

Abstract
Randomized clinical trial (RCT) is the gold standard study for the evaluation of health interventions and is considered the second level of evidence for clinical decision making. However, the quality of the evidence produced by these studies is dependent on the methodological rigor employed at every stage of their execution. The purpose of randomization is to create groups that are comparable independent of any known or unknown potential confounding factor. A critical evaluation of the literature reveals that, for many years, RCTs have been developed based on inaccurate methodological criteria, and empirical evidence began to accumulate. Thus, guidelines were developed to assist authors, reviewers, and editors in the task of developing and assessing the methodological consistency of this type of study. The objective of this article is to review key aspects to design a good-quality RCT, supporting the scientific community in the production of reliable evidence and favoring clinical decision making to allow the patient to receive the best health care.
Keywords: Clinical trial, evidence-based dentistry, evidence-based medicine, evidence-based nursing, methods
TO RANDOMIZE OR TO SAMPLE?
When properly designed, conducted, and reported, the randomized clinical trial (RCT) represents the gold standard study in the evaluation of health interventions. However, it can produce biased results if there is no methodological rigor.[1] The RCT design is considered the gold standard because the randomization of different groups can provide results without bias between groups exposed to different treatment conditions. The random assignment to treatment groups aims to ensure that the characteristics of the participants which may affect the results are balanced.[2]
Fifty years after, the publication of the first RCT, the technical meaning of the term randomization continues to confuse some researchers. Several journals continue to publish “randomized” trials without a full understanding of the procedure. The term random does not mean the same as haphazard but has a precise technical meaning. Random allocation presupposes that each patient has a known chance, usually an equal chance as other participants, of being given a treatment option to be tested, but the treatment to be given cannot be predicted. If there are two treatments, the simplest method of random allocation is the one that gives each patient an equal chance of receiving either treatment; it is equivalent to flipping a coin. A common approach is simply to randomize the treatments according to the dates of birth of patients, the hospital registration numbers or dates of enrollment in the study, for example, to provide one treatment to those with even dates and the other one to those with odd dates. Although all these approaches are in principle unbiased, since they are not related to patient characteristics, problems arise from the openness and knowledge of the allocation system. Because the treatment is known when a patient is considered for participation in the clinical trial, this knowledge may influence the decision to recruit that patient and thereby produce groups that are not comparable.[3,4]
Randomization depends on two processes: Generation of an unpredictable designation sequence and the concealment of this sequence until the intervention occurs. The generation or allocation of the sequence is appropriate if the sequences can prevent selection bias, for example, randomized computer-generated numbers, random number table, envelope drawing, coin flipping, card shuffling, dice throwing, etc.[2,3]
Randomization based on a single sequence of random assignments is known as simple randomization. Simple randomization can be trusted to generate similar numbers in the two trial groups and to generate groups that are roughly comparable in terms of known (and unknown) prognostic variables. Restricted randomization describes any procedure to control the randomization to achieve balance between groups in size or characteristics. Blocking is used to ensure that comparison groups will be of approximately the same size. Stratified randomization is achieved by performing a separate randomization procedure within each of two or more subsets of participants (e.g., those defining age, smoking, or disease severity). Stratification by the center is common in multicenter trials.[5]
HOW TO AVOID BIAS
According to Pannucci and Wilkins (2010),[4] bias can occur in the planning, data collection, analysis, and publication phases of research. Understanding research bias allows readers to critically and independently review the scientific literature and avoid treatments which are suboptimal or potentially harmful. A thorough understanding of bias and how it affects study results is essential for the practice of evidence-based science.
The concealment of the allocation sequence is appropriate if the patients and researchers involved cannot foresee the designation, for example, numbered or coded drug containers with identical appearance and prepared by an independent pharmacy, randomization performed in a location away from test location, sequentially numbered, sealed and opaque envelopes, etc. Many researchers mistakenly consider the sequence generation process as randomization and disregard blinding. However, without adequate concealment, even randomized, unpredictable assignment sequences can be corrupted. Moreover, the generation of unpredictable sequences is probably irrelevant if they are not hidden from those involved in the recruitment of patients.[1,6]
Blinding (or masking) should not be confused with allocation concealment. The allocation concealment is intended to prevent selection bias, protecting the designation sequence before and until allocation occurs. It can always be successfully implemented. However, blinding seeks to avoid determination bias, protects the sequence after allocation, and cannot always be implemented.[6] Performance bias may occur if additional treatment interventions are provided preferably for a group. Blinding of patients and of those involved in the application of interventions prevents this bias and also protects against placebo differences in responses between the groups. Detection bias arises if the knowledge of the patient's name influences the evaluation of the results. This is avoided by blinding those who assess the results.[1]
According to the Acceptance Program Guidelines,[7] the clinical trial should be double-blind, including a randomized selection of individuals using a parallel or a crossover design. The subjects may be admitted and must be fully informed of the study type. An informed consent should be obtained. According to Martínez-Ricarte et al.,[8] the use of a parallel control group allows to evaluate the effectiveness, equivalence or superiority of the study treatment.
