

Abstract
Previously considered a disease isolated to the pulmonary circulation, pulmonary arterial hypertension is now being recognized as a systemic disorder that is associated with significant metabolic dysfunction. Numerous animal models have demonstrated the development of pulmonary arterial hypertension following the onset of insulin resistance, indicating that insulin resistance may be causal. Recent publications highlighting alterations in aerobic glycolysis, fatty acid oxidation, and the tricarboxylic acid cycle in the pulmonary circulation and right ventricle have expanded our understanding of the complex pathobiology of this disease. By targeting these derangements in metabolism, numerous researchers are investigating noninvasive techniques to monitor disease activity and therapeutics that address the underlying metabolic condition. In the following review, we will explore pre-clinical and clinical studies investigating the metabolic dysfunction seen in pulmonary arterial hypertension.
Keywords: Pulmonary arterial hypertension, Insulin Resistance, Aerobic Glycolysis, BMPR2, PPARγ, Adiponectin, Apolipoprotein E, Right ventricular failure, Lipotoxicity, Metformin
Introduction
Pulmonary arterial hypertension (PAH) is a highly morbid and fatal illness defined by progressive pulmonary vascular obstruction, which ultimately leads to right ventricular failure and death [1–3]. While the disease is characterized histopathologically as an arteriopathy of small-to-medium-sized pulmonary arteries [4], PAH is increasingly being recognized as a systemic illness with a predilection for the pulmonary vasculature and right ventricle (RV) [5].
There is a growing body of evidence that a variety of systemic metabolic derangements are associated with, if not causative of, PAH. Much of the earlier research on this subject has focused on the role of insulin resistance (IR), glucose intolerance, and the metabolic syndrome (MS) on disease progression within the pulmonary circulation [6–9]. Several new discoveries over the past few years have illuminated more extensive metabolic dysfunction in PAH, as alterations in aerobic glycolysis, fatty acid oxidation (FAO), and the tricarboxylic acid (TCA) cycle [10] have been associated with PAH and the development of lipotoxicity in the RV [11]. Ongoing research will elucidate whether these metabolic alterations contribute to or result from pulmonary vascular disease.
As there are currently no known curative treatments for this devastating illness, pulmonary vasodilators, such as endothelin receptor antagonists, phosphodiesterase-5 inhibitors, and prostaglandins, are utilized to improve pulmonary vascular and right ventricular hemodynamics with varying degrees of success [12]. It is presently unclear whether treatments that target metabolic dysfunction in PAH will improve pulmonary vascular disease. Due to their ready availability and ease of administration, several FDA-approved metabolically-active drugs are being considered for the treatment of PAH, though none are known to have a beneficial effect in PAH. With improved understanding of the specific metabolic pathobiology of PAH, novel therapeutic modalities will hopefully emerge that treat the underlying disease state, rather than the secondary hemodynamic consequences. This review will focus on both basic science mechanisms of metabolic dysfunction and clinical knowledge regarding the potential role of altered metabolism in PAH.
Basic Mechanisms of Metabolic Disease in the Pulmonary Vasculature
BMPR2 Mutation and Insulin Resistance in Pulmonary Arterial Hypertension
Bone morphogenic protein receptor type 2 (BMPR2) is a protein kinase that, after binding its bone morphogenic protein (BMP) ligands of the transforming growth factor-β (TGF-β) superfamily, modulates tissue injury repair and many other signaling mechanisms including cytoskeletal function [13]. Germline mutations in BMPR2 are well-known causes of heritable PAH (HPAH) [14, 15], and are thought to induce PAH through increased apoptosis of pulmonary vascular endothelial cells [16] and uncontrolled proliferation of pulmonary vascular smooth muscle cells [17]. The inheritance pattern in HPAH is autosomal dominant with reduced penetrance (typically around 20%) [18]. While other novel genetic mutations associated with PAH have been discovered in recent years, approximately 80% of known HPAH is caused by BMPR2 mutations [5]. In fact, other forms of pulmonary hypertension have been associated with sporadic BMPR2 mutations, most notably idiopathic PAH (IPAH) [19], stimulant-associated PAH [20], and pulmonary veno-occlusive disease [21], and BMPR2 expression is reduced in IPAH even in the absence of germline BMPR2 mutations [22]. Given the clinical, histological, and genetic similarities between heritable and other forms of PAH [23], BMPR2 models of PAH are ideal for the study of molecular mechanisms of disease in PAH.
One known downstream target of BMPR2 is peroxisome proliferator–activated receptor-γ (PPARγ), a transcription factor that belongs to the nuclear receptor family, which has many downstream targets involved in vascular modeling and glucose homeostasis [24]. Two important downstream targets of PPARγ are apolipoprotein E (ApoE) [25] and adiponectin [26]. ApoE is predominantly involved in reverse cholesterol transport by macrophages [27], and adiponectin, a so-called adipokine as it is a cell-signaling protein secreted exclusively by adipocytes, is important for regulating endothelial inflammation [28]. Although their biochemical pathways are mostly unrelated, both proteins inhibit platelet-derived growth factor-β (PDGF-β) function.
While ApoE inhibits PDGF-β function by binding to lipoprotein-like receptor protein [29] and adiponectin binds the homodimer of PDGF-β [30], both decrease its bioavailability. The downstream consequences of heightened PDGF-β signaling on vascular smooth muscle cells are profound, as increased PDGF-β concentration leads to dysregulated vascular smooth muscle cell proliferation and survival due to upregulation of cell cycle-promoting genes via PDGF-β-dependent MAP kinase activation [31].
Circulating levels of PPARγ [32] and ApoE [33] have been noted to be reduced in PAH, either via BMPR2-mediated suppression or otherwise. It is important to note that aside from BMPR2 mutations or the presence of pulmonary vascular disease, reduced levels of PPARγ [34], ApoE [35], and adiponectin [36] have all been associated with a variety of pathologic metabolic states, such as obesity, type II diabetes mellitus (DMII), and IR.
