) and the recording electrode at the dorsolateral striatum. Time course of EPSC amplitudes in a control experiment (with and without glucose). Representative EPSCs for a glucose-deprived slice. Calibration bar: 100 pA, 100 ms. (b) Schematic representation of experiments using the whole-cell patch clamp configuration. During ascorbic acid (Asc) and Phospho-L-ascorbic acid (P-Asc) recordings 4-CIN was superfused and anti-SVCT2 antibody was included within the patch pipette. Bar plots for EPSC amplitudes after glucose deprivation (amplitude recovery) with the indicated treatments. Analysis of variance (ANOVA) followed by the Bonferroni post-test, n=3, *** p<0.001, ** p<0.01. (c) 3H-deoxyglucose (DOG) transport analysis using a 10-sec uptake assay (37 °C) in Q7 and Q111 cells. Cells were preincubated for 1 hour with non-radioactive 1 mM ascorbic acid (Asc), 1mM Phospho-L-ascorbic acid (P-Asc) or IB-DTT (Control). Analysis of variance (ANOVA) followed by the Bonferroni post-test, n=3, *** p<0.001, ** p<0.01
Discussion
Changes in neuronal metabolism occur between the resting state and periods of activity (Castro et al., 2009; Allaman et al., 2011). Under conditions of glutamatergic synaptic activity neurons preferentially use monocarboxylates, such as lactate and pyruvate, to sustain synapse function (Castro et al., 2007; Izumi et al., 1997) and thus they are metabolically dependent on mitochondrial function. Mitochondria are also responsible for the generation of substantial amounts of superoxide, caused by electron leakage from the oxidative phosphorylation pathway. ROS generated from mitochondria have been implicated in various forms of cell signalling in the vasculature and in other physiological functions (Zinkevich and Gutterman, 2012). However, redox balance may be disrupted by an imbalance between oxidant species generation and maintenance of the scavenging system, producing oxidative stress. A failure in brain energy metabolism has been described for several neurodegenerative disorders including HD (Beltrán et al., 2012). An increase in oxidative stress has also been documented in these pathologies (Przedborski and Ischiropoulos, 2005; Ischiropoulos and Beckman, 2003; Wang et al., 2013; Mochel et al., 2011), such that metabolic failure and redox imbalance would seem to be linked. Here, we describe a direct connection between ROS production and metabolic impairment in HD models through GLUT3 glucose transporters.
With respect to the neuronal glucose transporter, Q111 cells show a decreased GLUT3 expression, as R6/2 mice does. We demonstrated that glucose transport inhibition by ascorbic acid is impaired in Q111 cells. The decrease in GLUT3 expression may thus be an adaptive response mechanism. A lowered expression of GLUT3 has been described in post-mortem brains of HD patients and HD mice (Capurro et al., 2015; Gamberino & Brennan, 1994; Runne et al., 2008) and also in other disorders such as multiple sclerosis (Nijland et al., 2014). Furthermore, a decreased GLUT3 localisation at cellular surface has been associated with the expression of mutant Htt (mHtt; McClory et al., 2014). An inversely proportional relationship has been described between the dosage of copy-number variations of the gene coding for GLUT3 and age of onset of HD (Vittori et al., 2014). The dosage of copy-number variations of a gene is directly related to its expression (Zhou et al., 2011), whilst the age of onset has been inversely related to the number of CAG repeats in the exon 1 of the gene coding for Htt (Langbehn et al, 2010). The subcellular distribution of GLUT3 in Q111 cells does not seem to be altered in intracellular compartments. Conversely, GLUT3 localisation at the plasma membrane was markedly decreased in Q111 cells, suggesting an important role for Htt in the delivery of GLUT3 to the cell surface. A similar situation has been described in HD models for other surface proteins, such as the neuronal vitamin C transporter SVCT2 (Acuña et al., 2013), the GABA(A) receptor (Jacob et al., 2008; Twelvetrees et al., 2010; Yuen et al., 2012), the glutamate/cysteine transporter EAAC1 (Li et al., 2010) and the NMDA receptor (Yuen et al., 2012). Several regulatory mechanisms that control GLUT3 trafficking to the cell surface have been documented. GLUT3 delivery to the plasma membrane is stimulated by IGF-1 (Dimitriades et al., 2008), by activation of the Akt/PI3K pathway (Zambrano et al., 2010; Acuña et al., 2013b), by AMPK activation (Weisová et al., 2009), and by synaptic activity itself (Ferreira et al., 2011). Fusion of GLUT3-containing vesicles with the plasma membrane is SNAP-25- and syntaxin-1-dependent (Heather et al., 2003). Recently, it has been proposed that Rab11 modulates Glut3 trafficking to the cell surface (McClory et al., 2014) and a deficient activity of Rab11 has been related with hypometabolism in HD (Li et al., 2012). All of the aforementioned proteins or mechanisms may thus be regarded as potential therapeutic targets for enhancing the expression of GLUT3 at the plasma membrane.
