

SUMMARY
Primary prostate cancer almost always has a luminal phenotype. However, little is known about the stem/progenitor properties of transformed cells within tumors. Using the aggressive Pten/Tp53 null mouse model of prostate cancer, we show that two classes of luminal progenitors exist within a tumor. Not only did tumors contain previously-described multipotent progenitors but also a major population of committed luminal progenitors. Luminal cells, sorted directly from tumors or grown as organoids, initiated tumors of adenocarcinoma or multilineage histological phenotypes, consistent with luminal and multipotent differentiation potentials, respectively. Moreover, using organoids we show that the ability of luminal-committed progenitors to self-renew is a tumor-specific property, absent in benign luminal cells. Finally, a significant fraction of luminal progenitors survived in vivo castration. In all, these data reveal two luminal tumor populations with different stem/progenitor cell capacities, providing insight into prostate cancer cells that initiate tumors and can influence treatment response.
Keywords: Prostate cancer, stem/progenitor cells, heterogeneity, castration, luminal
Graphical Abstract
INTRODUCTION
Epithelial tissues have remarkable capacities to maintain repair damage and maintain homeostasis as a result of cell divisions mediated by stem cells located within special microenvironments (Morrison and Spradling, 2008). Particularly relevant to tumorigenesis are a variety of recent finding showing that the differentiation pathway of epithelial cells can be plastic. Even committed normal epithelial cells can dedifferentiate to a stem-like state in certain nonhomeostatic conditions of severe injury (summarized in (Blanpain and Fuchs, 2014)). This has implications for the “memory” of transformed epithelium relative to re-expression of stem cell lineage properties. An interesting example comes from mammary epithelium. There is evidence for multipotent stem cells in the developing and adult mammary gland (Rios et al., 2014; Van Keymeulen et al., 2011). Following inactivation of BRCA1 in luminal-committed mammary cells, progressive tumors demonstrate reacquisition of multipotent stem cell properties such as combined basal and luminal marker expression (Molyneux et al., 2010). Similarly, for prostate cancer, there are questions about how the cell of origin and/or characteristics of cancer stem/progenitor cell populations may affect various important properties including treatment resistance (Shibata and Shen, 2012; Wang and Shen, 2011; Zong and Goldstein, 2013).
Prostate glands are composed of an outer layer of basal cells expressing KRT5, KRT14, and TP63, an inner layer of secretory, luminal cells expressing KRT8, KRT18, and AR, and rare SYP and CHGA positive neuroendocrine cells (Shen and Abate-Shen, 2010). TP63 is a marker of prostate basal epithelial and stem cells and is required for prostate development (Pignon et al., 2013). Lineage tracing studies based upon cytokeratin drivers have established a number of principles for stem cell hierarchies in the developing and adult prostate (Choi et al., 2012; Ousset et al., 2012; Wang et al., 2013). The majority of regenerative adult stem cells appear to be unipotent (Choi et al., 2012; Wang et al., 2013). In addition, studies using other lineage tracing schemes have described minor populations of multipotent progenitor cells that have not been captured with KRT-specific drivers. Using an inducible NKX3.1-specific CRE driver, a rare (0.7%) population of bipotential luminal cells in the castrate prostate (CARNs) has been described (Wang et al., 2009). In addition, the existence of KRT5neg, KRT14neg, TP63+ cells has been observed, as well as the ability of TP63 lineage marked cells to generate luminal epithelial cells in the adult (Lee et al., 2014). Therefore, there are hints of multipotent stem/progenitor cells in the intact (non-castrate) prostate, as well.
Prostate cancer is almost always luminal in phenotype (Humphrey, 2011). Appropriate CRE drivers have been used to analyze the consequences of Pten deletion in basal, luminal, and castration-resistant NKX3.1-expressing (CARN) cells. Pten deletion in luminal cells and CARNs gave rise to prostatic intraepithelial neoplasia (PIN)/early cancer and microinvasive adenocarcinoma (Choi et al., 2012; Wang et al., 2009). In addition, Pten loss in basal cells led to PIN/early cancer associated with basal to luminal differentiation (Choi et al., 2012; Wang et al., 2013). These studies established that CARNs as well as broadly-defined basal and luminal cells can serve as experimental cells of origin for prostate cancer and strongly suggest that Pten deletion promotes prostatic epithelial transformation in the context of luminal lineage commitment.
Tumor initiating cells (TICs), defined by clonal tumor initiation from transplanted cells, have not been analyzed in primary prostate cancers, partly due to the poor transplantation ability of single cell suspensions of human prostate cancers and low grade mouse tumors (Toivanen et al., 2011). This may be due to the fragility of fractionated prostate tumor cells, to a high percentage of indolent cells in primary tumors, to a strict requirement for the proper microenvironment, or other unknown reasons. In Probasin-CRE (PB-CRE) driven Pten null tumors, fractionation and co-transplantation with embryonic urogenital mesenchyme (UGM) of bulk CD49fhi basal cells but not CD49flo luminal cells led to the development of histologically abnormal glands, suggesting that transformed cells initiating tumorigenesis exist in the basal cell fraction (Mulholland et al., 2009). However, to date, definitive evidence for clonal tumor initiating stem cells in primary prostate cancer is lacking (Wang and Shen, 2011).
Prior ex vivo prostate stem/progenitor studies have been constrained by culture conditions that promote basal but not luminal stem/progenitor cell growth (Xin et al., 2007). The recent development of organoid culture methods that support long-term propagation of luminal epithelium has extended our ability to phenotype and manipulate prostate stem/progenitor cells (Chua et al., 2014; Karthaus et al., 2014). Organoid cultures have revealed the presence of multipotent stem/progenitor cells, capable of reconstituting prostate glands in vivo following UGM recombination assays, within the luminal fraction of mouse and human prostates (Chua et al., 2014; Karthaus et al., 2014). In addition, populations of genetically modified, mouse multilineage organoids gave rise to histologically abnormal, hyperproliferative glands in recombination assays, suggesting an ability to serve as cells of origin for prostate cancer (Chua et al., 2014; Karthaus et al., 2014). There have been technical limitations to growing primary human prostate cancer in organoid cultures (Karthaus et al., 2014), and therefore, the expression of the multilineage stem/progenitor phenotype in primary human prostate cancer has yet to be determined.
Organoid cultures demonstrate a luminal stem/progenitor cell with multilineage potential, although the existence of such stem/progenitor cells has not been observed in adult mouse tissues with luminal KRT driver-dependent tracing schemes, suggesting important questions. First, is multipotentiality conditionally induced in culture or do organoid-defined multipotent luminal cells reflect their in vivo differentiation pathway? Second, is there a definable relationship between multipotent and TP63neg luminal cells, the latter of which are characteristic of prostate cancer?
