

Abstract
Purpose
This “3+3” phase I study evaluated the safety, biologic, and clinical activity of lenvatinib, an oral multikinase inhibitor, in patients with solid tumors.
Experimental Design
Ascending doses of lenvatinib were administered po bid in 28-day cycles. Safety and response were assessed for all patients. Angiogenic and apoptotic factors were tested as possible biomarkers in an expanded melanoma cohort.
Results
Seventy-seven patients were treated in 3 cohorts: 18 with intermittent bid dosing (7 days on, 7 days off) of 0.1–3.2 mg; 33 with bid dosing of 3.2–12 mg; and 26 with bid dosing of 10 mg (expanded melanoma cohort). Maximum tolerated dose was established at 10 mg po bid. Prominent drug-related toxicities included hypertension (43%), fatigue (42%), proteinuria (39%), and nausea (25%); dose-limiting toxicities included hypertension, fatigue, and proteinuria. Twelve patients (15.6%) achieved partial response (PR, n=9) or unconfirmed PR (uPR, n=3), and 19 (24.7%) achieved stable disease (SD) ≥23 weeks. Total PR/uPR/SD≥23 weeks was 40.3% (n=31). Responses (PR/uPR) by disease were: melanoma, 5/29 patients (includes 1 patient with NRAS mutation); thyroid, 3/6; pancreatic, 1/2; lung, 1/1; renal, 1/1; endometrial, 1/4; and ovarian, 1/5. AUC0-24 and Cmax increased dose-proportionally. In multivariate Cox proportional hazard model analyses, increased baseline systolic blood pressure and decreased angiopoietin-1 ratio (2 hours:baseline) were associated with longer progression-free survival (PFS) in the expanded melanoma cohort (P=0.041 and P=0.03, respectively).
Conclusions
The toxicity profile, pharmacokinetics, and antitumor activity of lenvatinib are encouraging. Decreases in the angiopoietin-1 ratio correlated with longer PFS in melanoma patients.
Keywords: lenvatinib, melanoma, pharmacodynamic, phase I, advanced solid tumors, E7080, thyroid cancer, VEGFR, FGFR, PDGFR, RET, KIT
Introduction
Angiogenesis is required for tumor growth, progression, and metastasis, making it a logical target for antitumor drug development. Several growth factors are positive regulators of angiogenesis, including vascular endothelial growth factor (VEGF), basic and acidic fibroblast growth factor (FGF), and platelet-derived growth factor (PDGF). Each of these factors signal through specific transmembrane tyrosine kinase receptors: VEGFRs 1 and 2 (FMS-like tyrosine kinase [FLT-1] and fetal liver kinase 1/kinase insert domain receptor [FLK-1/KDR]), FGFR, and PDGFR (1-3).
Lenvatinib is an oral, multi–tyrosine kinase inhibitor active against RET, VEGFR1–3, FGFR1–3, KIT, and PDGFRα (4-6). Lenvatinib inhibits VEGF-driven human umbilical vein endothelial cell proliferation and tube formation and significantly inhibits tumor growth in various murine tumor models, including human lung (H146) and breast cancer (MDA-MB-231) mouse xenograft models (4, 5). Based on a population pharmacokinetics/pharmacodynamics analysis of lenvatinib, agents that modify gastric pH levels do not have a significant effect on the absorption of lenvatinib (Eisai, data on file). Earlier studies have shown that lenvatinib exposure is neither affected by food intake (7) or co-administration of CYP3A4 inhibitors and inducers (8, 9). Lenvatinib is rapidly and well-absorbed; it is also extensively metabolized, with predominant excretion in feces, and to a smaller extent in urine (10). The primary objectives of this study were to identify the maximum tolerated dose (MTD), dose-limiting toxicities (DLTs), and the pharmacokinetic (PK) profile of lenvatinib.
Materials and Methods
Ethics
The study followed the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use guidelines and local Independent Ethics Committee standards. The study was approved by the Institutional Review Boards of The University of Texas MD Anderson Cancer Center and the Mary Crowley Cancer Research Center. All participants provided written informed consent prior to participation in the study.
Patients
Major inclusion criteria were age ≥18 years, histological and/or cytological diagnosis of a solid tumor/lymphoma not amenable to standard therapy, Eastern Committee Oncology Group (ECOG) performance status of ≤1, and adequate hematologic (hemoglobin ≥9 g/dL, neutrophils ≥1.5 × 109/L, platelets ≥100 × 109/L), hepatic (serum bilirubin ≤1.5 mg/dL and other liver parameters ≤3 × upper limit of normal [ULN]), and renal (serum creatinine ≤1.5 × ULN and creatinine clearance ≥60 mL/minute) function. Exclusion criteria included poor cardiac function, unstable ischemic heart disease, poorly controlled hypertension, and prolongation of the QT/QTc interval calculated using the Fridericia method (QTcF interval >450 msec for men or >470 msec for women). Patients who were pregnant, had untreated or unstable metastases to the central nervous system, or required chronic use of full-dose aspirin or nonsteroidal anti-inflammatory drugs were also excluded. Additional inclusion criteria for the expanded melanoma cohort included a histological or cytological diagnosis of advanced or metastatic melanoma untreatable by standard therapies, presence of melanoma lesions amenable to biopsy and willingness to undergo biopsies of malignant and adjacent nonmalignant tissue pretreatment and at the end of cycle 1.
Study Design
This open-label phase I study was conducted using a modified “3+3” dose-escalation design, which was adapted based on observed toxicities. Lenvatinib was administered orally as 0.1 mg, 1 mg, and 10 mg tablets. The study had 2 schedules. Schedule 1 (cohort 1) examined escalating doses of lenvatinib ranging from 0.1 mg to 3.2 mg administered twice daily (bid) using a schedule of 7 days on treatment followed by 7 days off. Schedule 2 (cohort 2) examined doses ranging from 3.2 mg to 12 mg administered bid by continuous daily administration. Schedule 2 was designed to supersede schedule 1, since a previous phase I study had indicated that continuous dosing with levatinib was tolerated among patients with advanced solid tumors (11). Once the MTD was determined, the safety and tolerability of a 10-mg bid dose was tested in an expanded melanoma cohort (cohort 3) composed of 26 patients with refractory melanoma. A 28-day treatment period constituted 1 treatment cycle. Only DLTs during the first 28 days of therapy (cycle 1) were assessed for dose-escalation purposes.