We have shown that Q7 cells and cultured striatal neurons express functional glucose transporters. The rate of NBDG uptake by Q7 cells (in the absence of ascorbic acid) was 1.5×10−3 arbitrary units/sec. Primary cultures of striatal neurons exhibited a 10 fold increase in the rate of NBDG uptake when compared to Q7 cells. This is to be expected, as neurons are more likely to have the highest GLUT3 expression of all cell types (Vannucci et al., 1997). GLUT3 overexpression induced an increased NBDG uptake rate in Q cells and cultured striatal neurons. This would suggest that GLUT3 overexpression is enough to improve the correct insertion of GLUT3 into the plasma membrane. Biotinylation assays may corroborate this suggestion. However, Q cells and primary cultures of neurons have a low transfection efficiency making it difficult to perform studies of this nature. If GLUT3 insertion into the plasma membrane is increased by GLUT3 overexpression, then drugs which are able to induce GLUT3 expression, such as endothelin-1 (Sanchez-Alvarez et al., 2004), might be considered for future development of therapies. However, if GLUT3 overexpression is sufficient to increase the proportion of GLUT3 at the plasma membrane, we may speculate that GLUT3 trafficking is not completely altered by mHtt expression. It must be noted that an alteration in protein folding has also been reported in HD (Labbadia and Morimoto, 2013). Thus, we cannot exclude the possibility that problems with protein folding may be responsible for the lack of GLUT3 insertion into the plasma membrane.
Ascorbic acid is used as a modulator of neuronal metabolism in the brain. We have proposed a mechanism to explain the neuronal modulation of glucose and lactate use between the resting state and activity periods (ascorbic acid metabolic switch, Castro et al., 2009). Over the last 20 years, increasing evidence has reinforced the idea that astrocytes support neurons metabolically through lactate production (Caesar et al. 2008; Izumi et al. 1997; Rouach et al. 2009). In fact, impaired astrocyte-neuron interaction has been proposed to be involved in early energy metabolic alterations in HD (Acuña et al., 2013; Boussicault, et al., 2014). Neurons are equipped for glucose metabolism. Activation of glycolysis produces an increase in pyruvate concentration and the NADH/NAD+ ratio. Under these conditions, lactate oxidation cannot occur. Therefore, neuronal lactate consumption would only be possible if glucose uptake or glucose metabolism were inhibited, bestowing importance to ascorbic acid metabolic switch. The ascorbic acid metabolic switch is impaired in presymptomatic HD models (Acuña et al, 2013) indicating that correct modulation of neuronal metabolism is essential for proper functioning of neuronal circuitry. In this context, ascorbic acid only inhibits the transport of hexokinase-hosphorylable glucose analogues such as DOG and 2-NBDG (Beltrán et al., 2011). At the same time we observed that GLUT3-negative cells are not affected by ascorbic acid when DOG uptake is measured. Therefore, we conclude that hexokinase and GLUT3 are important in the mechanism of glucose transport inhibition. However, neurons also express other isoforms of glucose transporters such as GLUT1 and GLUT8 (Acuña et al., 2013b). It is possible that glucose uptake through GLUT1 and GLUT8 might be involved in glucose oxidation through alternative pathways, such as the pentose phosphate pathway (PPP). Bolanos and colleagues (2010) have proposed a mechanism to explain why neuronal glucose should be directed mainly to the PPP to generate NADPH and regenerate GSH (Bolanos et al., 2010). As the PPP participates in regenerating scavenger species, it is plausible that the deregulation of glucose uptake in HD neurons may be associated with redox imbalance. Here we demonstrated an inversely proportion between ROS production and the inhibition of glucose uptake by GSH indicating that oxidative stress would be sequestering antioxidant molecules which are also able to modulate glucose uptake. In fact, in presence of GSH in Q111 cells we observed the same ROS production than the control groups suggesting which is consistent with high levels of ROS in those cells.