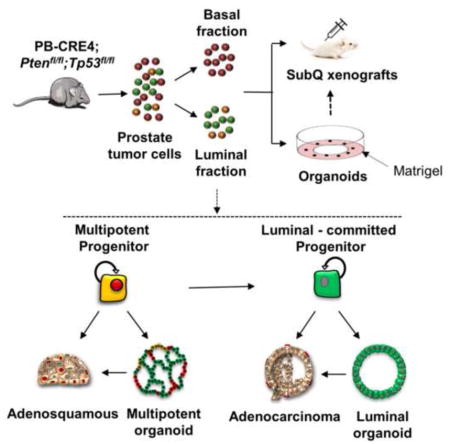
Here we use the aggressive Pten/Tp53 null model of mouse prostate cancer in combination with organoid cultures and clonal TIC assays to characterize luminal stem/progenitor cell populations and their relationship to tumorigenesis. PTEN and TP53 are two of the most frequently deleted or mutated genes in primary prostate cancers, which often are co-selected (Boutros et al., 2015; Taylor et al., 2010). In addition, TP53 is the most selectively enriched altered gene in metastatic castration resistant prostate cancer (Robinson et al., 2015), and therefore, insights into the functional consequences of Tp53 inactivation in prostate epithelium will inform the development of hypotheses related to mechanism of metastasis and acquired resistance. Compared to Pten null prostate cancer, the Pten/Tp53 null prostate cancer model produces significantly faster growing tumors and early mortality (Chen et al., 2005; Martin et al., 2011). Also, Pten/Tp53 tumors are more heterogeneous, being composed primarily of adenocarcinoma but also displaying adenosquamous as well as sarcomatoid differentiation at late stages of disease (Martin et al., 2011), suggesting that the PB-CRE4 driver is active in stem cells and/or that Tp53 deletion leads to a high level of differentiation plasticity. Here we present evidence delineating stem/progenitor phenotypes and their relationship to pathogenesis. We demonstrate the amplification of transformed, luminal stem/progenitor cells in aggressive primary prostate tumors, show that luminal multipotent and luminal-committed progenitors are serial stages in lineage differentiation, and identify the autonomous tumor-initiating cells in primary prostate tumors.
RESULTS
CD49f and Prominin-1 fractionate EpCAM+ Pten−/−, Tp53−/− prostate adenocarcinoma cells into basal and luminal populations
To interrogate discreet tumor subpopulations, we analyzed by flow cytometry the expression of various epithelial lineage and/or potential cancer stem cell markers in PB-CRE4, Ptenfl/fl Tp53fl/fl prostate tumors. After excluding hematopoietic and endothelial cells, flow cytometry was used to characterize combinations of markers on EpCAM+ tumor cell suspensions. Unlike normal prostate tissue, tumor epithelial populations were not fractionated into subpopulations by either Sca-1 or CD24 surface expression, both of which showed a continuum of relatively strong expression (Fig. S1). To separate basal and luminal cells respectively, EpCAM+ cells were separated into CD49fhi and CD49fmed/lo fractions respectively, detecting high and low ITGA6 levels (Fig. 1A) (Fig. S2A). Prominin-1 (PROM1), which is widely expressed on mouse luminal cells (Missol-Kolka et al., 2010), was used to further resolve cell subpopulations. The antibody used here is directed against a protein epitope and not against the controversial stem cell glycosylation epitope, AC133. More than half of the CD49fmed/lo cells labeled with PROM1 antibody (hereafter referred to as PROM1+) while there was no appreciable labeling of the CD49fhi basal population. Fluorescence activated cell sorting (FACS) was used to separate and recover three populations from the prostate tissue: CD49fhi, PROM1+, and CD49flo PROM1neg cells. The fractions corresponding to CD49fhi and PROM1+ each include 5–20% of the EpCAM+ fraction. Post-sort purity scans were performed on all three populations, and purities of >95% were routinely achieved (Fig. S2A). Similar FACS distribution profiles were observed for wild type (i.e. Ptenfl/fl;Tp53fl/fl;PB-CRE4neg littermates) (Fig. S3) and tumor-bearing prostates.
Figure 1. CD49f and PROM1 fractionate EpCAM+ Pten−/−, Tp53−/− prostate basal and luminal progenitor populations.

(A) Representative FACS plot of CD49f and PROM1 staining of primary EpCAM+ Pten−/−, Tp53−/− prostate cells. P7 (purple)= CD49fhi; P8 (orange)= PROM1+; P9 (green)=CD49flo PROM1neg. (B) qRT-PCR analysis of Itga6 and Prom1 gene expression from the indicated cell fractions. RNA levels were normalized to Gapdh. (C&D) qRT-PCR analysis of lineage marker genes in the indicated cell fractions of Pten−/−, Tp53−/− (C) and WT (D) prostates. The data is reported as mean ± SEM. (E&F) Quantification by immunofluorescent staining of TP63+ KRT5+, KRT8+, and KRT8+/KRT5+ cells in the indicated fractions isolated from primary Pten−/−, Tp53−/− (E) and WT (F) prostate tissue. The average of three independent experiments is shown. The data is reported as mean ± SEM. (G) Schematic showing that cells from Pten−/−, Tp53−/− prostates were fractionated and assayed for organoid formation or were transplanted directly into NOD/SCID mice for tumor initiation assays. G1 organoids were harvested, dissociated and transplanted in vivo to assess tumorigenesis. See also Figures S1 and S2.
Quantitative RT-PCR (qRT-PCR) was used to confirm the differential expression of Itga6 and Prom1 in the relevant fractions (Fig. 1B) and to interrogate the expression of prostate lineage markers, TP63, KRT5 and KRT18. Importantly, for both tumor and WT prostate, Tp63 RNA was expressed in the CD49fhi fraction but was almost undetectable in the CD49fmed/lo cells (Figs. 1C&D). Consistent with RNA data, nuclear TP63 immunofluorescence staining demonstrated a significant enrichment (>90%) in the CD49fhi fraction and <3% in CD49fmed/lo fractions (Figs. 1E&F). The distribution of cells expressing the definitive basal cell marker, TP63, indicates a distinct separation of basal and luminal cells.
Interestingly, an analysis of cytokeratin expression suggests different proportions of lineage subpopulations in tumor as compared to WT for both basal and luminal fractions. For the CD49fhi fraction, tumor cells expressed high levels of RNA encoding Krt5 as well as lower levels of Krt18. Immunofluorescent co-staining of KRT5 and KRT8 in the tumor basal fraction demonstrated about 90% KRT5+8neg and a small fraction of KRT5+8+ cells (Fig. 1E). WT CD49fhi cells expressed robust levels of Krt5 and Krt18. Greater than 95% of cells within the basal fraction stained strongly for KRT5, and a significant subpopulation (30–40%) of cells were composed of KRT5+, KRT8+, and TP63+ co-stained cells (Fig. 1F). These data are consistent with publications describing the presence of a substantial population of basal KRT5++/KRT8+ cells identified by in situ immunofluorescent staining of normal mouse (Peng et al., 2011) and human (van Leenders et al., 2000) prostate tissue as well as in transgenic KRT5 and KRT18 promoter reporter mice (Peng et al., 2011).