Hansmann et al sought out to determine whether reduced levels of PPARγ and ApoE seen in PAH are causative, or merely biologic spectators [6]. By using an ApoE (−/−) mouse model, they observed that mice fed a high fat diet were prone to develop PAH, with a much more severe phenotype observed in the male cohort. They determined that the degree of PAH was inversely proportional to adiponectin levels, and since females had higher adiponectin levels than males at baseline, they were relatively protected from the development of PAH. IR developed in the male Apo (−/−) mice fed a high fat diet, and not in the corresponding female mice, indicative of the protective role of adiponectin in the development of IR as well. A fundamental observation from these data was that PPARγ activation with rosiglitazone raised adiponectin levels, improved insulin sensitivity, and ultimately reversed PAH in these mice.
Further supporting a role for adipokines in PAH, adiponectin knockout mice have also demonstrated significant vascular remodeling and PAH. Adiponectin (−/−) mice developed PAH in an age-dependent manner [37], and the phenotype was intensified when these mice were exposed to acute allergic airway inflammation independent of hypoxia [38]. The mechanisms through which airway disease stimulates pulmonary vascular disease in this model are as yet unclear, but these data suggest that adipokines are active in the lungs and may influence pulmonary vascular disease.
Our group has demonstrated a high degree of IR in a transgenic rodent model of inducible mutant BMPR2 overexpression. In this model, transgenic mice developed early IR (within two weeks of transgene activation and prior to development of PAH) associated with rapid weight gain [39]. We further found tissue markers of lipid deposition, including fat staining in the peripheral muscles [40]. Similarly to Apo (−/−) mice, BMPR2 mutant mice fed a high-fat diet developed a more severe PAH phenotype with higher penetrance of the disease [40]. Furthermore, we demonstrated that BMPR2 mutant mice developed glucocorticoid receptor translocation abnormalities, which led to glucocorticoid resistance that may underlie some of the metabolic findings in this model.
These data taken together are interesting for several reasons. First, they demonstrate that BMPR2 signaling is linked downstream to PPARγ. Since PPARγ is a known “master regulator” of insulin sensitivity, dysfunctional BMPR2 expression may influence cellular glucose homeostasis. Additionally, in a model of BMPR2 mutation, IR develops before pulmonary hypertension, suggesting a causative role for IR in the development of pulmonary vascular disease. Finally, these studies show that traditional animal models of IR such as the ApoE or adiponectin (−/−) mice develop pulmonary hypertension, demonstrating that IR may promote pulmonary vascular disease independent of BMPR2 signaling (Figure 1).
Figure 1.
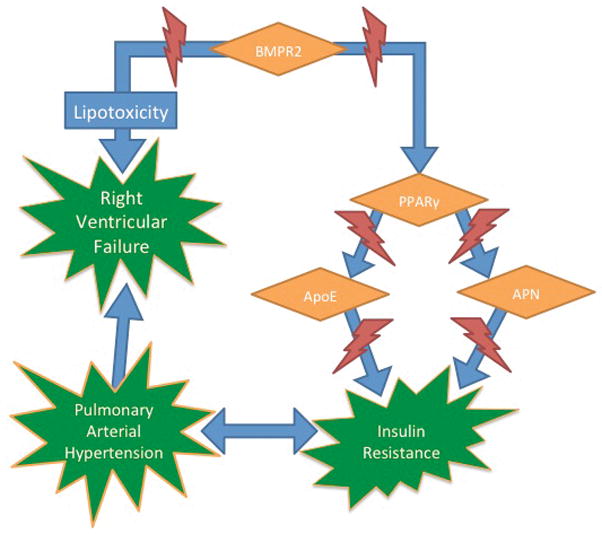
Insulin resistance and PAH. BMPR2, PPARγ, ApoE, and APN knockout mice all lead to the development of insulin resistance (IR) prior to pulmonary arterial hypertension (PAH), and IR is commonly associated with PAH in human studies. BMPR2 mutations also lead to right ventricular lipotoxicity. BMPR2 = bone morphogenic protein receptor 2, PPARγ = peroxisome proliferator–activated receptor-γ, ApoE = apolipoprotein E, APN = adiponectin.
Other Affected Metabolic Pathways in Pulmonary Arterial Hypertension
IR seems to play a major role in PAH and has thus far been the best studied, however observed derangements in glycolytic, TCA cycle, carnitine, fatty acid, and glutamate metabolism indicate a more widespread metabolic disease state beyond glucose homeostasis. It has been noted that aerobic glycolysis, otherwise known as the “Warburg effect”, is highly prevalent in patients with PAH [41]. The Warburg effect is considered an adaptive mechanism exhibited by rapidly proliferating cells (notably cancer cells) that allows for unrestrained growth [42]. It is also the fundamental principle behind positron emission tomography-computed tomography (PET-CT) detection of radioactively labeled fluorodeoxy-D-glucose (FDG) uptake in such cells [43]. While the ATP generated per glucose molecule in glycolysis is far less than via mitochondrial respiration, the reliance on less efficient catabolism may have some advantages when glucose uptake is not rate-limiting [44].
Marsboom et al explored alterations in glucose metabolism in two different mouse models of PAH, both the monocrotaline and SU5416 with chronic hypoxia models. FDG uptake in lung tissue as determined by PET-CT scan increased simultaneously with the development of PAH [45], suggesting that increased glucose turnover can be detected non-invasively by FDG uptake and is a relatively early feature of pulmonary vascular disease in these rodent models. The observed increase in aerobic glycolysis was found to be secondary to increased glucose transporter 1 expression in PA endothelial and smooth muscle cells in vivo and in vitro, partly due to increased hypoxia-inducible factor-1α (HIF-1α) expression.
These largely mechanistic studies have focused on the glucose metabolic pathway, but other fundamental areas of metabolism have been less explored. We studied the metabolic profile of pulmonary microvascular endothelial cells (PMVEC) in humans and BMPR2 mutant mice. Fessel et al showed that human PMVEC transfected with BMPR2 vector were noted to have extensive alterations in their gene expression when compared to empty vector controls [10]. Nearly one-half of the altered genes regulated small molecule metabolism, and there was evidence of increased aerobic glycolysis and decreased FAO. Furthermore, we observed increased pentose phosphate pathway activation, decreased carnitine metabolism, decreased TCA cycle enzymatic activity distal to citrate, and increased catabolism of peptides and amino acids. This unbiased discovery approach has demonstrated that multiple metabolic pathways are affected in cells with a mutation known to cause PAH. Although failure of adequate glucose homeostasis is an important feature of PAH, it is likely not the only metabolic derangement in this disease.