Following the initial dose for each of the first 2 cohorts, subsequent dose levels were selected according to an accelerated study design and toxicity-adapted model (12). There was an accelerated dose escalation phase (only during schedule 1) and a standard dose escalation phase. In the accelerated phase, a higher dose level could only be opened for patient accrual after 1 patient at the current dose level completed cycle 1 with no drug-related toxicity exceeding grade 1 (except alopecia, lymphopenia, and anemia). Dose increases were in 100% increments until the first patient experienced toxicity of grade ≥2. In the standard dose escalating phase, dose increases were 50% or less depending upon the seriousness of the toxicity, and the first 3 subjects at a dose level were followed for a full cycle before the next dose level was opened. Standard dose escalation was invoked when the first patient at any dose level experienced ≥ grade 2 toxicity. No intra-patient dose escalation was allowed and only DLTs experienced during the first cycle defined dose escalations. For example, if 1 of 3 patients experienced a DLT during the first cycle, an additional 3 patients were to be treated at that dose level. If no additional DLT was observed at the expanded dose level (i.e., 5 of 6 patients did not experience DLT during the first cycle), dose escalation continued. If more than 1 of the 6 patients experienced DLT during the first cycle of therapy at any dose level, dose escalation was stopped. Once the MTD was determined, the safety, tolerability, and efficacy of the MTD were tested in cohort 3, which enrolled patients with refractory melanoma.
Dose-Limiting Toxicity, Maximum Tolerated Dose
A DLT was defined as any of the following: any grade ≥3 hematologic toxicity; any grade ≥3 nonhematologic toxicity (except grade 3 hypertension controllable by more intensive antihypertensive monotherapy or by adding a second antihypertensive agent); any repeated nonhematologic toxicity grade ≥2 that represented at least a 2-grade increase over baseline and required reduction of the study drug dose (excluding repeated grade 3 hypertension controllable by antihypertensive treatment); or any failure to administer ≥75% of the planned dosage of lenvatinib during cycle 1 as a result of treatment-related toxicity. MTD was defined as the highest dose level at which no more than 1 of 6 patients developed a DLT.
Safety Assessments: Screening, Baseline, and Follow-Up
Screening and baseline assessments included evaluation of demographic data, medical and surgical history, prior medications, a complete physical examination, vital signs, ECOG performance status, electrocardiogram (ECG), and clinical laboratory tests (including urinalysis and a serum pregnancy test), tumor history, primary diagnosis, previous treatments, and biopsies of tumor tissues. AEs were recorded for patients who received at least 1 dose of lenvatinib.
Safety assessments were performed in each cycle, with additional weekly blood pressure (BP) monitoring. AE severity was assessed according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE), version 3.0 (13). Safety assessments consisted of monitoring and recording all AEs; documenting concomitant medications; regular monitoring of hematology, blood chemistry, and urine values; periodic measurement of vital signs and ECOG performance status, attainment of ECGs; and performance of physical examinations. An ECG was at minimum recorded for each cohort at screening, and then pre-dose, 1 hour and 2 hours after dosing on day 1 of cycle 1, and at the final visit. AE screenings and laboratory tests were performed weekly, or as clinically indicated.
Pharmacokinetics
Blood samples were collected for PK analysis immediately prior to the first dose of lenvatinib, and at 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, and 24 hours following the first dose of lenvatinib on day 1 of both cycle 1 and 2 of schedules 1 and 2. In schedule 2, lenvatinib was administered once daily on day 1 of both cycle 1 and cycle 2 (in which only 1 bid dose was administered on that day), then bid throughout the remainder of the cycles, to facilitate comparison of once‑daily and bid PK values. Trough samples were collected within 2 hours prior to the morning dose of lenvatinib or 12 hours after the last dose on days 8, 15, and 22 in cycle 1 (schedules 1 and 2), days 8 and 15 of all subsequent cycles (schedule 1), and day 1 of all subsequent cycles (schedules 1 and 2), as well as at the final visit.
Urine samples were collected during cycles 1 and 2 on days 1 and 8 (schedule 1) and during cycle 1 days 1 and 8 and cycle 2 day 1 (schedule 2). For both schedules, a 24-hour urine collection was performed following the dose of lenvatinib on day 1 of cycles 1 and 2. Three aliquots were collected: at 0–8 hours, 8–16 hours, and 16–24 hours.
Lenvatinib was extracted from plasma and urine with diethylether under alkaline conditions and then assayed by liquid chromatography with electrospray ionization tandem mass spectrometry (LC-MS/MS). The assay range was 0.800−400 ng/mL for plasma and was 0.400−400 mg/mL for urine. Prior to the analysis, assay sensitivity, specificity, linearity, reproducibility and stability were established. The interday accuracy (% bias) ranged from -8.9 to 4.8 and precision (% CV) ranged from 3.0 to 17.8 in plasma and urine.
Individual lenvatinib PK parameters were calculated from plasma and urine concentration time data using noncompartmental methods. Data analysis was conducted using WinNonlin (Phoenix version 6.2, Pharsight Corp., Mountain View, CA). Plasma concentrations reported as below the lower limit of quantitation were imputed as zero prior to the time of the peak concentration and as missing thereafter. For urine, concentrations below the lower limit of quantitation were also imputed as zero. The parameters Cmax and Tmax were determined from visual inspection of the data. AUC from 0 to 6 hours and 24 hours were determined by the linear up-log down trapezoidal rule. Oral clearance (CL/F) and terminal volume of distribution were obtained using standard equations at steady state (Vss/F) on cycle 2 day 1. Amount of lenvatinib recovered in urine was obtained from the analyte concentration and urine volumes. Using these amounts, fraction of the dose excreted in urine (fe%) was calculated.