KRT staining patterns for the PROM1+ fractions from tumor and WT demonstrated approximately 80% KRT8+ cells. Tumor derived cells also frequently expressed low levels of KRT5, suggesting a possible intermediate or transitional phenotype. The CD49flo PROM1neg fractions contained mostly KRT8+ and cytokeratin negative populations.
Tumor-derived organoids are observed in both basal and luminal fractions
To begin evaluating the composition of primary tumors with respect to their stem/progenitor phenotypes as well as clonal tumor initiating activity, we analyzed such properties in fractionated populations of basal and luminal cells (Fig. 1G). Using organoid culturing conditions that allow for in vitro growth of basal and luminal stem/progenitor cells (Karthaus et al., 2014), we compared the relative efficiencies of organoid formation from the prostates of WT mice as well as PB-CRE4; Ptenfl/fl;Tp53fl/fl and PB-CRE4; Ptenfl/fl tumor-bearing mice. Because there is a continuum in morphologies from spherical to more complex structures, we use the terms “organoids” to signify clonal stem/progenitor growth of >50 microns, regardless of morphology. WT CD49fhi and PROM1+ fractions showed approximately 20% and 2% organoid forming efficiency, respectively (Fig. 2A), consistent with other recently published reports describing relatively rare luminal fraction progenitors in normal mice (Chua et al., 2014; Karthaus et al., 2014). The organoid forming units (OFU) for the basal fraction of tumor-bearing prostates was between 20–40%, demonstrating that the culture conditions used here are highly efficient in promoting clonal basal cell growth. The CD49flo PROM1neg fraction rarely formed organoids and was not further analyzed. Notably, in comparing tumor and WT luminal fractions, organoid formation by the Pten−/− Tp53−/− fraction was about 10 fold greater than wild-type, while the Pten−/− tumors demonstrated relatively modest expansion of luminal OFU.
Figure 2. Tumor-derived basal and luminal progenitors are almost all PTEN null.

(A) Comparison of first generation organoid formation from wild type (WT), Pten−/−, and Pten−/−, Tp53−/− CD49fhi and PROM1+ cells. Data is reported as mean % OFU ± SEM. (B) IHC images of WT and Pten−/−, Tp53−/− CD49fhi and PROM1+ organoids stained for PTEN and quantification of PTEN+ organoids in each fraction. Data is reported as %PTEN+ organoids ± SEM. Scale bars = 100 μm. (C) IHC images of pS6240/242 expression in WT, and Pten−/−, Tp53−/,− PROM1+ organoids. Scale bars = 50 μm. (D) Representative IF (KRT5 and AR), and (KRT8 and TP63) images of G1 WT and Pten−/−, Tp53−/− CD49fhi organoids. * indicates a multilineage organoid. Scale bars = 50 μm. See also Figures S3 and S4.
To determine the relative abundance of WT (i.e. non-Pten deleted) progenitors found in Pten/Tp53 null tumors, organoids derived from the basal and luminal progenitors from tumor-bearing and WT mice were stained for PTEN expression. PTEN was expressed in nearly 100% of organoids from WT prostates (Fig. 2B) but was seen in ≤ 2% of either basal and luminal fraction of tumor-derived organoids. Thus, at 14 weeks of age in PB-CRE4-driven Pten/Tp53 deleted tumors, at which time the prostate is usually significantly enlarged, the majority of cells that grow in organoid cultures are tumor-derived and rarely contaminated with WT organoids. A lack of WT cells within the tumor fractions was validated further by expression of floxed Pten exon 5 in WT but not tumor organoids (Fig. S3). Further, to analyze pathways downstream of Pten deletion, we stained sections of organoids for pS6240/242, a target downstream of RPS6KB1 and mTORC1 activation. Luminal-derived organoids from WT prostates showed weak and patchy staining, while Pten/Tp53 were stained uniformly strongly (Fig. 2C).
The CD49fhi fractions from WT and Pten/Tp53 null tumor-derived prostates were characterized in organoid culture (Fig. 2D). Histologically in tumors, basal cells were a minor population occurring as scattered individual cells or within uncommon areas of adenosquamous carcinoma (Martin et al., 2011). Organoids from the CD49fhi fractions were TP63+ with KRT5+ peripheral cells and KRT8+ cells near lumen. Time lapse analyses of organoid development showed actively dividing cells in the outer cortex of the CD49fhi tumor organoids (Fig. S4 & Movie S1). The tumor-derived organoids were composed mostly of tightly-packed concentric layers with small lumen, which was an abundant phenotype observed in WT prostate organoids as well. One clear difference between WT and tumor organoids was easily observed AR staining in multiple layers of WT organoids and a lack of AR staining in tumor organoids (Fig. 2D). In addition, the WT organoids notably contained an infrequent (<10%) population characterized by two to four cell layers around a large lumen, containing basal, luminal and AR+ cells (denoted by * in Fig. 2D), similar to the phenotype of “multilineage organoids” that have recently been described (Karthaus et al., 2014). Taken together, these data suggest that basal cells in the Pten/Tp53 model are predominantly a tumor-specific, committed basal phenotype.
Luminal fraction tumor-derived organoids demonstrate luminal only and multilineage phenotypes
The PROM1+ fraction from WT mice produced organoids that were heterogeneous in size and translucent with discernable lumen (Fig. 3A). The majority of large organoids contained both KRT8+/AR+ and KRT5+/TP63+ cells (Fig. 3B), similar to the multilineage organoids described previously, and confirm multilineage organoids in both basal and luminal fractions of WT prostate (Karthaus et al., 2014). In addition, there was a minor number of small, single-layered organoids (denoted by arrow in Fig. 3b) containing luminal cells with limited proliferative and self-renewing capacity, as these were no longer observed upon serial passage (see Fig. 4A).
Figure 3. PROM1+ progenitors show heterogeneous phenotypes in organoid culture.

(A) Representative phase images of first generation translucent, acinar and twined organoids generated from PROM1+ cells. Organoid morphologies are observed in wild-type (WT) and Pten−/−, Tp53−/− tumor fractions as indicated. Scale bars = 50 μm. (B) Representative IF (KRT5 and AR), and (KRT8 and TP63) images of G1 WT and Pten−/−, Tp53−/− PROM1+ organoids. The arrow indicates a luminal WT organoid. Scale bars = 50 μm. (C) Representative phase, IF (KRT5+KRT8 or KRT5+TP63) and IHC (pS6240/242 ) images of G1 Pten−/− PROM1+ multi-lobulated structures. Scale bars = 50 μm. See also Figure S4.
Figure 4. Differentiation potential of multilineage and luminal organoids in vitro and in vivo.