Basic Mechanisms of Right Ventricular Metabolic Disease
Altered metabolism is not isolated to the PMVEC or PA smooth muscle cells in PAH, but is also seen in the RV. It is clear that different animal models of PAH have varying effects on the RV. Some models, such as the pulmonary artery banding (PAB) model, induce an early and adaptive model of RV hypertrophy soon after the intervention. Other models, such as those that utilize the endothelial cell toxin monocrotaline, stimulate early and dysfunctional changes in the RV, such as dilation and fibrosis [46, 47].
Exploiting this distinction between models, investigators have shown that the RV of mice treated with PAB as compared to a SU5416 and chronic hypoxia model demonstrate different metabolic profiles [48]. Specifically, the RV of mice treated with SU5416 and chronic hypoxia had decreased expression of PPARγ, its cofactors, and its target genes. There was also a qualitative and quantitative reduction in mitochondria associated with reduced oxidative capacity. These changes were not found in the corresponding mice treated with PAB.
In contrast, other groups have observed that even the hypertrophied RVs of mice that have undergone PAB demonstrated a shift away from glucose oxidation towards aerobic glycolysis and FAO [49]. While FAO is the preferred form of ATP-generation in cardiomyocytes in the healthy RV [50, 51], inhibitors of FAO (trimetazidine and ranolazine) induced an increase in cardiac output and exercise capacity, with a regression of established RV hypertrophy in the PAB model. Inhibitors of FAO may improve RV hemodynamics by shifting RV metabolism towards glucose oxidation, by the so-called “Randle cycle” [52], thus improving the oxidative metabolism of RV cardiomyocytes [47]. The long-term consequences of suppressing FAO in the heart, however, are presently unstudied and may not be beneficial based on the normal metabolic preferences of the heart.
In the case of PAH associated with germline BMPR2 mutations, the metabolic effects would be predicted to affect all tissues of the body to varying degrees. The RV changes seen in BMPR2 mutant mice are illustrative of the breadth of metabolic alterations in the context of PAH, as we have demonstrated a maladaptive RV response that is associated with marked myocardial lipotoxicity. The compensatory concentric RV hypertrophy that is seen in PAB-treated mice and smooth muscle-specific BMPR2 mutant mice is mitigated in mice with a systemic BMPR2 mutation, despite moderate increases in PA pressures [11]. The RVs of these mice demonstrated extensive lipid deposition (notably triglycerides and ceramides) within the cardiomyocytes. Circulating lipids were not elevated in these mice, and extracellular RV and left ventricle (LV) lipid deposition was minimal. This effect, though profound, could be reversed qualitatively and quantitatively by metformin administration.
The reason behind this observed lipotoxicity is under active investigation, however it is almost certainly multifactorial. We have shown that there is increased expression of CD36, a fatty acid transporter, in the RV of BMPR2 mutant mice and control mice fed a high-fat diet [53], which may be responsible for the increase in fatty acid entry into RV cardiomyocytes. Expression patterns in human RVs have suggested suppression of FAO [11], thus the utilization of fatty acids may be reduced at a mitochondrial level in cardiomyocytes with BMPR2 mutations [10]. The increase in fatty acid uptake, coupled with potentially reduced utilization, necessitates the conversion of these free fatty acids into complex lipids, such as triglycerides and ceramides [54]. Once in the cytoplasm of RV cardiomyocytes, these lipids fail to be transported out of the cells and may mediate the RV dysfunction seen in these mice. The mechanisms of how lipotoxicity alters RV hypertrophy and compensation are currently unknown and are active areas of exploration. Development of RV-specific metabolic therapies is the ultimate goal of these studies of lipotoxicity in the RV of patients with PAH.
Human Studies Involving Metabolic Disease and the Pulmonary Vasculature
Insulin Resistance in Pulmonary Arterial Hypertension
While IR can be broadly defined as an abnormal clinical response to a physiologic amount of insulin, there is a wide array of clinical conditions associated with it, ranging from frank DMII to the MS [55]. While the American Diabetes Association has identified several modalities for the diagnosis of DMII, notably a hemoglobin A1c greater than 6.5 [56], the diagnosis of the MS is less clearly defined. The most unified definition was jointly published by several American and international organizations in 2009, and includes the presence of or treatment for three out of the following five entities: elevated waist circumference, elevated triglycerides (TG), reduced high density lipoproteins (HDL), elevated blood pressure, or elevated fasting blood glucose [57].
DM and the MS have a steadily increasing prevalence around the USA and world, occurring in approximately 1/8 and 1/3 of American adults, respectively [58, 59]. IR is thought to be the underlying defect in both conditions [60], and several biomarkers have been validated as reasonable surrogates for IR, particularly an elevated TG/HDL ratio and the homeostasis model assessment (HOMA), which utilizes fasting glucose and insulin levels to estimate pancreatic beta cell function and insulin sensitivity [61–63].
Numerous investigators have demonstrated that high levels of pro-inflammatory biomarkers, such as C-reactive protein, interleukin 6, and myeloperoxidase, are seen in conditions associated with IR [64–67], and this inflammatory milieu is thought to underlie the associated endothelial dysfunction and vascular disease seen in patients with obesity, DM, and the MS. While much is known about the association of IR and vascular disease in the systemic circulation [68, 69], the role of IR and other metabolic derangements on the pulmonary circulation and RV is an emerging field (Table 1).
Table 1.