Pharmacodynamics
Pharmacodynamic assessments were conducted and analyzed in the expanded melanoma cohort. Serum samples were collected at baseline (pre-dose) and 2 hours after the first dose, and on days 8, 15, and 22 of cycle 1.
Serum samples were tested for angiogenesis-related (MDS Pharma/Clearstone Central Lab) and apoptosis-related (Pathway Diagnostics/Quest) markers. These angiogenesis-related markers were PDGF-homodimer BB, soluble tie-2 (receptor expressed by endothelial cells [sTie-2]), angiopoietin-1 (Tie-2 ligand), soluble E-selectin (mediates leukocyte and tumor cell rolling), and soluble c-kit. The apoptosis-related markers assessed were cytochrome C (a measure of intrinsic apoptotic pathway activation) and M30 neoantigen (caspase-cleaved cytokeratin-18, also a terminal apoptotic product for epithelial-derived tumors). sTie-2, angiopoietin-1, PDGF-BB, soluble e-selectin, soluble c-kit, cytochrome C, and M30 were measured by enzyme-linked immunosorbent assay. PDGF-BB was assayed by Luminex Technology using Growth Factor Buffer Reagent and a Human Custom Multiplex Antibody Bead Kit from BioSource Invitrogen (Frederick, MD).
The following vital signs, clinical chemistry, and hematology parameters were also evaluated at the corresponding time points for any association with clinical outcome: diastolic BP (dBP), systolic blood pressure (sBP), lactate dehydrogenase (LDH) level, hematocrit percentage, and blood hemoglobin.
Response
Tumor measurements were assessed by clinical examination and photography (for skin lesions), computed tomography, or magnetic resonance imaging, and evaluated using Response Evaluation Criteria in Solid Tumors (RECIST), version 1.0 (14). Assessments were conducted at baseline and then approximately every 2 cycles during treatment. Responses were confirmed at a follow-up examination after ≥30 days. Tumor response was defined as complete response (CR), partial response (PR), stable disease (SD), or disease progression (PD). SD was to be maintained for ≥7 weeks and durable SD for ≥23 weeks. Clinical benefit was defined as CR+PR+durable SD ≥23 weeks.
Mutation Analyses
Gene mutation analyses were conducted using DNA extracted from micro-dissected, archival paraffin-embedded tumor samples obtained from 26 patients in the expanded melanoma cohort. Molecular analyses were performed in the Clinical Laboratory Improvement Amendments (CLIA)–certified Molecular Diagnostics Laboratory at MD Anderson using standard operating procedures. Tumor DNA from the expanded melanoma cohort was extracted and processed independently by Sequenom (San Diego, CA) to genotype DNA sequence mutations (MelaCarta™ panel, Sequenom) and to identify any mutation(s) associated with clinical response to lenvatinib therapy.
Statistical Analyses
Demographic, safety, PK, and efficacy data were evaluated using descriptive statistics. Categorical data were summarized as frequency and percentages; continuous data were summarized as mean and standard deviation, median, or range, as appropriate. To explore the correlation of drug exposure with serum biomarkers, Pearson and Spearman correlation coefficients were calculated between PK parameters (AUC0-6, AUC0-24, Cmax or Ctrough) and serum biomarkers (PDGF-BB, sTie-2, soluble E-selectin, soluble c-kit, or angiopoietin-1) at baseline (cycle 1 day 1) and at different time points compared with baseline.
Pearson and Spearman correlation coefficients were calculated between percent maximum tumor shrinkage (defined as percent change in sum of longest diameter from baseline to nadir) based on RECIST and baseline levels, as well as change from baseline levels of angiogenic and apoptotic factors.
Progression-free survival (PFS), overall survival (OS), and duration of response were calculated. The relationship between PFS and the biomarkers of angiogenesis and apoptosis were evaluated with univariate and multivariate Cox proportional hazard models. The multivariate model was selected by the forward selection method (P<0.05) using covariates identified from univariate analysis (P<0.1).
Results
Patients
Patients were enrolled between 11 July 2005 and 11 August 2009. Seventy-seven patients received at least 1 dose of study drug and were included in the safety (and overall) population. In schedule 1 (bid, 7 days on and 7 days off), 18 subjects were enrolled (3 each in the lenvatinib 0.1-, 0.2-, 0.4-, 0.8-, 1.6-, and 3.2-mg dose groups). In schedule 2 (bid continuous dosing), 33 subjects were enrolled: 3 subjects in the 3.2-mg group, 7 in the 5.0-mg group, 16 in the 8.0-mg group, and 7 in the 12.0-mg group. Twenty-six subjects were included in the expanded melanoma cohort (10-mg bid continuous dosing).
The median age of the overall study population (n=77) was 61.0 years (range, 28–85). Forty (52%) patients were male, and 75 (97.4%) patients had a baseline ECOG performance status of ≤1. The most frequently reported tumor types were melanoma (29 [38%]), colorectal (10 [13%]), thyroid (6 [8%]), and ovarian (5 [6%]). Twenty-six (33.7%) patients had ≥3 prior chemotherapy regimens.
In the expanded melanoma cohort, the median age was 69.0 years (range, 38–85). Sixteen (61.5%) patients were male. Five (19.2%) patients had ≥3 prior chemotherapy regimens. Based on independent or on-site assessment, BRAF mutations were detected in 9 (34.6%) patients and NRAS mutations in 8 (30.8%) patients (Table 1). No patient with melanoma in this study had received prior BRAF V600-targeted treatment, and only 1 patient had received prior ipilimumab treatment. When this study was initiated, the currently approved BRAF-targeted therapies were still in investigational stages.