During synaptic activity at the glutamatergic corticostriatal pathway, astrocytes remove glutamate from the synaptic cleft (Fig. 8a). Glutamate uptake in astrocytes stimulates lactate (Pellerin et al., 1994) and ascorbic acid release from these cells (Wilson, 2000). Neuronal cells take up ascorbic acid through the neuronal vitamin C transporter, SVCT2 (Castro et al., 2001). Neuronal intracellular ascorbic acid inhibits glucose uptake through GLUT3 inhibition favouring lactate uptake and optimal energy production. Ascorbic acid is also used to maintain redox balance. Synaptic activity produces oxidant species (Bindokas et al., 1996, Carriedo et al., 2000) that should oxidise ascorbic acid to dehydroascorbic acid (DHA). Intracellular DHA is unable to inhibit glucose uptake (Castro et al., 2007). In HD, the release of ascorbic acid from astrocytes and ascorbic acid uptake by neurons does not occur efficiently (Acuña et al., 2013; Fig. 8b). This should result in a decrease in neuronal intracellular ascorbic acid concentration, affecting both redox balance and metabolic modulation. HD neurons are shown to have increased quantities of oxidative species, so intracellular ascorbic acid may be sequestered to act as an antioxidant, rather than just as a metabolic modulator. GLUT3 has been postulated participating in neuronal DHA efflux (Nualart et al., 2014). So DHA could be accumulated intracellularly affecting neuronal viability. Our results demonstrate that oxidative stress contributes to metabolic failure in HD. This is exacerbated by a decreased GLUT3 expression at the plasma membrane in HD cells, which probably affects glucose uptake during resting periods.
Figure 8. Oxidative stress and a low GLUT3 expression contribute to metabolic failure in HD.

(a) During corticostriatal pathway activation glutamate is released into the synaptic cleft. Astrocytes surrounding the synaptic cleft take up glutamate, which stimulates lactate and ascorbic acid release from these cells. Neuronal cells uptake ascorbic acid via the neuronal ascorbic acid transporter, SVCT2. Intracellular ascorbic acid is utilised to maintain the redox balance within neurons. Synaptic activity produces ROS that are reduced by ascorbic acid. Ascorbic acid also inhibits glucose uptake via specific GLUT3 inhibition and stimulates lactate transport. (b) Ascorbic acid release from astrocytes and ascorbic acid uptake is impaired in HD, promoting a decrease in neuronal intracellular ascorbic acid concentration and redox imbalance. Ascorbic acid is sequestered to act more as an antioxidant molecule than a metabolic modulator. Decreased GLUT3 expression at the plasma membrane in HD cells contributes to metabolic failure.
Acknowledgments
Electrophysiological experiments were performed at Michael S. Levine’s laboratory, Intellectual and Developmental Disabilities Research Center, Semel Institute for Neuroscience and Human Behavior, Brain Research Institute, The David Geffen School of Medicine, UCLA. This work was supported by Chilean FONDECYT grant 1110571 (MAC), 1151206 (MAC), 1110906 (SB), 1151430 (SB), 1110508 (IIC), Chilean CONICYT grant 21110592 (ACP), DID-UACh University Research Grant from the Universidad Austral de Chile, Valdivia, Chile (MAC) and International SfA grant (MAC).
Footnotes
Conflict of Interest Statement
None declared.