Widespread zmetabolic dysfunction in PAH. The following systemic and right ventricular defects in metabolism have been identified in humans with pulmonary arterial hypertension (PAH). While very few targeted therapeutics have been studied in humans, animal models and case reports have demonstrated potential metabolic targets.
| Tissue(s) Affected | Metabolic Defect | Supporting Data and References | Investigative Therapeutics |
|---|---|---|---|
| Systemic Changes |
 IR IR |
|
|
 Glucose Tolerance Glucose Tolerance |
|||
|
HDL |
|
||
| Altered TCA Cycle |
|
|
|
|
Adiponectin |
|
|
|
|
Leptin |
|
||
|
Ghrelin |
|
||
| Right Ventricle |
Aerobic Glycolysis |
|
|
|
FAO | |||
| Altered TCA cycle | |||
| Lipid Deposition |
|
|
Validating the associations seen between IR and PAH in animal models, there is a growing body of data supporting these findings in humans. When compared to age-matched controls from the National Health and Nutrition Examination Survey (NHANES) database, non-diabetic female patients with PAH had significantly more IR assessed by the TG/HDL ratio [7]. Interestingly, when compared with insulin sensitive PAH patients, IR PAH patients had the same age and weight profile; however, when compared with the IR control patients, the IR PAH patients were both younger and less frequently overweight. Our group has shown that as many as 2/3 of non-diabetic patients with PAH will demonstrate some degree of glucose intolerance, defined as a hemoglobin A1c greater than 6.0 [8]. In fact, 15% of the patients evaluated were newly diagnosed with DMII because of the analysis. Similar to what Zamanian et al demonstrated, glucose intolerance in this population seems to be independent of other risk factors for IR, as age and BMI between PAH patients with and without glucose intolerance were equal. While IR and the MS are highly associated with PAH, the association is even stronger for pulmonary venous hypertension, with two of the three features of the MS seen in nearly all patients with PVH [70].
IR and disordered glucose metabolism also appear to impact survival in PAH. In both prior studies, while their degree of IR did not correlate with individual metrics of disease severity, IR PAH patients, as determined by both TG/HDL and hemoglobin A1c, have decreased short- and long-term survival [7, 9]. Aside from the TG/HDL ratio, a low HDL level was independently associated with worse outcomes in PAH [71].
While there are no published therapeutic trials in humans aimed at improving IR in PAH, we reported a potentially illustrative case of a female with IPAH and morbid obesity treated with laparoscopic Roux-en-Y gastric bypass surgery [72], an experiment of nature involving surgical correction of IR in PAH. Following surgery, and prior to any substantial weight loss, her pulmonary vascular and RV hemodynamics greatly improved. This trend continued as she sustained significant weight loss, and was paralleled with improved insulin sensitivity and lipid metabolism. Whether this pulmonary vascular improvement can be achieved by pharmacologic adjustments of metabolism or solely by gastric bypass is presently unknown. Moreover, as this is only a single case, bariatric surgery cannot presently be recommended as a therapy for PAH.
Other Affected Metabolic Pathways in Pulmonary Arterial Hypertension
Our group has shown that humans with HPAH have altered gene expression profiles in cultured lymphocytes from peripheral blood that predominantly affect the metabolic pathway [11]. In examining the RVs and serum from HPAH patients, we have also demonstrated reduced FAO and TCA cycle enzymatic expression compared with healthy controls [10, 11] and patients with dilated cardiomyopathy [11], favoring aerobic glycolysis instead. These data show conserved alterations in metabolic pathways across tissue types.
On the other hand, when the explanted lungs of patients with severe PAH following lung transplant were analyzed, Zhao et al noted an overall decrease in glycolysis and increase in TCA cycle activity [73]. The reason for this discrepancy is not entirely clear, however our patients all had HPAH at a presumed time of steady state, whereas the patients evaluated by Zhao et al had very advanced disease without mention of BMPR2 status.
Adipokines have recently been explored in PAH patients as biomarkers, if not mediators, of PAH. Adiponectin has been shown to be elevated in patients with PAH compared to age-, sex-, and BMI-matched controls in a large series [74]. At first glance this seems counterintuitive, as animal models of the disease demonstrate a protective role of adiponectin [6, 75, 76]. However, there is evidence of reduced survival in patients with left-sided heart failure and elevated adiponectin [77], so there may be a developed resistance to the once-protective protein similar to IR seen in the MS and DMII. Humbert et al investigated the role of another adipokine, leptin, in PAH. Compared to controls, PAH patients had increased serum leptin levels, with much of the leptin secretion occurring in the pulmonary endothelial cells [78]. Furthermore, the percentage of regulatory T cells expressing the leptin receptor was increased in PAH, with reduced function of these cells, indicating a leptin-dependent immunomodulatory effect in PAH [78]. Other reports have noted reduced levels of ghrelin, and reduced ghrelin-to-obestatin levels, in patients with PAH secondary to an ASD [79], further demonstrating the complex interplay between PAH and the digestive hormones.
Human Studies Involving Metabolic Disease and the Right Ventricle
Clinical data in humans has confirmed increased lipotoxicity and glycolytic activation in the RVs of patients with PAH. Similar to the RVs of BMPR2 mutant mice, humans with HPAH have evidence of lipid deposition limited to the cardiomyocytes of the RV, with sparing of the extracellular space and LV [11]. We have recently demonstrated that in HPAH and IPAH, patients with a diagnosis of DM had worse outcomes that appeared to correlate with markers of worse RV function, suggesting a negative impact of DM on RV stress response [80]. Interestingly, while the presence of IR was not associated with echocardiographic RV dysfunction in PAH in Brunner et al’s recent analysis, diastolic dysfunction of the LV was noted [81].
Building upon the PET-CT studies done on the lung tissue of mice with PAH, several authors have shown that PET-CT scans can also detect an increase in FDG uptake in the RV of patients with PAH [82, 83]. With the exception of patients receiving β-adrenergic receptor blockade, RV FDG uptake correlated well with worsening pulmonary hemodynamics and echocardiographic changes, and appeared to be HIF-1α-mediated. It appears that at least some of the increased RV glucose uptake could be attributed to a corresponding reduction in LV glucose uptake secondary to worsening cardiac output from LV under-filling [83].
Conclusions
Long known to be causal of an inflammatory cytokine profile, endothelial dysfunction, and systemic cardiovascular disease, IR and dysregulated glucose metabolism are now being recognized as major contributors to the development of pulmonary vascular disease. Through translational research involving animal models and human studies, we now recognize that alterations in glucose and fatty acid metabolism due to IR and aerobic glycolysis are associated with the development of uncontrolled vasoconstriction in the pulmonary circulation and RV lipotoxicity and dysfunction in patients with PAH.