(A) Representative phase images of G1 organoids isolated from WT and Pten−/−, Tp53−/− PROM1+ cells, and IF (KRT5+KRT8) confocal images of daughter organoids generated from the cloned organoids. Scale bars = 100 μm. Note that there are relative differences in magnification for the panels shown. (B) Representative H&E, IHC (TP63 and AR), and IF (KRT5+KRT8) images of regions of adenosquamous (left panels) and adenocarcinoma (right panels) in tumors generated from G1 PROM1+ organoids. The black arrow indicates nuclear TP63 labeling of cells situated along the basement membrane adjacent to an adenocarcinoma gland. Scale bars = 50 μm.
PROM1+ Pten/Tp53 null tumor organoids were morphologically heterogeneous and notable for the presence of large, complex twined structures, not usually observed in organoids derived from WT prostates (Fig. 3A). Sections from fixed organoids as well as time lapse microscopy suggested that a continuously enlarging central lumen twisted and folded back upon itself to produce twined single lumen-containing structures (Fig. S4, Movie S2). Histological analyses revealed two types of organoids: multilineage and luminal only. The multilineage organoids usually displayed a twined structure, suggesting a hyper-proliferative phenotype relative to WT multilineage organoids. Interestingly, large luminal only organoids were not observed in WT prostates suggesting a tumor specific characteristic. Tumor specific luminal only organoids have not been specifically described previously in mouse models of prostate cancer, although luminal organoids have been derived from clinical metastatic adenocarcinoma prostate cancer biopsies (Gao et al., 2014). Luminal only organoids predominated in frequency at 16–18 weeks of age, and the relative proportion varied between about 60–90% in individual tumors. The levels of AR expression in PROM1+ Pten/Tp53 null organoids were generally lower than WT organoids, consistent with relatively low levels of AR in Pten/Tp53 null tumors (Fig. 3B).
Luminal progenitor phenotypes are enriched in Pten/Tp53 co-deleted tumors relative to Pten-null only tumors
Because PB-CRE4;Ptenfl/fl;Tp53fl/fl and PB-CRE4;Ptenfl/fl prostates demonstrated distinct distributions of organoids relative to the basal and luminal fractions (Fig. 2A) as well as previously characterized differences in prostate cancer pathogenesis (Chen et al., 2005; Martin et al., 2011; Wang et al., 2006), we performed histological analyses on organoids from age-matched (16–18 weeks) prostates for comparison. As anticipated, the Pten null luminal fraction contained relatively few (<10%) luminal (KRT8+/TP63neg) organoids >50 μm, consistent with total organoid plating efficiency (Fig. 2A). Pten null only prostates contained large, solid lobulated structures present in both luminal and basal fractions, which displayed high pS6240/242 staining (Fig. 3C). Lobules were organized with KRT5+/TP63+/AR+ cells on the periphery and KRT8+ cells near the centers, demonstrating histological and morphological characteristics distinct from Pten/Tp53 null stem/progenitor cells and consistent with deregulated growth of basal multipotent progenitors. These data imply that the loss of Tp53 in the context of Pten deletion specifically supports survival or amplification of luminal-committed progenitors. Our findings are consistent with the described expansion of basal stem/progenitor cells in PB-CRE4;Ptenfl/fl prostates (Wang et al., 2006) and with the significantly more rapid development of adenocarcinoma in PB-CRE4;Ptenfl/fl;Tp53fl/fl compared to PB-CRE4;Ptenfl/fl prostates (Chen et al., 2005; Martin et al., 2011).
Multilineage and luminal progenitors are serial stages in luminal commitment
Serial passage of Pten/Tp53 null PROM1+ organoids demonstrated continued presence of both multilineage and luminal phenotypes. In order to analyze the self-renewal and differentiation properties of the progenitors giving rise to the two organoid phenotypes, individual organoids were clonally isolated from G1 organoid populations, serially passaged as a single cell suspension and subsequently analyzed histologically at G2 or G4 for organoid phenotype (Fig. 4A). Individual, tumor-derived organoids passaged at nearly 100% efficiency and were observed for a minimum of 6 passages. Two types of differentiation patterns were observed from individual tumor organoids: one pattern showed a mixture of multilineage and luminal phenotyped organoids, and the other pattern showed luminal phenotyped organoids only. The former pattern generally correlated with large, twined organoids and the latter with acinar organoids. No cultures contained only multilineage organoids. This data strongly implies that multipotent tumor progenitors produce multilineage and luminal organoids, while luminal-committed tumor organoids renew themselves and maintain a luminal phenotype.
Similar analyses carried out with individual WT PROM1+ organoids showed that large organoids could be efficiently passaged for at least 6 generations and gave rise to a combination of multilineage and basal organoid phenotypes (Fig. 4A), in contrast to multilineage and luminal phenotypes derived from multilineage tumor organoids. These data suggest that Pten/Tp53 loss in multipotent progenitors may increase luminal and decrease basal commitment or survival of daughter progenitors. In addition, we were unable to serially propagate WT luminal only organoids, unlike tumor-derived luminal organoids.
PROM1+ tumor-organoids display luminal and bipotential differentiation in vivo
To investigate the tumorigenicity and resulting histopathology of organoids, we performed subcutaneous injection of single cell suspensions derived from G1 tumor organoids. WT organoids did not grow under these conditions. PROM1+ G1 tumor organoids produced tumors, which contained multiple foci displaying one of two possible phenotypes, embedded in stroma and disorganized tumor cells. One type of focus consisted of irregular, mostly small, adenocarcinoma glands suggesting initiation from a luminal-committed progenitor (Fig. 4B, right panels). These glands were composed of KRT8+/KRT5neg/AR+ cells with no TP63+ cells within or adjacent to the glands. The other type of focus demonstrated a consistent adenosquamous histology indicating bipotent lineage differentiation (Fig. 4B, left panels). Adenosquamous transformed foci were composed of variably-sized glandular structures lined with a layer of luminal cells that often transitioned into an area of disorganized piling at the edge of the lumen with continuation into an adjacent area of squamous differentiation. This histology clearly forms a continuum in lineage transition (Fig. S5) and is pathologically distinct from the pattern anticipated for tumors initiated from two or more lineage-committed cells. In addition, squamous histology alone was never observed, providing evidence that tumorigenic basal-committed stem/progenitor cells are exceedingly rare or absent. TP63 staining was strong in the squamous histology and contained in intermittent basilar located cells adjacent to the luminal glandular regions (denoted by an arrow, Fig. 4B). A high proportion of cells within adenocarcinoma and squamous histological phenotypes were nuclear AR+. These data show that the foci observed in vivo parallel the lineage composition of multilineage (adenosquamous) and luminal (adenocarcinoma) organoids.
By contrast, tumor-derived, CD49fhi organoids from several independent cultures did not produce detectable tumor growth after in vivo transplantation. We hypothesize that the majority of Pten/Tp53 null CD49fhi cells are committed basal cells, consistent with the organoid morphologies shown in Figs. 2B & D. Such cells proliferate well in organoid culture conditions (Fig. 2A), but basal cells have been shown in models of basal keratin CRE-driven Pten/Tp53 deletion to be relatively resistant to oncogenic growth (Choi et al., 2012).