References
- Acuna AI, Esparza M, Kramm C, Beltran FA, Parra AV, Cepeda C, Toro CA, Vidal RL, Hetz C, Concha II, Brauchi S, Levine MS, Castro MA. A failure in energy metabolism and antioxidant uptake precede symptoms of Huntington’s disease in mice. Nat Commun. 2013;4:2917. doi: 10.1038/ncomms3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaman I, Belanger M, Magistretti PJ. Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci. 2011;34:76–87. doi: 10.1016/j.tins.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Beltran FA, Acuna AI, Miro MP, Angulo C, Concha II, Castro MA. Ascorbic acid-dependent GLUT3 inhibition is a critical step for switching neuronal metabolism. J Cell Physiol. 2011;226:3286–3294. doi: 10.1002/jcp.22674. [DOI] [PubMed] [Google Scholar]
- Beltran FA, Acuna AI, Miró MP, Castro MA. Carlos Dr, Contreras M., editors. Brain Energy Metabolism in Health and Disease, Neuroscience - Dealing With Frontiers. 2012 [Google Scholar]
- Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block RC, Dorsey ER, Beck CA, Brenna JT, Shoulson I. Altered cholesteroñ and fatty acid metabolism in Huntington disease. J Clin Lipidol. 2010;4:17–23. doi: 10.1016/j.jacl.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolanos JP, Almeida A, Moncada S. Glycolysis: a bioenergetic or a survival pathway? Trends Biochem Sci. 2010;35:145–149. doi: 10.1016/j.tibs.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Boussicault L, Herard AS, Calingasan N, Petit F, Malgorn C, Merienne N, Jan C, Gaillard MC, Lerchundi R, Barros LF, Escartin C, Delzescaux T, Mariani J, Hantraye P, Beal MF, Brouillet E, Vega C, Bonvento G. Impaired brain energy metabolism in the BACHD mouse model of Huntington’s disease: critical role of astrocyte-neuron interactions. J Cereb Blood Flow Metab. 2014;34:1500–1510. doi: 10.1038/jcbfm.2014.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caesar K, Hashemi P, Douhou A, Bonvento G, Boutelle MG, Walls AB, Lauritzen M. Glutamate receptor-dependent increments in lactate, glucose and oxygen metabolism evoked in rat cerebellum in vivo. J Physiol. 2008;586:1337–1349. doi: 10.1113/jphysiol.2007.144154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capurro A, Bodea LG, Schaefer P, Luthi-Carter R, Perreau VM. Computational deconvolution of genome wide expression data from Parkinson’s and Huntington’s disease brain tissues using population-specific expression analysis. Front Neurosci. 2014;8:441. doi: 10.3389/fnins.2014.00441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro M, Caprile T, Astuya A, Millan C, Reinicke K, Vera JC, Vasquez O, Aguayo LG, Nualart F. High-affinity sodium-vitamin C co-transporters (SVCT) expression in embryonic mouse neurons. J Neurochem. 2001;78:815–823. doi: 10.1046/j.1471-4159.2001.00461.x. [DOI] [PubMed] [Google Scholar]
- Castro MA, Angulo C, Brauchi S, Nualart F, Concha II. Ascorbic acid participates in a general mechanism for concerted glucose transport inhibition and lactate transport stimulation. Pflugers Arch. 2008;457:519–528. doi: 10.1007/s00424-008-0526-1. [DOI] [PubMed] [Google Scholar]
- Castro MA, Beltran FA, Brauchi S, Concha II. A metabolic switch in brain: glucose and lactate metabolism modulation by ascorbic acid. J Neurochem. 2009;110:423–440. doi: 10.1111/j.1471-4159.2009.06151.x. [DOI] [PubMed] [Google Scholar]
- Castro MA, Pozo M, Cortes C, Garcia Mde L, Concha II, Nualart F. Intracellular ascorbic acid inhibits transport of glucose by neurons, but not by astrocytes. J Neurochem. 2007;102:773–782. doi: 10.1111/j.1471-4159.2007.04631.x. [DOI] [PubMed] [Google Scholar]