While inherited or sporadic BMPR2 mutations seem to be partially responsible, as BMPR2 and its downstream targets are intimately involved in glucose homeostasis, many of the PAH animal models have demonstrated that the disease phenotype worsens with certain modifications, such as a high-fat diet. This seems to indicate a “second hit” phenomenon for the development of PAH, with the penetrance and severity of the disease dependent on environmental factors (such as poor diet or a sedentary lifestyle) in the presence of altered BMPR2 signaling.
Exploiting the altered glucose metabolism in PAH has opened the door for noninvasive measures to diagnose and follow the disease. The use of PET-CT scans may eventually minimize the need for direct hemodynamic data to monitor response to therapy or stratify patients at greater risk of developing RV failure. While it seems promising, this noninvasive modality has yet to replace the right heart catheterization as the gold standard for the diagnosis of PAH.
Therapeutically targeting this pathologic metabolic condition with medications such as thiazolidediones, metformin, or inhibitors of FAO is an area of active research by numerous investigators. While pulmonary vasodilators will have a role in treating this condition for the foreseeable future, modifying the metabolic dysfunction of PAH in the pulmonary vasculature and RV is an exciting prospect for the treatment of this devastating illness and the goal of future pre-clinical and human studies.
Acknowledgments
Funding Sources: 1P01HL108800
Footnotes
Disclosure: No potential conflicts of interest relevant to this article were reported.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, et al. Updated clinical classification of pulmonary hypertension. Journal of the American College of Cardiology. 2013;62(25 Suppl):D34–41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 2.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) Circulation. 2010;122(2):164–72. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- 3.Farber HW, Loscalzo J. Pulmonary Arterial Hypertension. New England Journal of Medicine. 2004;351(16):1655–65. doi: 10.1056/NEJMra035488. [DOI] [PubMed] [Google Scholar]
- 4.Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM, et al. Pathologic assessment of vasculopathies in pulmonary hypertension. Journal of the American College of Cardiology. 2004;43(12 Suppl S):25S–32S. doi: 10.1016/j.jacc.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 5.Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res. 2014;115(1):189–202. doi: 10.1161/circresaha.115.303404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, et al. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation. 2007;115(10):1275–84. doi: 10.1161/CIRCULATIONAHA.106.663120. [DOI] [PubMed] [Google Scholar]
- 7.Zamanian RT, Hansmann G, Snook S, Lilienfeld D, Rappaport KM, Reaven GM, et al. Insulin resistance in pulmonary arterial hypertension. The European respiratory journal. 2009;33(2):318–24. doi: 10.1183/09031936.00000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8•.Pugh ME, Robbins IM, Rice TW, West J, Newman JH, Hemnes AR. Unrecognized glucose intolerance is common in pulmonary arterial hypertension. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2011;30(8):904–11. doi: 10.1016/j.healun.2011.02.016. This was one of the first clinical studies that demonstrated glucose intolerance was very prevalent in patients with PAH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belly MJ, Tiede H, Morty RE, Schulz R, Voswinckel R, Tanislav C, et al. HbA1c in pulmonary arterial hypertension: a marker of prognostic relevance? The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2012;31(10):1109–14. doi: 10.1016/j.healun.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 10••.Fessel JP, Hamid R, Wittmann BM, Robinson LJ, Blackwell T, Tada Y, et al. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulmonary circulation. 2012;2(2):201–13. doi: 10.4103/2045-8932.97606. This paper highlighted the extent and diversity of metabolic dysfunction seen in patients with PAH, notably an increase in aerobic glycolysis, pentose phosphate pathway activation, and peptide catabolism, along with a decrease in fatty acid oxidation, carnitine metabolism, and TCA enzymatic activity distal to citrate. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11••.Hemnes AR, Brittain EL, Trammell AW, Fessel JP, Austin ED, Penner N, et al. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. American journal of respiratory and critical care medicine. 2014;189(3):325–34. doi: 10.1164/rccm.201306-1086OC. This paper demonstrated that BMPR2 mutations, in a mouse model of PAH and in humans with HPAH, leads to lipid deposition in peripheral muscles and the right ventricle. This paper also demonstrated that a high fat diet in a BMPR2 mouse model amplifies the degree of PAH seen. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT) European heart journal. 2009;30(20):2493–537. doi: 10.1093/eurheartj/ehp297. [DOI] [PubMed] [Google Scholar]
- 13.West J. Cross talk between Smad, MAPK, and actin in the etiology of pulmonary arterial hypertension. Advances in experimental medicine and biology. 2010;661:265–78. doi: 10.1007/978-1-60761-500-2_17. [DOI] [PubMed] [Google Scholar]
- 14.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, et al. Familial Primary Pulmonary Hypertension (Gene PPH1) Is Caused by Mutations in the Bone Morphogenetic Protein Receptor–II Gene. The American Journal of Human Genetics. 2000;67(3):737–44. doi: 10.1086/303059. doi: http://dx.doi.org/10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.International PPHC, Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nature genetics. 2000;26(1):81–4. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 16.Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, Karoubi G, Courtman DW, Zucco L, et al. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res. 2006;98(2):209–17. doi: 10.1161/01.RES.0000200180.01710.e6. [DOI] [PubMed] [Google Scholar]