Assessing the quality of randomization of 250 controlled trials and 33 meta-analyses and analyzing the association between these evaluations and the estimated effects of treatment, Schulz[6] concluded that trials in which the allocation sequence was inadequately concealed produced higher estimates of treatment effects than trials in which authors reported adequate concealment (odds ratio exaggerated, on average, by 30–40%). However, trials without proper generation sequence led to an estimation of treatment effects similar to those of trials with adequate generation. Thus, the procedure for generating sequence has a lower overall role in preventing bias than the procedure for concealment. This observation makes sense, since having a random sequence (unpredictable) should make little difference without an adequate concealment. In their review, the authors also concluded that studies that were not double-blind yielded larger estimates of effects than double-blind trials (odds ratio exaggerated, on average, by 17%). Double-blinding and avoidance of exclusions after trial entry are the most important methods for reducing bias. Although the strength of this effect falls short of that for allocation concealment, double blinding appears to prevent bias.[6] Randomization controls the selection bias, and the double-blind design controls the observer bias.
USING THE CONSOLIDATED STANDARDS OF REPORTING TRIAL
To understand the results of a randomized controlled trial, readers should understand its design, conduction, analysis, and interpretation.[9] This goal can be achieved only through the complete transparency of the authors. Despite several decades of educational efforts, RCT and reports need to be improved. Researchers and editors developed the Consolidated Standards of Reporting Trials (CONSORT) report to help authors improve reporting using a checklist and a flow diagram.[10] The items on the checklist relate to the content of the Title, Abstract, Introduction, Methods, Results, and Discussion. The revised checklist [Table 1] includes 22 items that were selected because empirical evidence indicates that not reporting the information is associated with biased estimation, (biased) effects of treatment or because the information is essential to judge the reliability or relevance of the results. The flow diagram is intended to represent the progress of all participants through the RCT [Figure 1]. It describes information for four stages of a trial: Enrollment, intervention allocation, monitoring, and analysis. It explicitly includes the number of participants according to each intervention group, each group behavior during the study and the participants who were included in the primary analysis of data.[9,10]
Table 1.
Consolidated standards of reporting trials checklist
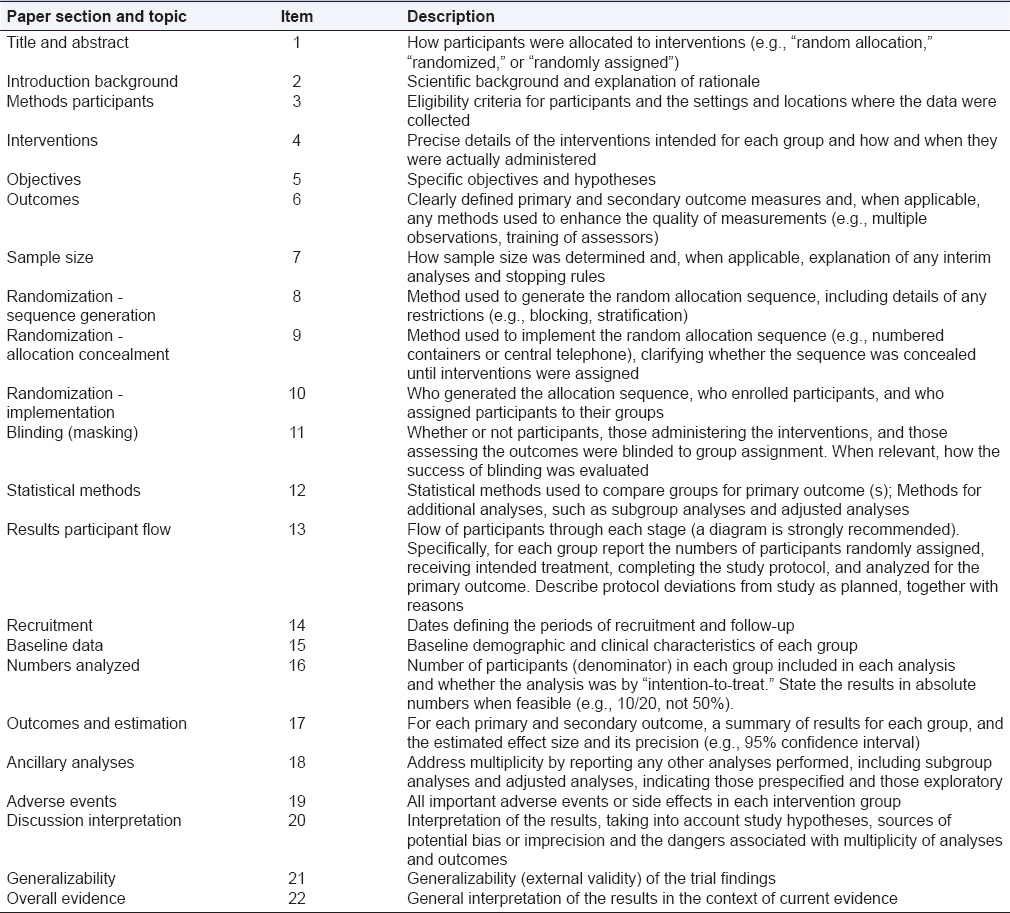
Figure 1.