- Caviston JP, Holzbaur EL. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 2009;19:147–155. doi: 10.1016/j.tcb.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Hurst RS, Calvert CR, Hernandez-Echeagaray E, Nguyen OK, Jocoy E, Christian LJ, Ariano MA, Levine MS. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington’s disease. J Neurosci. 2003;23:961–969. doi: 10.1523/JNEUROSCI.23-03-00961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda-Prado E, Popp S, Khan U, Stefanov D, Rodriguez J, Menalled LB, Dow-Edwards D, Small SA, Moreno H. R6/2 Huntington’s disease mice develop early and progressive abnormal brain metabolism and seizures. J Neurosci. 2012;32:6456–6467. doi: 10.1523/JNEUROSCI.0388-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang MC, Chen CM, Lee MR, Chen HW, Chen HM, Wu YS, Hung CH, Kang JJ, Chang CP, Chang C, Wu YR, Tsai YS, Chern Y. Modulation of energy deficiency in Huntington’s disease via activation of the peroxisome proliferator-activated receptor gamma. Hum Mol Genet. 2010;19:4043–4058. doi: 10.1093/hmg/ddq322. [DOI] [PubMed] [Google Scholar]
- Dimitriadis G, Maratou E, Boutati E, Kollias A, Tsegka K, Alevizaki M, Peppa M, Raptis SA, Hadjidakis DJ. IGF-I increases the recruitment of GLUT4 and GLUT3 glucose transporters on cell surface in hyperthyroidism. Eur J Endocrinol. 2008;158:361–366. doi: 10.1530/EJE-07-0532. [DOI] [PubMed] [Google Scholar]
- Ferreira JM, Burnett AL, Rameau GA. Activity-dependent regulation of surface glucose transporter-3. J Neurosci. 2011;31:1991–1999. doi: 10.1523/JNEUROSCI.1850-09.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamberino WC, Brennan WA., Jr Glucose transporter isoform expression in Huntington’s disease brain. J Neurochem. 1994;63:1392–1397. doi: 10.1046/j.1471-4159.1994.63041392.x. [DOI] [PubMed] [Google Scholar]
- Gamberino WC, Brennan WA., Jr Glucose transporter isoform expression in Huntington’s disease brain. J Neurochem. 1994;63:1392–1397. doi: 10.1046/j.1471-4159.1994.63041392.x. [DOI] [PubMed] [Google Scholar]
- Group THsDCR. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- He H, Farnell MB, Kogut MH. Inflammatory agonist stimulation and signal pathway of oxidative burst in neonatal chicken heterophils. Comp Biochem Physiol A Mol Integr Physiol. 2003;135:177–184. doi: 10.1016/s1095-6433(03)00049-7. [DOI] [PubMed] [Google Scholar]
- Heather West Greenlee M, Uemura E, Carpenter SL, Doyle RT, Buss JE. Glucose uptake in PC12 cells: GLUT3 vesicle trafficking and fusion as revealed with a novel GLUT3-GFP fusion protein. J Neurosci Res. 2003;73:518–525. doi: 10.1002/jnr.10684. [DOI] [PubMed] [Google Scholar]
- Hornig D. Distribution of ascorbic acid, metabolites and analogues in man and animals. Ann N Y Acad Sci. 1975;258:103–118. doi: 10.1111/j.1749-6632.1975.tb29271.x. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J Clin Invest. 2003;111:163–169. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Katsuki H, Zorumski CF. Monocarboxylates (pyruvate and lactate) as alternative energy substrates for the induction of long-term potentiation in rat hippocampal slices. Neurosci Lett. 1997;232:17–20. doi: 10.1016/s0304-3940(97)00567-3. [DOI] [PubMed] [Google Scholar]
- Jacob TC, Moss SJ, Jurd R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci. 2008;9:331–343. doi: 10.1038/nrn2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kc S, Carcamo JM, Golde DW. Vitamin C enters mitochondria via facilitative glucose transporter 1 (Glut1) and confers mitochondrial protection against oxidative injury. FASEB J. 2005;19:1657–1667. doi: 10.1096/fj.05-4107com. [DOI] [PubMed] [Google Scholar]