- 17.Yang X, Long L, Reynolds PN, Morrell NW. Expression of mutant BMPR-II in pulmonary endothelial cells promotes apoptosis and a release of factors that stimulate proliferation of pulmonary arterial smooth muscle cells. Pulmonary circulation. 2011;1(1):103–10. doi: 10.4103/2045-8932.78100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nichols WC, Koller DL, Slovis B, Foroud T, Terry VH, Arnold ND, et al. Localization of the gene for familial primary pulmonary hypertension to chromosome 2q31-32. Nature genetics. 1997;15(3):277–80. doi: 10.1038/ng0397-277. [DOI] [PubMed] [Google Scholar]
- 19.Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. Journal of medical genetics. 2000;37(10):741–5. doi: 10.1136/jmg.37.10.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Humbert M, Deng Z, Simonneau G, Barst RJ, Sitbon O, Wolf M, et al. BMPR2 germline mutations in pulmonary hypertension associated with fenfluramine derivatives. The European respiratory journal. 2002;20(3):518–23. doi: 10.1183/09031936.02.01762002. [DOI] [PubMed] [Google Scholar]
- 21.Runo JR, Vnencak-Jones CL, Prince M, Loyd JE, Wheeler L, Robbins IM, et al. Pulmonary Veno-occlusive Disease Caused by an Inherited Mutation in Bone Morphogenetic Protein Receptor II. American journal of respiratory and critical care medicine. 2003;167(6):889–94. doi: 10.1164/rccm.200208-861OC. [DOI] [PubMed] [Google Scholar]
- 22.Dewachter L, Adnot S, Guignabert C, Tu L, Marcos E, Fadel E, et al. Bone morphogenetic protein signalling in heritable versus idiopathic pulmonary hypertension. The European respiratory journal. 2009;34(5):1100–10. doi: 10.1183/09031936.00183008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Austin ED, Menon S, Hemnes AR, Robinson LR, Talati M, Fox KL, et al. Idiopathic and heritable PAH perturb common molecular pathways, correlated with increased MSX1 expression. Pulmonary circulation. 2011;1(3):389–98. doi: 10.4103/2045-8932.87308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansmann G, Zamanian RT. PPARgamma activation: a potential treatment for pulmonary hypertension. Science translational medicine. 2009;1(12):12ps4. doi: 10.1126/scitranslmed.3000267. [DOI] [PubMed] [Google Scholar]
- 25.Akiyama TE, Sakai S, Lambert G, Nicol CJ, Matsusue K, Pimprale S, et al. Conditional disruption of the peroxisome proliferator-activated receptor gamma gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Molecular and cellular biology. 2002;22(8):2607–19. doi: 10.1128/MCB.22.8.2607-2619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang WS, Jeng CY, Wu TJ, Tanaka S, Funahashi T, Matsuzawa Y, et al. Synthetic peroxisome proliferator-activated receptor-gamma agonist, rosiglitazone, increases plasma levels of adiponectin in type 2 diabetic patients. Diabetes care. 2002;25(2):376–80. doi: 10.2337/diacare.25.2.376. [DOI] [PubMed] [Google Scholar]
- 27.Galetto R, Albajar M, Polanco JI, Zakin MM, Rodriguez-Rey JC. Identification of a peroxisome-proliferator-activated-receptor response element in the apolipoprotein E gene control region. The Biochemical journal. 2001;357(Pt 2):521–7. doi: 10.1042/0264-6021:3570521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ouchi N, Kihara S, Arita Y, Maeda K, Kuriyama H, Okamoto Y, et al. Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation. 1999;100(25):2473–6. doi: 10.1161/01.cir.100.25.2473. [DOI] [PubMed] [Google Scholar]
- 29.Swertfeger DK, Bu G, Hui DY. Low density lipoprotein receptor-related protein mediates apolipoprotein E inhibition of smooth muscle cell migration. The Journal of biological chemistry. 2002;277(6):4141–6. doi: 10.1074/jbc.M109124200. [DOI] [PubMed] [Google Scholar]
- 30.Arita Y, Kihara S, Ouchi N, Maeda K, Kuriyama H, Okamoto Y, et al. Adipocyte-derived plasma protein adiponectin acts as a platelet-derived growth factor-BB-binding protein and regulates growth factor-induced common postreceptor signal in vascular smooth muscle cell. Circulation. 2002;105(24):2893–8. doi: 10.1161/01.cir.0000018622.84402.ff. [DOI] [PubMed] [Google Scholar]
- 31.Heldin CH, Westermark B. Platelet-derived growth factor: mechanism of action and possible in vivo function. Cell regulation. 1990;1(8):555–66. doi: 10.1091/mbc.1.8.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ameshima S, Golpon H, Cool CD, Chan D, Vandivier RW, Gardai SJ, et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ Res. 2003;92(10):1162–9. doi: 10.1161/01.RES.0000073585.50092.14. [DOI] [PubMed] [Google Scholar]
- 33.Geraci MW, Moore M, Gesell T, Yeager ME, Alger L, Golpon H, et al. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: a gene microarray analysis. Circ Res. 2001;88(6):555–62. doi: 10.1161/01.res.88.6.555. [DOI] [PubMed] [Google Scholar]
- 34.Park KS, Ciaraldi TP, Abrams-Carter L, Mudaliar S, Nikoulina SE, Henry RR. PPAR-gamma gene expression is elevated in skeletal muscle of obese and type II diabetic subjects. Diabetes. 1997;46(7):1230–4. doi: 10.2337/diab.46.7.1230. [DOI] [PubMed] [Google Scholar]
- 35.Eto M, Watanabe K, Ishii K. Apolipoprotein E polymorphism and hyperlipoproteinemia in obesity. International journal of obesity. 1989;13(4):433–40. [PubMed] [Google Scholar]
- 36.Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochemical and biophysical research communications. 1999;257(1):79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 37.Summer R, Fiack CA, Ikeda Y, Sato K, Dwyer D, Ouchi N, et al. Adiponectin deficiency: a model of pulmonary hypertension associated with pulmonary vascular disease. American journal of physiology Lung cellular and molecular physiology. 2009;297(3):L432–8. doi: 10.1152/ajplung.90599.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Medoff BD, Okamoto Y, Leyton P, Weng M, Sandall BP, Raher MJ, et al. Adiponectin deficiency increases allergic airway inflammation and pulmonary vascular remodeling. American journal of respiratory cell and molecular biology. 2009;41(4):397–406. doi: 10.1165/rcmb.2008-0415OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.West J, Fagan K, Steudel W, Fouty B, Lane K, Harral J, et al. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res. 2004;94(8):1109–14. doi: 10.1161/01.res.0000126047.82846.20. [DOI] [PubMed] [Google Scholar]