Table 1. Baseline patient characteristics.
| Characteristic | Melanoma Expansion Cohort (n=26) | Entire Study Population (N=77) |
|---|---|---|
| Age, ya | ||
| Mean (SD) | 65.5 (11.66) | 61.3 (12.85) |
| Median | 69.0 | 61.0 |
| Range (min, max) | (38, 85) | (28, 85) |
| Sex, n (%) | ||
| Male | 16 (61.5) | 40 (51.9) |
| Female | 10 (38.5) | 37 (48.1) |
| Race, n (%) | ||
| Non-Hispanic White | 26 (100) | 64 (83.1) |
| Hispanic | 0 | 9 (11.7) |
| African-American | 0 | 4 (5.2) |
| ECOG status, n (%) | ||
| 0 | 8 (30.8) | 30 (39.0) |
| 1 | 17 (65.4) | 45 (58.4) |
| 2 | 1 (3.8) | 2 (2.6) |
| Tumor type, n (%) | ||
| Colorectal | 0 | 10 |
| Thyroid | 0 | 6 |
| Ovarian | 0 | 5 |
| Endometrial | 0 | 4 |
| Breast | 0 | 3 |
| Melanoma | 26 (100) | 3 |
| Lung | 0 | 2 |
| Pancreatic | 0 | 2 |
| Prostate | 0 | 2 |
| Other | 0 | 14 |
| Previous anticancer treatment, n (%) | ||
| Chemotherapy | 22 (84.6) | 70 (90.9) |
| Radiotherapy | 13 (50.0) | 42 (54.5) |
| Surgery | 26 (100) | 77 (100.0) |
| Other anticancer treatment regimens | 18 (69.2) | 47 (61.0) |
| Previous systemic treatment (chemotherapy), n (%) | ||
| 0 | 4 (15.4) | 7 (9.1) |
| 1 | 8 (30.8) | 19 (24.7) |
| 2 | 9 (34.6) | 25 (32.5) |
| ≥3 | 5 (19.2) | 26 (33.8) |
| Mutation statusb, n (%) | ||
| BRAF | 9 (34.6) | ― |
| NRAS | 8 (30.8) | ― |
Age calculated as (date of informed consent – date of birth + 1)/365.25.
Mutations were assessed only in patients in the melanoma expansion cohort.
Abbreviation: SD, standard deviation.
Reasons for discontinuation of study treatment included disease progression or clinical deterioration (43 [55.8%]), adverse events (17 [22.1%]), and withdrawal of consent (5 [6.5%]). Thirteen (16.9%) patients died either during therapy or within 30 days of the last treatment. In the schedule 1 cohort (n=18), 11 (61.1%) patients discontinued due to disease progression or clinical deterioration, and 6 (33.3%) patients due to adverse events. In the schedule 2 cohort (n=33), 17 (51.5%) patients terminated due to disease progression or clinical deterioration, and 9 (27.3%) due to adverse events. In the melanoma expansion cohort, 15 (57.7%) patients discontinued due to disease progression or clinical deterioration, and 2 (7.7%) due to adverse events. Of note, 8 (30.8%) patients in the expanded melanoma cohort died during therapy or within 30 days of the last treatment.
Dose Escalation, Dose-Limiting Toxicities, and Maximum Tolerated Dose
Since no DLTs were observed in the schedule 1 cohort, schedule 2 was initiated and dose levels of 3.2 to 12.0 mg bid were examined. At 12 mg, DLTs of hypertension (n=3, all grade 3), fatigue (n=1, grade 3), and proteinuria (n=1, grade 2) were observed in 5 of 7 treated patients. Therefore, the previous lower dose of 8 mg bid was initially established as the MTD. However, an intermediate dose of 10 mg bid was evaluated in the expanded melanoma cohort and judged as well tolerated, with no DLTs observed in the first 6 subjects who received the 10-mg bid dose. Therefore, the MTD was determined to be 10 mg bid.
Safety
The most frequently reported study drug–related toxicities were hypertension (43%), fatigue (42%), proteinuria (39%), nausea (25%), body weight reduction (25%), anorexia (25%), and diarrhea (22%) (Table 2). The majority of study drug–related toxicities of all grades were observed in patients who had received dose levels ≥10 mg.
Table 2.
Study drug–related (possibly/probably, as deemed by investigator) treatment-emergent adverse events (TEAEs) by CTCAE grade/severity in the safety population, in descending order of total frequency. (Includes all TEAEs for which the total of all grades of the particular TEAE exceeded 20%.)
| MedDRA SOC CTCAE Gradea/Severity (2) | Melanoma Expansion Cohortb 10.0 mg bid (n=26) n (%) | Entire Study Population (N=77) n (%) |
|---|---|---|
| Any AE | ||
| Total | 26 (100.0) | 68 (88.3) |
| 1 | 0 | 6 (7.8) |
| 2 | 10 (38.5) | 30 (39.0) |
| 3 | 13 (50.0) | 28 (36.4) |
| 4 | 3 (11.5) | 4 (5.2) |
| 3+4 | 16 (61.5) | 32 (41.6) |
| Hypertension | ||
| Total | 16 (61.5) | 33 (42.9) |
| 1 | 1 (3.8) | 2 (2.6) |
| 2 | 9 (34.6) | 18 (23.4) |
| 3 | 6 (23.1) | 13 (16.9) |
| 4 | 0 | 0 |
| 3+4 | 6 (23.1) | 13 (16.9) |
| Fatigue | ||
| Total | 13 (50.0) | 32 (41.6) |
| 1 | 3 (11.5) | 10 (13.0) |
| 2 | 7 (26.9) | 17 (22.1) |
| 3 | 3 (11.5) | 5 (6.5) |
| 4 | 0 | 0 |
| 3+4 | 3 (11.5) | 5 (6.5) |
| Proteinuria | ||
| Total | 15 (57.7) | 30 (39.0) |
| 1 | 5 (19.2) | 7 (9.1) |
| 2 | 7 (26.9) | 18 (23.4) |
| 3 | 2 (7.7) | 4 (5.2) |
| 4 | 1 (3.8) | 1 (1.3) |
| 3+4 | 3 (11.5) | 5 (6.5) |
| Nausea | ||
| Total | 6 (23.1) | 19 (24.7) |
| 1 | 1 (3.8) | 9 (11.7) |
| 2 | 3 (11.5) | 8 (10.4) |
| 3 | 2 (7.7) | 2 (2.6) |
| 4 | 0 | 0 |
| 3+4 | 2 (7.7) | 2 (2.6) |
| Weight decreased | ||
| Total | 10 (38.5) | 19 (24.7) |
| 1 | 2 (7.7) | 6 (7.8) |
| 2 | 4 (15.4) | 7 (9.1) |
| 3 | 4 (15.4) | 6 (7.8) |
| 4 | 0 | 0 |
| 3+4 | 4 (15.4) | 6 (7.8) |
| Anorexia | ||
| Total | 12 (46.2) | 19 (24.7) |
| 1 | 3 (11.5) | 8 (10.4) |
| 2 | 9 (34.6) | 11 (14.3) |
| 3 | 0 | 0 |
| 4 | 0 | 0 |
| 3+4 | 0 | 0 |
| Diarrhea | ||
| Total | 8 (30.8) | 17 (22.1) |
| 1 | 3 (11.5) | 7 (9.1) |
| 2 | 2 (7.7) | 6 (7.8) |
| 3 | 3 (11.5) | 4 (5.2) |
| 4 | 0 | 0 |
| 3+4 | 3 (11.5) | 4 (5.2) |
The grade of AE was assigned based on NCI-CTCAE criteria.