- Labbadia J, Morimoto RI. Huntington’s disease: underlying molecular mechanisms and emerging concepts. Trends Biochem Sci. 2013;38:378–385. doi: 10.1016/j.tibs.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langbehn DR, Hayden MR, Paulsen JS, Group P-HIotHS. CAG-repeat length and the age of onset in Huntington disease (HD): a review and validation study of statistical approaches. Am J Med Genet B Neuropsychiatr Genet. 2010;153:397–408. doi: 10.1002/ajmg.b.30992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Valencia A, McClory H, Sapp E, Kegel KB, Difiglia M. Deficient Rab11 activity underlies glucose hypometabolism in primary neurons of Huntington’s disease mice. Biochem Biophys Res Commun. 2012;421:727–730. doi: 10.1016/j.bbrc.2012.04.070. [DOI] [PubMed] [Google Scholar]
- Li X, Valencia A, Sapp E, Masso N, Alexander J, Reeves P, Kegel KB, Aronin N, Difiglia M. Aberrant Rab11-dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington’s disease. J Neurosci. 2010;30:4552–4561. doi: 10.1523/JNEUROSCI.5865-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher F, Davies-Hill TM, Simpson IA. Substrate specificity and kinetic parameters of GLUT3 in rat cerebellar granule neurons. Biochem J. 1996;315(Pt 3):827–831. doi: 10.1042/bj3150827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher F, Vannucci S, Takeda J, Simpson IA. Expression of mouse-GLUT3 and human-GLUT3 glucose transporter proteins in brain. Biochem Biophys Res Commun. 1992;182:703–711. doi: 10.1016/0006-291x(92)91789-s. [DOI] [PubMed] [Google Scholar]
- McClory H, Williams D, Sapp E, Gatune LW, Wang P, DiFiglia M, Li X. Glucose transporter 3 is a rab11-dependent trafficking cargo and its transport to the cell surface is reduced in neurons of CAG140 Huntington’s disease mice. Acta Neuropathol Commun. 2014;2:179. doi: 10.1186/s40478-014-0178-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochel F, Haller RG. Energy deficit in Huntington disease: why it matters. J Clin Invest. 2011;121:493–499. doi: 10.1172/JCI45691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijland PG, Michailidou I, Witte ME, Mizee MR, van der Pol SM, van Het Hof B, Reijerkerk A, Pellerin L, van der Valk P, de Vries HE, van Horssen J. Cellular distribution of glucose and monocarboxylate transporters in human brain white matter and multiple sclerosis lesions. Glia. 2014;62:1125–1141. doi: 10.1002/glia.22667. [DOI] [PubMed] [Google Scholar]
- Nualart F, Mack L, Garcia A, Cisternas P, Bongarzone ER, Heitzer M, Jara N, Martinez F, Ferrada L, Espinoza F, Baeza V, Salazar K. Vitamin C Transporters, Recycling and the Bystander Effect in the Nervous System: SVCT2 versus Gluts. J Stem Cell Res Ther. 2014;4:209. doi: 10.4172/2157-7633.1000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padh H. Cellular functions of ascorbic acid. Biochem Cell Biol. 1990;68:1166–1173. doi: 10.1139/o90-173. [DOI] [PubMed] [Google Scholar]
- Patel M, McIntosh L, Bliss T, Ho D, Sapolsky R. Interactions among ascorbate, dehydroascorbate and glucose transport in cultured hippocampal neurons and glia. Brain Res. 2001;916:127–135. doi: 10.1016/s0006-8993(01)02877-3. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przedborski S, Ischiropoulos H. Reactive oxygen and nitrogen species: weapons of neuronal destruction in models of Parkinson’s disease. Antioxid Redox Signal. 2005;7:685–693. doi: 10.1089/ars.2005.7.685. [DOI] [PubMed] [Google Scholar]
- Ramirez O, Garcia A, Rojas R, Couve A, Hartel S. Confined displacement algorithm determines true and random colocalization in fluorescence microscopy. J Microsc. 2010;239:173–183. doi: 10.1111/j.1365-2818.2010.03369.x. [DOI] [PubMed] [Google Scholar]