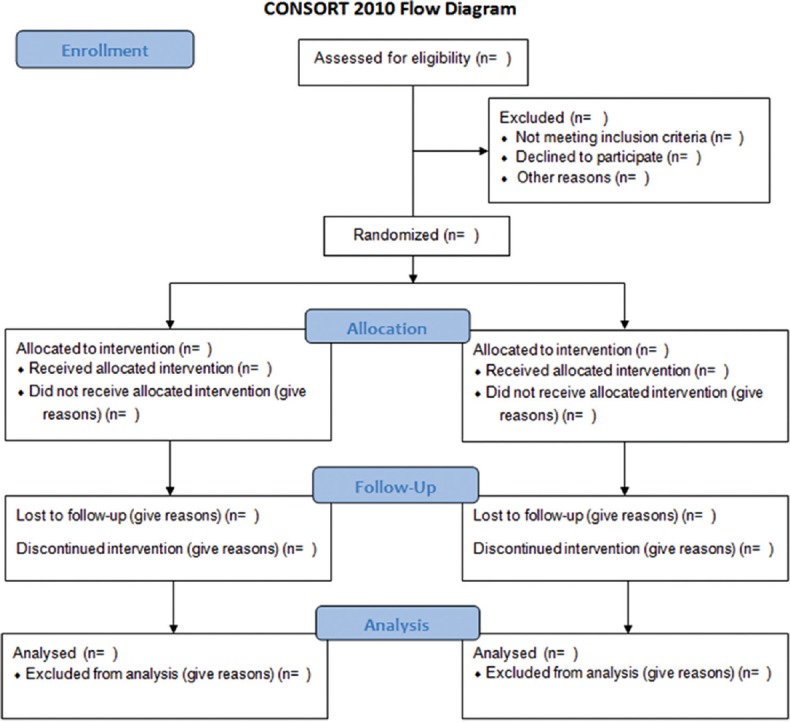
Consolidated Standards of Reporting Trials flow diagram
HOW TO MANAGE DEVIATIONS AND FOLLOW-UP LOSS
The random assignment to treatment groups aims to ensure that the characteristics of the participants which may affect the results are balanced. However, the advantages of this experimental design can be affected by absences, withdrawals and losses of participants, which may void the initial equivalence of control and experimental groups.[2] To manage these deviations, two strategies are commonly used: (1) The principle of analysis by intention to treat (ITT) states that any person should be analyzed as if he/she had completely followed the planned project and (2) Per protocol analysis, which proposes to include only those volunteers who joined the designated intervention and concluded the default action, without any deviation from the original protocol.[11]
The analytical strategy known as ITT has been considered the best approach to preserve the integrity of randomization and strengthen the internal validity of the test and is defended by CONSORT[2] guidelines. ITT is technical analysis of RCTs in which the final results of patients are compared within the groups to which they were randomized at baseline, regardless of whether they have been treated, left the study (drop-outs) or for any reason have not obeyed the initial protocol. Thus, analysis of ITT preserves the similarity between the treatment groups with respect to prognosis, except for the causal variation. For example: In an assay in which medical and surgical treatments for chronic stable angina pectoris are compared, some patients assigned for surgical intervention died before surgery. If these deaths are not attributed to surgical intervention, using an analysis by ITT, surgery may seem to have a false low mortality.[12]
It is also necessary to consider that in clinical trials of substance abuse, many of the missing data are due to loss of follow-up, that is, individuals who discontinued the study after randomization to treatment and whose data were lost later. Using the last observation or the worst possible outcome in substance abuse testing may be valid because much of the observed abandonment may be due to recurrence, adverse effects or no change in the response. A high rate of discontinuation of the drug poses a problem because it can dilute the true treatment effect, reducing the power of comparison.[13,14]
When the treatment is effective, but the losses are substantial, the analysis following the ITT principle underestimates the magnitude of the effect of the treatment that occurs in adherent patients.[15] The subsequent potential loss of study power can be cut by increasing the sample size.[16] Losses smaller than 10% of the total number of randomized patients reduce the chances of bias in the analysis.[17]
SAMPLE SIZE
The estimation of sample size is a key issue in RCTs. Its purpose is to enroll an adequate number of subjects with a given confidence on the number that may be affected by sampling error.[18] Thus, the researcher will get the data in a shorter period, cost-effectively and following ethical principles. The estimation of sample size is essential to avoid the occurrence of errors Types I and II.[18] The size can be estimated using a mathematical formula which will depend on the purpose, nature and parameters investigated in the RCT.[19] However, the decision to choose the appropriate values of the parameters required in the formula is not always simple.[20] It is, therefore, crucial that the authors present the estimated sample size through statistical principles.