Subtotal of the full study population.
Abbreviations: SOC, System Order Class; MedDRA, Medical Dictionary for Regulatory Activities.
There were few treatment-related grade 4 toxicities (4/77 [5.2%]), which consisted of 1 event each of grade 4 thrombocytopenia, thrombotic thrombocytopenic purpura, reversible posterior leukoencephalopathy syndrome, and proteinuria. The most common grade 3 study drug–related toxicities in the overall study population were hypertension (17%), body weight reduction (8%), fatigue (7%), proteinuria (5%), and diarrhea (5%) (Table 2). Most grade 3 toxicities were seen in the 5- to 12-mg dose range (5 mg, 71.4%; 8 mg, 56.3%; 12 mg, 71.4%). The incidence of grade 3 hypertension was 86% at 12 mg and 23% at 10 mg (MTD dose); for most patients, these hypertensive events were resolved through either dose adjustments/delays, antihypertensive therapy, or both.
Response
The best overall tumor responses evaluated based on RECIST (version 1.0) for patients from the dose-escalation phase and patients from the expanded melanoma cohort are shown in Figure 1A and Figure 1B, respectively. Overall, 9 (11.7%) patients achieved a confirmed PR. Forty (51.9%) patients had SD, including 19 (24.7%) with SD ≥23 weeks. Eighteen (23.4%) patients experienced PD, 17 by cycle 2. Response was not assessable in 10 (13.0%) patients: 4 withdrew consent after the screening stage, and 6 had events that precluded treatment assessment. The confirmed PRs were most frequent in patients with melanoma (n=3) and medullary thyroid cancer (n=3); these PRs were determined after a median of 18 cycles (range, 4–46 cycles). Three patients (2 melanoma, 1 non–small cell lung cancer [NSCLC]) had unconfirmed PRs (uPR), for a total of 12 (15.6%) patients with PR/uPR as their best response.
Figure 1.
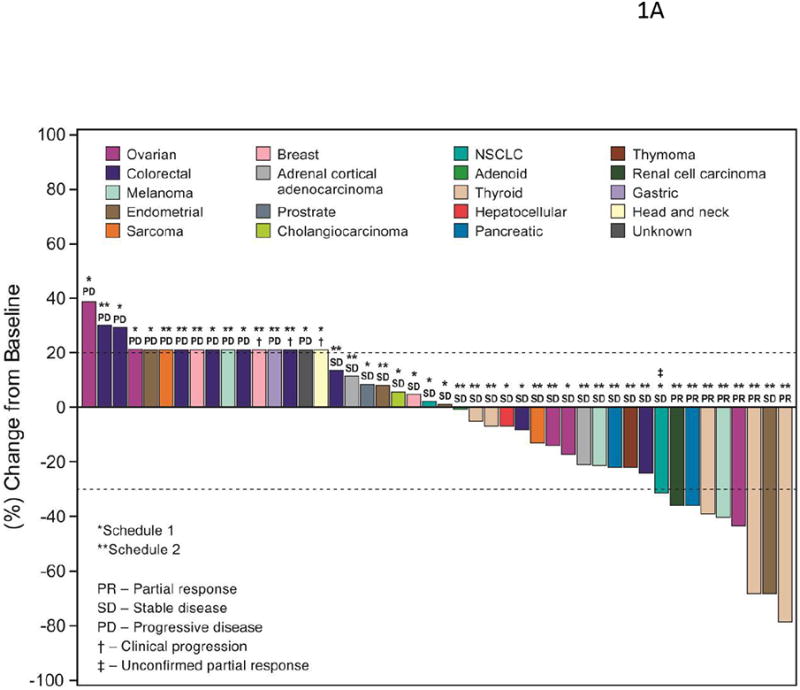
(A) Waterfall graph showing best response to lenvatinib treatment by RECIST criteria. An arbitrary value of 21% (indicated by †) was assigned for patients who failed early due to clinical progression or new metastatic lesions. The ‡ indicates unconfirmed PR (i.e., initial PR was not confirmed in subsequent tumor assessments, or no further assessments were available). Note that 1 patient's initial designation of SD was changed to PR after the database lock due to overall responses. (B) Waterfall graph for patients in the melanoma expansion cohort: best response to treatment by BRAF and NRAS mutation status. An arbitrary value of 21% (indicated by †) was assigned for patients who failed early due to clinical progression or new metastatic lesions. The ‡ indicates unconfirmed PR, as defined above. Identifiers a, b and c represent patients for whom there was discordance between on-site and independent assessments of either BRAF or NRAS mutation status.