Luminal and basal TICs demonstrate luminal-committed and multilineage tumor-initiating activity
We next analyzed the tumor-initiating activity of CD49fhi and PROM1+ fractions isolated directly from primary tumors. Limiting dilution analysis was performed using subcutaneous injection of cells with Matrigel™ into NOD/SCID mice in the absence of embryonic urogenital mesenchyme (UGM) in order to evaluate autonomous tumorigenic and lineage differentiation activity without exogenous differentiating signals. Cell numbers used for limiting dilution injections ranged from 100 to 100,000 cells per injection (Table 1). For the PROM1+ fraction, tumor formation was relatively uncommon, reflecting an autonomous tumor-initiating cell frequency of ~1/100,000. Importantly, two independent tumor histologies were observed: adenosquamous carcinoma and adenocarcinoma with variable amounts of sarcomatoid carcinoma, consistent with histologies found in primary GEM tumors (Martin et al., 2011). Squamous only tumors were not observed. EMT trans-differentiation of EpCAM+ Pten/Tp53 null adenocarcinoma to sarcomatoid morphology occurs in Pten/Tp53 null tumors and has been previously characterized (Liu et al., 2012; Martin et al., 2011).
Table 1.
Tumor-propagating cell (TPC) frequency in each population was determined by limiting dilution analysis using L-Calc™ software with the transplanted cell numbers shown. Pooled fractions obtained from 2 to 4 tumor bearing animals were used in each experiment. The number of independent experiments are shown in brackets below. CD49hi (9), and PROM1+ (8). Summary of histological phenotypes of primary transplant tumors is shown.
| CD49fhi | PROM1+ | |
|---|---|---|
| Tumor Histology | ||
| Adenocarcinoma | n=2 | n=4 |
| Adenosquamous | n=1 | n=2 |
| # Cells/Injection | # Mice with Tumors/# Mice Transplanted | |
| 100 – 1,200 | 0/6 | 0/4 |
| 1,200 – 10,000 | 1/8 | 3/8 |
| 10,000 – 50,000 | 1/7 | 0/3 |
| 50,000 – 100,000 | 1/1 | 3/8 |
| > 100,000 | 0/0 | 0/0 |
| 3/22 | 6/23 | |
| Frequency of TPC | 1/98,060 | 1/103,032 |
| 0.4724 |
||
Adenocarcinoma was KRT8+/TP63neg/KRT5rare with glands demonstrating a high frequency of AR+ cells (Fig. 5, right panels). For the adenosquamous tumors, there was clear continuity between adenocarcinoma and squamous histological phenotypes, showing a common origin (Fig. 5, left panels). Both adenocarcinoma and basal components were AR+. The glandular component was composed of KRT8+/TP63neg cells with intermittent TP63+ cells in a basal position, while the squamous tumor cells were strongly KRT5+/TP63+. These data imply that a multipotent stem cell produced prostatic acini organized with basal and luminal components as well as areas of committed basal cell (squamous) tumors. In addition, we observed in one clonally-initiated adenosquamous tumor from the PROM1+ fraction that serial passage of the initial tumor gave rise to adenocarcinoma tumors in three recipients, demonstrating that luminal-committed TICs can arise from adenosquamous tumors, consistent with the lineages produced by serially-passaged individual, mutilineage organoids (summarized in Fig. 5B).
Figure 5. PROM1+ cells demonstrate luminal-committed and multilineage tumor-initiating activity in vivo.

Representative H&E, IHC (AR) and IF (KRT5+KRT8 or KRT5+TP63) staining of regions of adenosquamous (left panels) or adenocarcinoma (right panels) from tumors initiated from Pten−/−, Tp53−/− PROM1+ cells. White arrows indicate nuclear TP63 staining in adenosquamous tumors. TP63 staining was not observed in adenocarcinoma. Ad = adenocarcinoma region, Sq = squamous carcinoma. Scale bars = 50 μm. (B) Schematic representation of clonally-initiated in vitro progenitor activity and tumor phenotypes originating from luminal and basal fractions of Pten−/−Tp53−/− tumors. See also Figure S5.
Tumor initiating cell assays were performed with the CD49fhi fraction. We observed tumor initiation leading to adenocarcinoma and adenosquamous histological phenotypes at a frequency of ~1/100,000 (Table 1). The existence of cells with tumorigenic potential in the basal fraction is consistent with previous analyses demonstrating tumor initiation by bulk populations of PB-CRE4 Ptenfl/fl basal cells (Mulholland et al., 2009). The presence of TICs contrasts with the lack of tumorigenicity for organoids derived from the CD49fhi fraction. Although various explanations are possible, we favor the interpretation that endogenous CD49fhi TICs in this model are relatively rare compared to non-tumorigenic Pten/Tp53 deleted basal cells, and therefore, are not routinely recovered in organoid cultures. This would seem inconsistent with a similar TIC frequency between basal and luminal fractions. However, because the relative efficiencies of tumor initiation by luminal as compared to basal cells under the conditions used here are not known, direct quantitative comparisons between the two fractions cannot be made.
To summarize (Fig. 5B), there are multiple autonomous TICs in primary prostate tumors, included within both the CD49fhi basal and the PROM1+ luminal fractions that give rise to either adenosquamous or adenocarcinoma. These data, based upon analyses of organoid phenotypes and tumorigenicity as well as clonal TIC phenotypes in vivo, strongly support the existence of transformed multipotent and luminal-committed progenitors resulting from PB-CRE4-initiated deletion of Pten and Tp53. Importantly, the existence of multipotent TICs implies that multilineage organoids reflect endogenous progenitors and are not solely a conditional phenotype induced by ex vivo culture.
A fraction of PROM1+ stem/progenitor cells are castration-tolerant
A central question in prostate cancer biology is the cellular origins of castration resistance. To begin addressing this question, we have evaluated the in vivo castration sensitivity or tolerance (survival) of progenitors giving rise to organoids The histopathology of castration in 14–16 week old Pten/Tp53 null tumors was analyzed between 3 days and 6 weeks. Histologically, androgen deprivation led to attenuation in the size of tumor cells, increased vacuolation of luminal-appearing cells, and a subpopulation of cells with increased cytoplasmic basophilia/anaplasia, especially in the ventral prostate. Apoptosis, assayed by nuclear cleaved CASP3, was increased at day 3 and reached plateau levels by day 12, which were maintained for several weeks (Fig. S6A). Nuclear AR localization was used as a measure of AR activity in tissue sections (Fig. 6A). Following castration, AR staining mostly was diffuse and cytoplasmic, but by 7 days post-castration, clusters of cells with weak nuclear AR staining and representing less than 10% of total tumor cells were observed in all prostate lobes. Two weeks was chosen as a short-term castration time point to assay the in situ tolerance of stem/progenitor populations for androgen deprivation. Due to the stoichastic potential of this model for sarcomatoid transformation, long time points were not feasible.