PLANNING STATISTICAL ANALYSIS
The importance of the correct use of statistical analysis lies in the researcher's effort to better interpret, organize and analyze data from his/her search. In addition, through the statistics, it is possible to draw conclusions, make predictions for the population and assist in decision making. In a clinical trial, after identifying the groups to be compared, it is necessary to define the dependent variable response that will be submitted to hypothesis testing. It is usual to set as the hypothesis of interest the lack of difference between groups, known as the null hypothesis. The alternative hypothesis is a second statement which contradicts the null hypothesis, that is, the alternative hypothesis is the lack of equality between the groups. These two cases cover all possible values (0–1) for the statistical hypothesis test, and then one of the two statements is true. The null hypothesis is rejected if the P value is “large.” The P value indicates the probability that Type I error has occurred. In the medical field, it is considered that a P value lower than 0.05 indicates that there are significant differences between the groups. However, this statistical significance does not necessarily imply clinical significance.[21]
An alternative to statistical analyses that are based on the P value is the size effect analysis that aims to determine the clinical significance of the effect found. It is not limited to dichotomous outcomes (significant or not significant).[22] In other words, this statistical model is an appropriate measure to determine the clinical significance of the clinical procedure proposed by the RCT. In addition, it will enable to determine whether the sample size was adequate to get enough statistical power.[23] Thus, through the use of size effect analysis, it is possible to identify whether the observed differences are small, medium, or large.[24]
FINAL CONSIDERATIONS
Certain characteristics that produce consequences for modern reviews must be taken into account when developing secondary studies, such as systematic reviews, and assessing the quality of primary studies, such as RCTs. One of these is that former clinical trial results may be less reliable. The majority of these studies were conducted at a time when the methodology of the assays was less stringent than now. Thus, they cannot be compared with more modern studies regarding the results and how to measure them.[25]
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Jüni P, Altman DG, Egger M. Systematic reviews in health care: Assessing the quality of controlled clinical trials. BMJ. 2001;323:42–6. doi: 10.1136/bmj.323.7303.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Polit DF, Gillespie BM. Intention-to-treat in randomized controlled trials: Recommendations for a total trial strategy. Res Nurs Health. 2010;33:355–68. doi: 10.1002/nur.20386. [DOI] [PubMed] [Google Scholar]
- 3.Altman DG, Bland JM. Statistics notes. Treatment allocation in controlled trials: Why randomise? BMJ. 1999;318:1209. doi: 10.1136/bmj.318.7192.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pannucci CJ, Wilkins EG. Identifying and avoiding bias in research. Plast Reconstr Surg. 2010;126:619–25. doi: 10.1097/PRS.0b013e3181de24bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altman DG, Schulz KF, Moher D, Egger M, Davidoff F, Elbourne D, et al. The revised CONSORT statement for reporting randomized trials: Explanation and elaboration. Ann Intern Med. 2001;134:663–94. doi: 10.7326/0003-4819-134-8-200104170-00012. [DOI] [PubMed] [Google Scholar]
- 6.Schulz KF. Randomised trials, human nature, and reporting guidelines. Lancet. 1996;348:596–8. doi: 10.1016/S0140-6736(96)01201-9. [DOI] [PubMed] [Google Scholar]
- 7.American Dental Association, Council on Scientific Affairs. American Dental Association. Program Guidelines: Products for the Treatment of Dentinal Hypersensitivity. 2012. [Last accessed on 2015 Oct 15]. Available from: www.ada.org .