Of the 18 patients treated on schedule 1, no patient achieved a confirmed PR. Nine (50.0%) had SD (including 1 patient each with breast, hepatocellular, ovarian cancer, or NSCLC who had durable SD ≥23 weeks), and 8 (44.4%) had PD. One patient with NSCLC had a uPR. Of the 33 patients treated on schedule 2, 7 (21.2%) patients achieved a confirmed PR, including 3 with medullary thyroid cancer, 1 with melanoma, and 1 each with ovarian, pancreatic, or renal cell cancer. Fifteen patients (45.5%) had SD; 9 patients had durable SD, including 2 patients with endometrial adenocarcinoma, and 1 patient each with epithelial thymoma, synovial sarcoma, adrenal cortical carcinoma, colon adenocarcioma, pancreatic cancer, melanoma, or thyroid cancer. Six (18.2%) patients had PD.
Of the 26 patients in the expanded melanoma cohort, 2 (7.7%) achieved a confirmed PR, 16 (61.5%) had SD (including 6 [23.1%] patients who had durable SD ≥23 weeks) and 4 (15.4%) had PD. Two patients in this cohort had uPRs, for a total of 4 patients (15.4%) with PRs/uPRs as their best response.
Twenty-nine patients in the study had melanoma: 3 were on schedule 2 of dose escalation, and 26 were in the expanded melanoma cohort. Overall, 3 (10.3%) achieved a PR, 17 (58.6%) had SD (including 7 [24.1%] who had durable SD ≥23 weeks), and 5 (17.2%) had PD. A total of 5 (17.2%) patients with melanoma had PRs/uPRs as their best response.
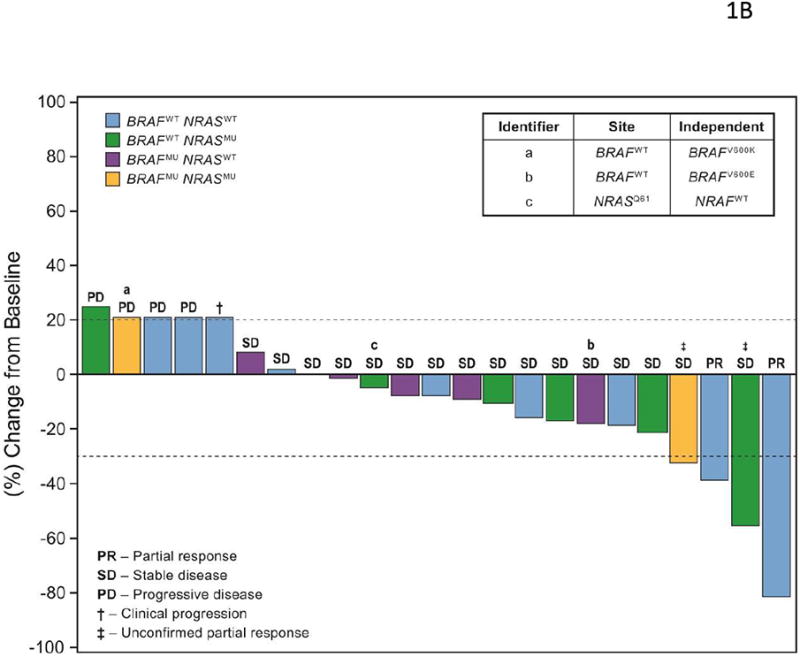
Figure 1B is a waterfall graph showing response by mutation status in patients from the expanded melanoma cohort. As noted in Table 1, BRAF and NRAS mutations were detected in 9 (34.6%) and 8 (30.8%) patients, respectively. Possibly reflecting the heterogeneity in tumor samples, discordance between the on-site and independent assessment was noted for 2 patients with respect to BRAF mutation status and 1 patient with respect to NRAS mutation status (Figure 1B inset). Two (7.7%) patients had coexisting BRAF and NRAS mutations, and 11 (42.3%) patients had both BRAF and NRAS wild-type tumors (Figure 1B). Three of 17 patients (17.6%) with wild-type BRAF had either PR (n=2) or uPR (n=1; Figure 1B).
Pharmacokinetics
Lenvatinib PK parameters are summarized by dose in Table 3. The PK population was equivalent to the overall population (n=77). Overall, lenvatinib's single-dose and steady-state PK parameters (Cmax, AUC0–6, and AUC0–24) increased proportionately over the entire dose range evaluated in this study. Median tmax was similar across all dose levels and ranged from 1.5 to 3 hours (excluding the 0.1-mg and 0.2-mg daily doses at 24 hours and 6 hours of cycle 1 day 1, respectively). The mean terminal elimination half-life (t1/2) has been shown to be approximately 28 to 29 hours in previous phase 1 studies (7, 8). On cycle 2 day 1, the apparent oral clearance was 8.05 and 6.09 L/hr for doses of 8-mg and 16-mg, respectively. The apparent terminal volume was 86.8 L for the 8-mg dose, and 68.5 L for the 16-mg dose. Regardless of dose, the fraction of the lenvatinib dose excreted unchanged in the urine (fe) during the 24 hours following the dose was low (<1.5%), indicating that excretion via urine is a minor pathway for lenvatinib. A comparison of PK parameters at 8-mg, 10-mg, and 12-mg doses after a single dose and at steady state demonstrated relatively low accumulation (approximately 1.5- to 2.2-fold; Figure 2A). A rapid absorption phase, followed by a distribution phase and then an elimination phase, were generally observed following increasing doses of lenvatinib after a single dose and at steady state (Figure 2A).
Table 3.