Figure 6. A fraction of PROM1+ stem/progenitor cells are castration-tolerant.

(A) IHC images of AR staining of Pten−/−, Tp53−/− prostate tissue from intact animals or 7 days post-castration. Scale bars = 50 μm. (B) Histological comparison of PROM1+organoids generated from intact and castrated Pten−/−, Tp53−/− prostate. (C) Representative IHC AR staining of PROM1+ organoids generated from castrated Pten−/−, Tp53−/− prostate. Scale bars = 50 μm. (D) Representative IHC AR staining in tumors generated from castrate Pten−/−, Tp53−/− G1 PROM1+ organoids. Insert a shows invasive cells; insert b shows nuclear atypia. Scale bars = 50 μm. (E) Histological comparison of PROM1+organoids generated from castrated Pten−/−, Tp53−/− prostate following Enzalutamide treatment and growth for 7 days. ** = p< 0.01, *= p< 0.05. See also Figure S6.
FACS analysis of tumors from mice castrated two weeks earlier demonstrated CD49fhi, PROM1+, and CD49flo PROM1neg fractions in approximately similar proportions to intact tumors (Fig. S6B). RT-PCR analyses of the fractions obtained from castrate as compared to intact tumors revealed population-specific transcription responses (Fig. S6C) such as increased levels of Tp63, Krt5, and Krt18 in the basal fraction and increased Clu in the luminal fraction. Clu expression previously has been described as a luminal progenitor marker in PSA-CRE;Ptenflox/flox mice (Korsten et al., 2009).
Castrated tumor fractions were characterized for the growth and differentiation potential of surviving progenitors in organoid cultures. The CD49fhi fraction showed no significant changes in OFU (Fig. S6D) and no obvious morphological/lineage changes following castration, consistent with undetectable AR expression in most tumor-derived basal organoids (Fig. 2D). Histological analyses of harvested PROM1+ fraction organoids demonstrated that both multilineage and luminal organoids were reduced following castration (Fig. 6B). Because multipotent progenitors can give rise to luminal progenitors, the inhibition or loss of multipotent progenitors in vivo following castration may be reflected in in vitro numbers of both multilineage and luminal organoids. Interestingly, the PROM1+ organoids initiated from castrated tumors contained a high percentage of nuclear AR+ cells in the presence of DHT, showing that castrate-tolerant luminal cells retained the potential for AR signaling (Fig. 6C). Consistent with this, transplantation of cell suspensions of PROM1+ G1 organoids derived from castrated tumors resulted in tumor foci with glandular morphologies (KRT8+/AR+/TP63rare) embedded in stroma (Fig. 6D). Neoplastic epithelial cells formed both low-grade mPIN lesions as well as invasive adenocarcinoma (insert a). Neoplastic cells had expected features of nuclear atypia including a euchromatin pattern and 1 to 2 prominent nucleoli (insert b). Therefore, a fraction of luminal progenitor cells appeared to be castration tolerant, some of which were capable of autonomously producing AR+ adenocarcinoma in vivo.
To compare castration to autonomous AR inhibition, we added 10 μM Enzalutamide in the absence of DHT at the time of organoid initiation from the luminal fraction of intact (non-castrated) tumors (Fig. 6E). Enzalutamide noticeably slowed but did not prevent the growth of PROM1+ organoids. Histological analyses showed a statistically significant decrease in numbers of multilineage organoids and a smaller, statistically insignificant decrease in luminal organoids. Overall, Enzalutamide treatment confirms the AR-dependence of organoid initiation by a fraction of Pten/Tp53 null multilineage progenitors, but also revealed the resistance to AR inhibition of a significant population of progenitors in the luminal fraction.
DISCUSSION
This study characterizes primary prostate tumors initiated by loss of the common tumor suppressors, Pten and Tp53, for stem/progenitor phenotypes as assayed by in vitro organoid cultures and in vivo tumor-initiating activity. It has not been routinely possible to culture luminal stem/progenitor cells, which has prevented ex vivo analysis of these important cells in primary prostate tumors, biasing most studies toward primary basal cells or human prostate cancer cell lines (Wang and Shen, 2011). We have observed two classes of self-renewing luminal progenitors in Pten/Tp53 null tumors, a minor population giving rise to multilineage organoids (multipotent progenitors) and a major population producing luminal-only organoids (luminal committed progenitors) (Fig. 5B). Of particular interest is the observation that multilineage organoids give rise to self-renewing luminal organoids, providing additional insight into progenitor subpopulations, lineage stages leading to luminal commitment, and one route of prostate adenocarcinoma histogenesis. We suggest that combined loss of Pten and Tp53 either in the luminal multipotent progenitor or a precursor has revealed a naturally transient population, possibly by inhibiting the normal rate of differentiation. This interpretation is consistent with considerable evidence linking Tp53 to the regulation of differentiation in stem cells (Bonizzi et al., 2011; Cicalese et al., 2009).
To date, luminal multipotent progenitor cells have not been observed in lineage tracing experiments (Choi et al., 2012; Ousset et al., 2012; Wang et al., 2013), except in the case of rare CARN’s (Wang et al., 2009), prompting questions about the significance of the multipotent progenitors revealed in organoid cultures. We show the existence of multipotent and luminal-committed TICs isolated directly from tumors, producing either adenosquamous carcinoma or adenocarcinoma, respectively. Importantly, the TIC assays used here measured autonomous differentiation potential in the absence of inductive embryonic urogenital mesenchyme (Neubauer et al., 1983; Taylor et al., 2009). Endogenous adenosqamous prostate carcinoma is observed in a fraction of PB-CRE4; Ptenfl/fl;Tp53fl/fl mice (Martin et al., 2011), supporting the concept that transformed multipotent progenitors exist in vivo and can differentiate to both basal and luminal lineages in tumors in situ. It seems likely that the microenvironment will influence lineage commitment, and we note that organoids and TIC assays are performed in the absence of stromal cells. Therefore, it is possible in these assays that the extent of basal cell commitment by multilineage progenitors may be increased relative to the endogenous microenvironment.
Although engineered models of prostate cancer are often used to analyze the consequences of combined genetic mutations (Shen and Abate-Shen, 2010), the effect upon stem/progenitor populations has not been commonly considered. We show here for the PB-CRE4-initiated genetic changes that Tp53 in combination with Pten loss demonstrated significantly different stem/progenitor populations compared to Pten loss alone. Specifically, Tp53 loss leads to the presence of luminal multipotent stem/progenitor cells and a self-renewing luminal population, correlated with accelerated adenocarcinoma development, that is absent in Pten null prostates. In addition, it is possible that Tp53 loss primes for lineage plasticity, similarly to the phenotypic dedifferentiation of luminal mammary epithelium following Brca1 loss (Molyneux et al., 2010). Analyses of stem/progenitor populations contribute fundamental knowledge for molecular and pathological comparisons of GEM models and for interpretation of target populations responding to therapeutics, as exemplified for castration/Enzalutamide sensitivity in Figure 6.