- 40.West J, Niswender KD, Johnson JA, Pugh ME, Gleaves L, Fessel JP, et al. A potential role for insulin resistance in experimental pulmonary hypertension. The European respiratory journal. 2013;41(4):861–71. doi: 10.1183/09031936.00030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rehman J, Archer SL. A proposed mitochondrial-metabolic mechanism for initiation and maintenance of pulmonary arterial hypertension in fawn-hooded rats: the Warburg model of pulmonary arterial hypertension. Advances in experimental medicine and biology. 2010;661:171–85. doi: 10.1007/978-1-60761-500-2_11. [DOI] [PubMed] [Google Scholar]
- 42.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. American journal of physiology Heart and circulatory physiology. 2008;294(2):H570–8. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 43.Kelloff GJ, Hoffman JM, Johnson B, Scher HI, Siegel BA, Cheng EY, et al. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11(8):2785–808. doi: 10.1158/1078-0432.ccr-04-2626. [DOI] [PubMed] [Google Scholar]
- 44.Vazquez A, Liu J, Zhou Y, Oltvai ZN. Catabolic efficiency of aerobic glycolysis: the Warburg effect revisited. BMC systems biology. 2010;4:58. doi: 10.1186/1752-0509-4-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45••.Marsboom G, Wietholt C, Haney CR, Toth PT, Ryan JJ, Morrow E, et al. Lung (1)(8)F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. American journal of respiratory and critical care medicine. 2012;185(6):670–9. doi: 10.1164/rccm.201108-1562OC. This study demonstrated that PET-CT scans can detect an increase in aerobic glycosis soon after the development of PAH in a mouse model of PAH. They determined that this was largely due to normoxic HIF-1α activation leading to increased glucose transporter 1 expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Das M, Fessel J, Tang H, West J. A process-based review of mouse models of pulmonary hypertension. Pulmonary circulation. 2012;2(4):415–33. doi: 10.4103/2045-8932.105030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Archer SL, Fang YH, Ryan JJ, Piao L. Metabolism and bioenergetics in the right ventricle and pulmonary vasculature in pulmonary hypertension. Pulmonary circulation. 2013;3(1):144–52. doi: 10.4103/2045-8932.109960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gomez-Arroyo J, Mizuno S, Szczepanek K, Van Tassell B, Natarajan R, dos Remedios CG, et al. Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circulation Heart failure. 2013;6(1):136–44. doi: 10.1161/CIRCHEARTFAILURE.111.966127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fang YH, Piao L, Hong Z, Toth PT, Marsboom G, Bache-Wiig P, et al. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle’s cycle. Journal of molecular medicine. 2012;90(1):31–43. doi: 10.1007/s00109-011-0804-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stanley WC, Lopaschuk GD, Hall JL, McCormack JG. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovascular research. 1997;33(2):243–57. doi: 10.1016/s0008-6363(96)00245-3. [DOI] [PubMed] [Google Scholar]
- 51.Randle PJ, Priestman DA, Mistry SC, Halsall A. Glucose fatty acid interactions and the regulation of glucose disposal. Journal of cellular biochemistry. 1994;55(Suppl):1–11. doi: 10.1002/jcb.240550002. [DOI] [PubMed] [Google Scholar]
- 52.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1(7285):785–9. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 53.Megha T, Niki P, Mitch F, Aaron WT, Joshua PF, James DW, et al. BMPR2 Mutation And Western Diet Are Associated With Altered Lipid Transport And Lipotoxicity In The Right Ventricle. A106. MOLECULAR MECHANISMS OF RIGHT VENTRICULAR DYSFUNCTION. American Thoracic Society International Conference Abstracts: American Thoracic Society; 2014; p. A2338-A. [Google Scholar]
- 54.Holloway GP, Jain SS, Bezaire V, Han XX, Glatz JF, Luiken JJ, et al. FAT/CD36-null mice reveal that mitochondrial FAT/CD36 is required to upregulate mitochondrial fatty acid oxidation in contracting muscle. American journal of physiology Regulatory, integrative and comparative physiology. 2009;297(4):R960–7. doi: 10.1152/ajpregu.91021.2008. [DOI] [PubMed] [Google Scholar]
- 55.Moller DE, Flier JS. Insulin Resistance — Mechanisms, Syndromes, and Implications. New England Journal of Medicine. 1991;325(13):938–48. doi: 10.1056/NEJM199109263251307. [DOI] [PubMed] [Google Scholar]
- 56.American Diabetes A. Diagnosis and classification of diabetes mellitus. Diabetes care. 2011;34(Suppl 1):S62–9. doi: 10.2337/dc11-S062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120(16):1640–5. doi: 10.1161/circulationaha.109.192644. [DOI] [PubMed] [Google Scholar]
- 58.Selvin E, Parrinello CM, Sacks DB, Coresh J. Trends in prevalence and control of diabetes in the United States, 1988–1994 and 1999–2010. Annals of internal medicine. 2014;160(8):517–25. doi: 10.7326/m13-2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ervin RB. Prevalence of metabolic syndrome among adults 20 years of age and over, by sex, age, race and ethnicity, and body mass index: United States, 2003–2006. National health statistics reports. 2009;(13):1–7. [PubMed] [Google Scholar]
- 60.DeFronzo RA, Ferrannini E. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes care. 1991;14(3):173–94. doi: 10.2337/diacare.14.3.173. [DOI] [PubMed] [Google Scholar]
- 61.McLaughlin T, Abbasi F, Cheal K, Chu J, Lamendola C, Reaven G. Use of metabolic markers to identify overweight individuals who are insulin resistant. Annals of internal medicine. 2003;139(10):802–9. doi: 10.7326/0003-4819-139-10-200311180-00007. [DOI] [PubMed] [Google Scholar]