- Rice ME, Russo-Menna I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience. 1998;82:1213–1223. doi: 10.1016/s0306-4522(97)00347-3. [DOI] [PubMed] [Google Scholar]
- Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. 2008;322:1551–1555. doi: 10.1126/science.1164022. [DOI] [PubMed] [Google Scholar]
- Runne H, Regulier E, Kuhn A, Zala D, Gokce O, Perrin V, Sick B, Aebischer P, Deglon N, Luthi-Carter R. Dysregulation of gene expression in primary neuron models of Huntington’s disease shows that polyglutamine-related effects on the striatal transcriptome may not be dependent on brain circuitry. J Neurosci. 2008;28:9723–9731. doi: 10.1523/JNEUROSCI.3044-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Alvarez R, Tabernero A, Medina JM. Endothelin-1 stimulates the translocation and upregulation of both glucose transporter and hexokinase in astrocytes: relationship with gap junctional communication. J Neurochem. 2004;89:703–714. doi: 10.1046/j.1471-4159.2004.02398.x. [DOI] [PubMed] [Google Scholar]
- Shulman RG, Rothman DL, Hyder F. Stimulated changes in localized cerebral energy consumption under anesthesia. Proc Natl Acad Sci U S A. 1999;96:3245–3250. doi: 10.1073/pnas.96.6.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Twelvetrees AE, Yuen EY, Arancibia-Carcamo IL, MacAskill AF, Rostaing P, Lumb MJ, Humbert S, Triller A, Saudou F, Yan Z, Kittler JT. Delivery of GABAARs to synapses is mediated by HAP1-KIF5 and disrupted by mutant huntingtin. Neuron. 2010;65:53–65. doi: 10.1016/j.neuron.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannucci SJ, Maher F, Simpson IA. Glucose transporter proteins in brain: delivery of glucose to neurons and glia. Glia. 1997;21:2–21. doi: 10.1002/(sici)1098-1136(199709)21:1<2::aid-glia2>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Vittori A, Breda C, Repici M, Orth M, Roos RA, Outeiro TF, Giorgini F, Hollox EJ Network RiotEHsD. Copy-number variation of the neuronal glucose transporter gene SLC2A3 and age of onset in Huntington’s disease. Hum Mol Genet. 2014;23:3129–3137. doi: 10.1093/hmg/ddu022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta. 2014;1842:1240–1247. doi: 10.1016/j.bbadis.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisova P, Concannon CG, Devocelle M, Prehn JH, Ward MW. Regulation of glucose transporter 3 surface expression by the AMP-activated protein kinase mediates tolerance to glutamate excitation in neurons. J Neurosci. 2009;29:2997–3008. doi: 10.1523/JNEUROSCI.0354-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JX, Peters CE, Sitar SM, Daoust P, Gelb AW. Glutamate stimulates ascorbate transport by astrocytes. Brain Res. 2000;858:61–66. doi: 10.1016/s0006-8993(99)02433-6. [DOI] [PubMed] [Google Scholar]
- Yuen EY, Wei J, Zhong P, Yan Z. Disrupted GABAAR trafficking and synaptic inhibition in a mouse model of Huntington’s disease. Neurobiol Dis. 2012;46:497–502. doi: 10.1016/j.nbd.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano A, Jara E, Murgas P, Jara C, Castro MA, Angulo C, Concha II. Cytokine stimulation promotes increased glucose uptake via translocation at the plasma membrane of GLUT1 in HEK293 cells. J Cell Biochem. 2010;110:1471–1480. doi: 10.1002/jcb.22711. [DOI] [PubMed] [Google Scholar]
- Zhou J, Lemos B, Dopman EB, Hartl DL. Copy-number variation: the balance between gene dosage and expression in Drosophila melanogaster. Genome Biol Evol. 2011;3:1014–1024. doi: 10.1093/gbe/evr023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkevich NS, Gutterman DD. ROS-induced ROS release in vascular biology: redox-redox signaling. Am J Physiol Heart Circ Physiol. 2011;301:H647–H653. doi: 10.1152/ajpheart.01271.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]