Summary of PK parameters by dose level during schedule 2 and at the selected dose of 10 mg bid in the melanoma expansion cohort. Data are presented as mean (SD) values, except when noted otherwise.
| Schedule 2 (Continuous Daily Dosing) | Melanoma Expansion Cohort | |||||||
|---|---|---|---|---|---|---|---|---|
| Cycle 1 Day 1 | Cycle 2 Day 1 | Cycle 1 Day 1 | Cycle 2 Day 1 | |||||
| Dose (Daily Dose)a | 3.2 mg | 5 mg | 8 mg | 12 mg | 8 mg | 16 mg | 10 mg | 10 mg |
| n | 3 | 7 | 10 | 4 | 6 | 6 | 25 | 16 |
| tmax, hrb | 2.00 (2.00−2.50) | 2.50 (2.00−24.50) | 1.50 (1.00−4.00) | 1.50 (1.00−1.50) | 2.50 (1.50−4.00) | 2.03 (1.00−4.00) | 2.00 (1.00−4.00) | 3.00 (0.00−6.00) |
| CL/F, L/hr | ND | ND | ND | ND | 8.05 (3.89) | 6.09 (1.18) (n=4) | ND | 7.77 (3.02) (n=14) |
| Vss/F, L | ND | ND | ND | ND | 86.8 (35.7) (n=5) | 68.5 (19.0) (n=3) | ND | 75.4 (24.6) (n=10) |
| Cmax, ng/mL | 51.4 (30.5) | 62.6 (25.5) | 177 (60.6) | 232 (60.0) | 168 (73.4) | 341(176) | 167(68.8) | 209 (107) |
| AUC0-6, ng·hr/mL | 180 (68.6) | 245(115) | 613 (229) | 913 (250) | 678 (262) | 1510(677) | 608 (241) | 815(320) |
| AUC0-24, ng·hr/mL | 407.38 (120.797) | 649.70 (256.151) | 1302.75 (441.924) | 1812.17 (649.112) | 1819.57 (735.252) | 3557.51 (688.828) | 1365.39 (647.136) | 1984.94 (971.427) |
| fe, % | 0.37 (0.102) | 1.47 (0.685) | 0.83a (0.351) | 0.68 (0.283) | 1.26 (1.168) | 1.35 (0.593) | 0.45 (0.282) | 0.95 (0.987) |
During schedule 2, on PK days cycle 1 day 1 and cycle 2 day 1, only 1 dose of lenvatinib bid was administered.
Reported as median, (range)
Abbreviation: ND, not determined.
Figure 2.
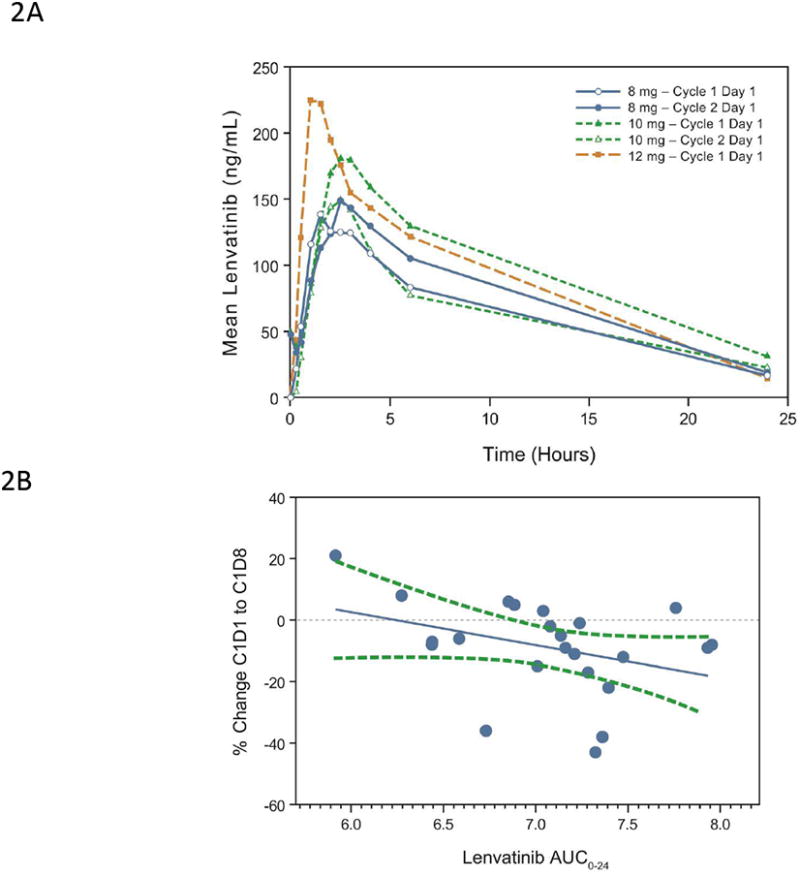
(A) Single-dose (cycle 1 day 1) and steady-state (cycle 2 day 1) mean observed plasma lenvatinib concentration vs time curve for dose levels 8 mg and 12 mg bid in schedule 2 and 10 mg bid in the expanded melanoma cohort, wherein only 1 dose of the bid dose was administered on cycle 1 day 1 and on cycle 2 day 1. (B) Association of PK exposure with changes in levels of serum sTie-2 protein on cycle 1 day 8 relative to baseline. Shown here is the correlation between AUC0-24 (natural logarithm transformed) and changes in sTie-2 for the expanded melanoma cohort (Spearman correlation R=-0.45, P=0.0322; n=23). Each blue dot represents 1 subject; the solid blue line symbolizes the regression line; and the dashed green lines represent 95% confidence limits.
Pharmacodynamics
In the expanded melanoma cohort, changes in sTie-2 levels following lenvatinib treatment correlated with lenvatinib exposure as assessed by AUC0-24 (Figure 2B; Spearman correlation R=-0.45, P=0.032, n=23). Baseline and change from baseline levels of serum apoptosis and angiogenic markers were also analyzed for possible correlations with clinical outcomes (maximum tumor shrinkage and PFS). High baseline cytochrome C (Pearson correlation R=-0.64, P =0.001, n=22) and a higher ratio of M30 on cycle 1 day 8 to baseline (Pearson correlation R=-0.44, P=0.05, n=20) were associated with greater tumor shrinkage (Figures 3A and 3B).