- 8.Martínez-Ricarte J, Faus-Matoses V, Faus-Llácer VJ, Flichy-Fernández AJ, Mateos-Moreno B. Dentinal sensitivity: Concept and methodology for its objective evaluation. Med Oral Patol Oral Cir Bucal. 2008;13:E201–6. [PubMed] [Google Scholar]
- 9.McQuay H, Carroll D, Moore A. Variation in the placebo effect in randomised controlled trials of analgesics: All is as blind as it seems. Pain. 1996;64:331–5. doi: 10.1016/0304-3959(95)00116-6. [DOI] [PubMed] [Google Scholar]
- 10.Moher D, Hopewell S, Schulz KF, Montori V, Gøtzsche PC, Devereaux PJ, et al. CONSORT 2010 explanation and elaboration: Updated guidelines for reporting parallel group randomised trials. J Clin Epidemiol. 2010;63:e1–37. doi: 10.1016/j.jclinepi.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Porta N, Bonet C, Cobo E. Discordance between reported intention-to-treat and per protocol analyses. J Clin Epidemiol. 2007;60:663–9. doi: 10.1016/j.jclinepi.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 12.Soares I, Carneiro AV. Intention-to-treat analysis in clinical trials: Principles and practical importance. Rev Port Cardiol. 2002;21:1191–8. [PubMed] [Google Scholar]
- 13.Chêne G, Morlat P, Leport C, Hafner R, Dequae L, Charreau I, et al. Intention-to-treat vs. on-treatment analyses of clinical trial data: Experience from a study of pyrimethamine in the primary prophylaxis of toxoplasmosis in HIV-infected patients. ANRS 005/ACTG 154 Trial Group. Control Clin Trials. 1998;19:233–48. doi: 10.1016/s0197-2456(97)00145-1. [DOI] [PubMed] [Google Scholar]
- 14.Hedden SL, Woolson RF, Carter RE, Palesch Y, Upadhyaya HP, Malcolm RJ. The impact of loss to follow-up on hypothesis tests of the treatment effect for several statistical methods in substance abuse clinical trials. J Subst Abuse Treat. 2009;37:54–63. doi: 10.1016/j.jsat.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montori VM, Guyatt GH. Intention-to-treat principle. CMAJ. 2001;165:1339–41. [PMC free article] [PubMed] [Google Scholar]
- 16.Heritier SR, Gebski VJ, Keech AC. Inclusion of patients in clinical trial analysis: The intention-to-treat principle. Med J Aust. 2003;179:438–40. doi: 10.5694/j.1326-5377.2003.tb05627.x. [DOI] [PubMed] [Google Scholar]
- 17.Silva Filho CR, Saconato H, Conterno LO, Marques I, Atallah AN. Assessment of clinical trial quality and its impact on meta-analyses. Rev Saude Publica. 2005;39:865–73. doi: 10.1590/s0034-89102005000600001. [DOI] [PubMed] [Google Scholar]
- 18.Arya R, Antonisamy B, Kumar S. Sample size estimation in prevalence studies. Indian J Pediatr. 2012;79:1482–8. doi: 10.1007/s12098-012-0763-3. [DOI] [PubMed] [Google Scholar]
- 19.Lwanga SK, Lemeshow S. Sample Size Determination in Health Studies. Geneva: WHO; 1991. [Google Scholar]
- 20.Naing L, Winn T, Rusli BN. Practical issues in calculating the sample size for prevalence studies. Arch Orofac Sci. 2006;1:9–14. [Google Scholar]
- 21.Pagano M, Gauverau K. Principles of Biostatistics. São Paulo: Cengage Learning; 2012. [Google Scholar]
- 22.Cohen J. Statistical Power Analysis for the Behavioral Sciences. 2nd ed. New York: Academic Press; 1988. [Google Scholar]
- 23.Cohen J. A power primer. Psychol Bull. 1992;112:155–9. doi: 10.1037//0033-2909.112.1.155. [DOI] [PubMed] [Google Scholar]
- 24.Steinberg L, Thissen D. Using effect sizes for research reporting: Examples using item response theory to analyze differential item functioning. Psychol Methods. 2006;11:402–15. doi: 10.1037/1082-989X.11.4.402. [DOI] [PubMed] [Google Scholar]
- 25.Hopayian K. The need for caution in interpreting high quality systematic reviews. BMJ. 2001;323:681–4. doi: 10.1136/bmj.323.7314.681. [DOI] [PMC free article] [PubMed] [Google Scholar]