- 62.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28(7):412–9. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 63.Hill NR, Levy JC, Matthews DR. Expansion of the homeostasis model assessment of beta-cell function and insulin resistance to enable clinical trial outcome modeling through the interactive adjustment of physiology and treatment effects: iHOMA2. Diabetes care. 2013;36(8):2324–30. doi: 10.2337/dc12-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gelaye B, Revilla L, Lopez T, Suarez L, Sanchez SE, Hevner K, et al. Association between insulin resistance and c-reactive protein among Peruvian adults. Diabetology & metabolic syndrome. 2010;2(1):30. doi: 10.1186/1758-5996-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA : the journal of the American Medical Association. 2001;286(3):327–34. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- 66.Yudkin JS, Stehouwer CD, Emeis JJ, Coppack SW. C-reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue? Arteriosclerosis, thrombosis, and vascular biology. 1999;19(4):972–8. doi: 10.1161/01.atv.19.4.972. [DOI] [PubMed] [Google Scholar]
- 67.Barzilay JI, Abraham L, Heckbert SR, Cushman M, Kuller LH, Resnick HE, et al. The relation of markers of inflammation to the development of glucose disorders in the elderly: the Cardiovascular Health Study. Diabetes. 2001;50(10):2384–9. doi: 10.2337/diabetes.50.10.2384. [DOI] [PubMed] [Google Scholar]
- 68.Ginsberg HN. Insulin resistance and cardiovascular disease. The Journal of Clinical Investigation. 2000;106(4):453–8. doi: 10.1172/JCI10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Steinberger J, Daniels SR. Obesity, insulin resistance, diabetes, and cardiovascular risk in children: an American Heart Association scientific statement from the Atherosclerosis, Hypertension, and Obesity in the Young Committee (Council on Cardiovascular Disease in the Young) and the Diabetes Committee (Council on Nutrition, Physical Activity, and Metabolism) Circulation. 2003;107(10):1448–53. doi: 10.1161/01.cir.0000060923.07573.f2. [DOI] [PubMed] [Google Scholar]
- 70.Robbins IM, Newman JH, Johnson RF, Hemnes AR, Fremont RD, Piana RN, et al. Association of the metabolic syndrome with pulmonary venous hypertension. Chest. 2009;136(1):31–6. doi: 10.1378/chest.08-2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Heresi GA, Aytekin M, Newman J, DiDonato J, Dweik RA. Plasma levels of high-density lipoprotein cholesterol and outcomes in pulmonary arterial hypertension. American journal of respiratory and critical care medicine. 2010;182(5):661–8. doi: 10.1164/rccm.201001-0007OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pugh ME, Newman JH, Williams DB, Brittain E, Robbins IM, Hemnes AR. Hemodynamic improvement of pulmonary arterial hypertension after bariatric surgery: potential role for metabolic regulation. Diabetes care. 2013;36(3):e32–3. doi: 10.2337/dc12-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhao Y, Peng J, Lu C, Hsin M, Mura M, Wu L, et al. Metabolomic heterogeneity of pulmonary arterial hypertension. PloS one. 2014;9(2):e88727. doi: 10.1371/journal.pone.0088727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Santos M, Reis A, Goncalves F, Ferreira-Pinto MJ, Cabral S, Torres S, et al. Adiponectin levels are elevated in patients with pulmonary arterial hypertension. Clinical cardiology. 2014;37(1):21–5. doi: 10.1002/clc.22210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hansmann G, Rabinovitch M. The protective role of adiponectin in pulmonary vascular disease. American journal of physiology Lung cellular and molecular physiology. 2010;298(1):L1–2. doi: 10.1152/ajplung.00367.2009. [DOI] [PubMed] [Google Scholar]
- 76.Summer R, Walsh K, Medoff BD. Obesity and pulmonary arterial hypertension: Is adiponectin the molecular link between these conditions? Pulmonary circulation. 2011;1(4):440–7. doi: 10.4103/2045-8932.93542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kistorp C, Faber J, Galatius S, Gustafsson F, Frystyk J, Flyvbjerg A, et al. Plasma adiponectin, body mass index, and mortality in patients with chronic heart failure. Circulation. 2005;112(12):1756–62. doi: 10.1161/CIRCULATIONAHA.104.530972. [DOI] [PubMed] [Google Scholar]
- 78.Huertas A, Tu L, Gambaryan N, Girerd B, Perros F, Montani D, et al. Leptin and regulatory T-lymphocytes in idiopathic pulmonary arterial hypertension. The European respiratory journal. 2012;40(4):895–904. doi: 10.1183/09031936.00159911. [DOI] [PubMed] [Google Scholar]
- 79.Li ZF, Zhou DX, Pan WZ, Zhang L, Ge JB. Circulating ghrelin was negatively correlated with pulmonary arterial pressure in atrial septal defect patients. Chinese medical journal. 2013;126(20):3936–9. [PubMed] [Google Scholar]
- 80.Benson L, Brittain EL, Pugh ME, Austin ED, Fox K, Wheeler L, et al. Impact of diabetes on survival and right ventricular compensation in pulmonary arterial hypertension. Pulmonary circulation. 2014;4(2):311–8. doi: 10.1086/675994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brunner NW, Skhiri M, Fortenko O, Hsi A, Haddad F, Khazeni N, et al. Impact of insulin resistance on ventricular function in pulmonary arterial hypertension. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2014 doi: 10.1016/j.healun.2014.02.016. [DOI] [PubMed] [Google Scholar]
- 82•.Lundgrin EL, Park MM, Sharp J, Tang WH, Thomas JD, Asosingh K, et al. Fasting 2-deoxy-2-[18F]fluoro-D-glucose positron emission tomography to detect metabolic changes in pulmonary arterial hypertension hearts over 1 year. Annals of the American Thoracic Society. 2013;10(1):1–9. doi: 10.1513/AnnalsATS.201206-029OC. This report explored the use of fasting PET-CT scans in humas with PAH, and they determined that pathologic glycolysis was detected in the right ventricles of patients with PAH compared to health controls that appeared to be HIF-1-α-mediated. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kluge R, Barthel H, Pankau H, Seese A, Schauer J, Wirtz H, et al. Different mechanisms for changes in glucose uptake of the right and left ventricular myocardium in pulmonary hypertension. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2005;46(1):25–31. [PubMed] [Google Scholar]
- 84.Kawut SM, Bagiella E, Lederer DJ, Shimbo D, Horn EM, Roberts KE, et al. Randomized clinical trial of aspirin and simvastatin for pulmonary arterial hypertension: ASA-STAT. Circulation. 2011;123(25):2985–93. doi: 10.1161/circulationaha.110.015693. [DOI] [PMC free article] [PubMed] [Google Scholar]