Due to a lack of biomarkers, the extent of innate stem/progenitor subpopulation heterogeneity in human prostate cancer is not known. However, histological heterogeneity is common in castrate resistant metastatic prostate cancer (Roudier et al., 2003; Shah et al., 2004), suggesting expression of “stemness” properties through pre-existing or acquired mechanisms. With respect to the multipotent progenitor described here, the existence of prostate adenosquamous carcinoma in humans is exceedingly rare (Giannico et al., 2013; Humphrey, 2011). However, the temporal accumulation of mutations in human prostate cancer or the microenvironment may favor luminal differentiation by multipotent progenitors. It remains to be determined whether a luminal multipotent stem/progenitor cell may be a cell of origin or similar to a cancer stem cell population in human prostate cancer. Another population described here, castration-tolerant luminal progenitor cells, are candidates as potential cellular targets for the accumulation of acquired resistance to androgen deprivation therapies (Goldstein et al., 2010). Similarly, castration-tolerant luminal cells have been observed in primary human prostate cancer xenografts (Toivanen et al., 2013). The ability to isolate and assay specific stem/progenitor populations from mice is an important resource for future investigations into molecular and proteomic markers that uniquely distinguish various stem/progenitor cell populations, which will enable refined lineage tracing approaches and histopathological analyses of clinical samples.
EXPERIMENAL PROCEDURES
Isolation, Labeling, and FACS Sorting of Primary Prostate Epithelial Cells
Single cell suspensions of WT and Pten−/−, Tp53−/− prostate tissue were prepared as previously described (Abou-Kheir et al., 2010). For labeling reactions, cells were resuspended in PBS (without Mg2+ or Ca2+) containing 1% heat-inactivated FBS. All antibodies and reagents were purchased from BD Pharmingen unless otherwise stated. Fcγ III/II receptors were blocked using anti-CD16/CD32 antibody for 15 min at 4°C. Cells were stained with CD45-FITC, CD31-FITC, Ter-119-FITC, EpCAM-APC-Cy7 (Biolegend), CD49f-PE and Prominin-1-APC (Miltenyi Biotech) for 30 min at 4°C. 7-aminoactinomycin D (Sigma) 100 μg ml−1 was added prior to analysis. Cell sorting was performed on FACSVantage and FACSAria cell sorters (Becton Dickinson) using FACSDiva software. The FACS gating strategy for CD49f/PROM1 fractionation is shown in (Fig. S2A).
Organoid culture, passage, and clone isolation
Single cell suspensions were cultured in organoid culture conditions, consisting of embedding cells within a Matrigel™ matrix and incubating with modified ENR media as described in Karthaus et. al (Karthaus et al., 2014), except that Matrigel™ containing cells was spread in a thin ring around the circumference of a six-well plate instead of as a drop. For passage, organoids were harvested and processed to single cells as described (Abou-Kheir et al., 2010) prior to being replated. For isolation of clonal organoids, 1000 PROM1+ cells were seeded on top of a Matrigel™ coated well of a six-well plate. Cells were allowed to attach for 30 mins at 37 degrees. Subsequently, cells were covered with 1 ml of media containing 5% Matrigel™. After 6–7 days, organoids were individually isolated under the microscope, deposited into a single well of a 96-well plate, visually checked for purity, and then trypsinized (0.05%) and seeded as single cells embedded within Matrigel™ into a 12 well plate. Organoids were then passaged over several generations at least through G6.
Additional methods are included in supplemental methods.
Supplementary Material
Acknowledgments
We thank Subhadra Banerjee, Barbara Taylor and Karen Wolcott at the NCI FACS Core Facility and Dr. Yvona Ward at the Center for Cancer Research, LGCP Microscopy Core Facility.
Footnotes
AUTHOR CONTRIBUTIONS
P.H., S.A., W.A.-K. and K.K. designed experiments. P.H., S.A., R.L., W.A-K., L.F., O.C., A.A., and J.J.Y. performed experiments and analyzed data. H.T. and P.M. performed pathology analyses. P.I., W.K., H.C., and C.S. provided reagents and analyzed data. P.H., S.A. and K.K. wrote the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abou-Kheir WG, Hynes PG, Martin PL, Pierce R, Kelly K. Characterizing the contribution of stem/progenitor cells to tumorigenesis in the Pten−/−TP53−/− prostate cancer model. Stem Cells. 2010;28:2129–2140. doi: 10.1002/stem.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C, Fuchs E. Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science. 2014;344:1242281. doi: 10.1126/science.1242281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzi G, Cicalese A, Insinga A, Pelicci PG. The emerging role of p53 in stem cells. Trends Mol Med. 2011;18:6–12. doi: 10.1016/j.molmed.2011.08.002. [DOI] [PubMed] [Google Scholar]
- Boutros PC, Fraser M, Harding NJ, de Borja R, Trudel D, Lalonde E, Meng A, Hennings-Yeomans PH, McPherson A, Sabelnykova VY, et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat Genet. 2015;47:736–745. doi: 10.1038/ng.3315. [DOI] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi N, Zhang B, Zhang L, Ittmann M, Xin L. Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell. 2012;21:253–265. doi: 10.1016/j.ccr.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua CW, Shibata M, Lei M, Toivanen R, Barlow LJ, Bergren SK, Badani KK, McKiernan JM, Benson MC, Hibshoosh H, et al. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat Cell Biol. 2014;16:951–961. 951–954. doi: 10.1038/ncb3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B, Brisken C, Minucci S, Di Fiore PP, Pelicci PG. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138:1083–1095. doi: 10.1016/j.cell.2009.06.048. [DOI] [PubMed] [Google Scholar]
- Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159:176–187. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannico GA, Ross HM, Lotan T, Epstein JI. Aberrant expression of p63 in adenocarcinoma of the prostate: a radical prostatectomy study. Am J Surg Pathol. 2013;37:1401–1406. doi: 10.1097/PAS.0b013e31828d5c32. [DOI] [PubMed] [Google Scholar]
- Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science. 2010;329:568–571. doi: 10.1126/science.1189992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey PA. Histological variants of prostatic carcinoma and their significance. Histopathology. 2011;60:59–74. doi: 10.1111/j.1365-2559.2011.04039.x. [DOI] [PubMed] [Google Scholar]
- Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, Dowling CM, Gao D, Begthel H, Sachs N, et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell. 2014;159:163–175. doi: 10.1016/j.cell.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsten H, Ziel-van der Made A, Ma X, van der Kwast T, Trapman J. Accumulating progenitor cells in the luminal epithelial cell layer are candidate tumor initiating cells in a Pten knockout mouse prostate cancer model. PLoS One. 2009;4:e5662. doi: 10.1371/journal.pone.0005662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DK, Liu Y, Liao L, Wang F, Xu J. The prostate basal cell (BC) heterogeneity and the p63-positive BC differentiation spectrum in mice. Int J Biol Sci. 2014;10:1007–1017. doi: 10.7150/ijbs.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YN, Abou-Kheir W, Yin JJ, Fang L, Hynes P, Casey O, Hu D, Wan Y, Seng V, Sheppard-Tillman H, et al. Critical and reciprocal regulation of KLF4 and SLUG in transforming growth factor beta-initiated prostate cancer epithelial-mesenchymal transition. Mol Cell Biol. 2012;32:941–953. doi: 10.1128/MCB.06306-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P, Liu YN, Pierce R, Abou-Kheir W, Casey O, Seng V, Camacho D, Simpson RM, Kelly K. Prostate epithelial Pten/TP53 loss leads to transformation of multipotential progenitors and epithelial to mesenchymal transition. Am J Pathol. 2011;179:422–435. doi: 10.1016/j.ajpath.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missol-Kolka E, Karbanova J, Janich P, Haase M, Fargeas CA, Huttner WB, Corbeil D. Prominin-1 (CD133) is not restricted to stem cells located in the basal compartment of murine and human prostate. Prostate. 2010;71:254–267. doi: 10.1002/pros.21239. [DOI] [PubMed] [Google Scholar]
- Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R, Mackay A, Grigoriadis A, Tutt A, Ashworth A, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010;7:403–417. doi: 10.1016/j.stem.2010.07.010. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland DJ, Xin L, Morim A, Lawson D, Witte O, Wu H. Lin-Sca-1+CD49fhigh stem/progenitors are tumor-initiating cells in the Pten-null prostate cancer model. Cancer Res. 2009;69:8555–8562. doi: 10.1158/0008-5472.CAN-08-4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer BL, Chung LW, McCormick KA, Taguchi O, Thompson TC, Cunha GR. Epithelial-mesenchymal interactions in prostatic development. II. Biochemical observations of prostatic induction by urogenital sinus mesenchyme in epithelium of the adult rodent urinary bladder. J Cell Biol. 1983;96:1671–1676. doi: 10.1083/jcb.96.6.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousset M, Van Keymeulen A, Bouvencourt G, Sharma N, Achouri Y, Simons BD, Blanpain C. Multipotent and unipotent progenitors contribute to prostate postnatal development. Nat Cell Biol. 2012;14:1131–1138. doi: 10.1038/ncb2600. [DOI] [PubMed] [Google Scholar]
- Peng W, Bao Y, Sawicki JA. Epithelial cell-targeted transgene expression enables isolation of cyan fluorescent protein (CFP)-expressing prostate stem/progenitor cells. Transgenic research. 2011;20:1073–1086. doi: 10.1007/s11248-010-9478-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignon JC, Grisanzio C, Geng Y, Song J, Shivdasani RA, Signoretti S. p63-expressing cells are the stem cells of developing prostate, bladder, and colorectal epithelia. Proc Natl Acad Sci U S A. 2013;110:8105–8110. doi: 10.1073/pnas.1221216110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios AC, Fu NY, Lindeman GJ, Visvader JE. In situ identification of bipotent stem cells in the mammary gland. Nature. 2014;506:322–327. doi: 10.1038/nature12948. [DOI] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roudier MP, True LD, Higano CS, Vesselle H, Ellis W, Lange P, Vessella RL. Phenotypic heterogeneity of end-stage prostate carcinoma metastatic to bone. Hum Pathol. 2003;34:646–653. doi: 10.1016/s0046-8177(03)00190-4. [DOI] [PubMed] [Google Scholar]
- Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, Macvicar GR, Varambally S, Harwood J, Bismar TA, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64:9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010;24:1967–2000. doi: 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M, Shen MM. The roots of cancer: stem cells and the basis for tumor heterogeneity. Bioessays. 2012;35:253–260. doi: 10.1002/bies.201200101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RA, Wang H, Wilkinson SE, Richards MG, Britt KL, Vaillant F, Lindeman GJ, Visvader JE, Cunha GR, St John J, et al. Lineage enforcement by inductive mesenchyme on adult epithelial stem cells across developmental germ layers. Stem Cells. 2009;27:3032–3042. doi: 10.1002/stem.244. [DOI] [PubMed] [Google Scholar]
- Toivanen R, Berman DM, Wang H, Pedersen J, Frydenberg M, Meeker AK, Ellem SJ, Risbridger GP, Taylor RA. Brief report: a bioassay to identify primary human prostate cancer repopulating cells. Stem Cells. 2011;29:1310–1314. doi: 10.1002/stem.668. [DOI] [PubMed] [Google Scholar]
- Toivanen R, Frydenberg M, Murphy D, Pedersen J, Ryan A, Pook D, Berman DM, Taylor RA, Risbridger GP. A preclinical xenograft model identifies castration-tolerant cancer-repopulating cells in localized prostate tumors. Sci Transl Med. 2013;5:187ra171. doi: 10.1126/scitranslmed.3005688. [DOI] [PubMed] [Google Scholar]
- Van Keymeulen A, Rocha AS, Ousset M, Beck B, Bouvencourt G, Rock J, Sharma N, Dekoninck S, Blanpain C. Distinct stem cells contribute to mammary gland development and maintenance. Nature. 2011;479:189–193. doi: 10.1038/nature10573. [DOI] [PubMed] [Google Scholar]
- van Leenders G, Dijkman H, Hulsbergen-van de Kaa C, Ruiter D, Schalken J. Demonstration of intermediate cells during human prostate epithelial differentiation in situ and in vitro using triple-staining confocal scanning microscopy. Laboratory investigation; a journal of technical methods and pathology. 2000;80:1251–1258. doi: 10.1038/labinvest.3780133. [DOI] [PubMed] [Google Scholar]
- Wang S, Garcia AJ, Wu M, Lawson DA, Witte ON, Wu H. Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc Natl Acad Sci U S A. 2006;103:1480–1485. doi: 10.1073/pnas.0510652103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Kruithof-de Julio M, Economides KD, Walker D, Yu H, Halili MV, Hu YP, Price SM, Abate-Shen C, Shen MM. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461:495–500. doi: 10.1038/nature08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZA, Mitrofanova A, Bergren SK, Abate-Shen C, Cardiff RD, Califano A, Shen MM. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat Cell Biol. 2013;15:274–283. doi: 10.1038/ncb2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZA, Shen MM. Revisiting the concept of cancer stem cells in prostate cancer. Oncogene. 2011;30:1261–1271. doi: 10.1038/onc.2010.530. [DOI] [PubMed] [Google Scholar]
- Xin L, Lukacs RU, Lawson DA, Cheng D, Witte ON. Self-renewal and multilineage differentiation in vitro from murine prostate stem cells. Stem Cells. 2007;25:2760–2769. doi: 10.1634/stemcells.2007-0355. [DOI] [PubMed] [Google Scholar]
- Zong Y, Goldstein AS. Adaptation or selection--mechanisms of castration-resistant prostate cancer. Nat Rev Urol. 2013;10:90–98. doi: 10.1038/nrurol.2012.237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.