Figure 3.
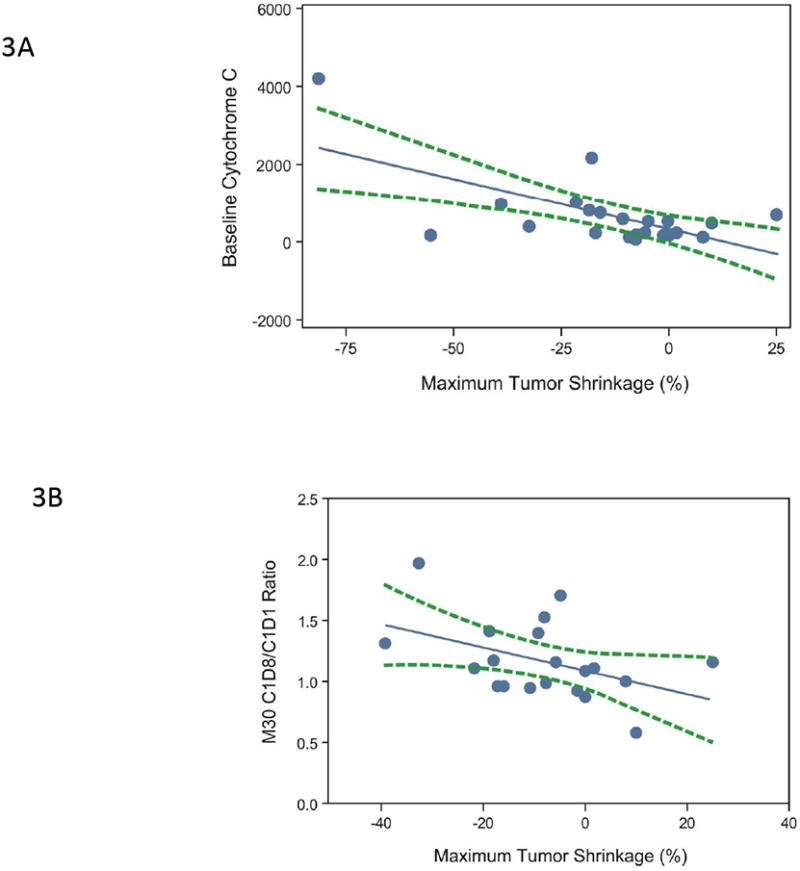
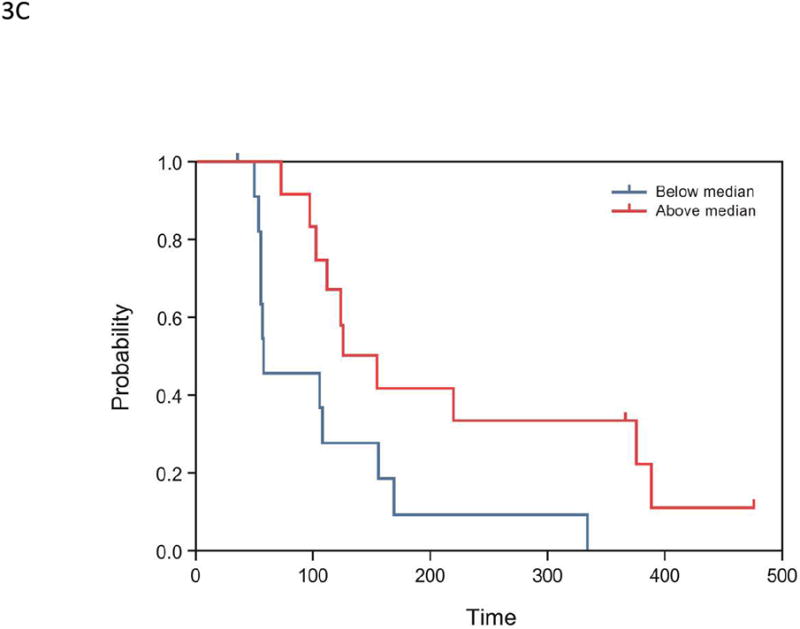
(A) Correlation of maximum tumor shrinkage (%) with baseline cytochrome C levels (n=22): Pearson correlation R=-0.64, P=0.001; Spearman correlation R=-0.45, P=0.0368. (B) Correlation between maximum tumor shrinkage (%) and M30 C1D8/C1D1 ratio (n=20): Pearson correlation R=-0.44, P=0.050; Spearman correlation R=-0.39, P=0.0894. Each blue dot represents 1 subject; the solid blue symbolizes the regression line; and the dashed green lines represent 95% confidence limits. Percent maximum tumor shrinkage was defined as the percentage of reduction in tumor size from baseline to post-baseline nadir. (C) Kaplan-Meier survival estimates of PFS, stratified by early changes in angiopoietin-1 levels, in the expanded melanoma cohort (n=24). Subjects were dichotomized by median ratios of angiopoietin-1 levels (2 hours postdose:baseline, P=0.007).
In univariate Cox proportional hazard model analyses, higher baseline sBP and dBP were associated with longer PFS (P=0.004 and P=0.028, respectively; Supplementary Table 1), whereas acute decreases in angiopoietin-1 (at 2 hours after the first dose of lenvatinib relative to baseline levels) were associated with shorter PFS (P=0.007; Figure 3C). Changes from baseline to day 8 in sTie-2, M30, or sBP levels were not found to be associated with PFS outcomes. In a multivariate Cox proportional hazard model analyses, baseline sBP and a ratio of angiopoietin-1 at 2 hours to baseline were significantly associated with PFS (P=0.041 and P=0.03, respectively; Supplementary Table 2) using covariates that were identified by univariate analysis (P<0.01, Supplementary Table 1).
Discussion
In this phase I dose-escalation study, the MTD for lenvatinib was established at 10 mg po bid. The drug was well tolerated, with most AEs being those known to be associated with VEGFR inhibition, such as hypertension, fatigue, and proteinuria (15, 16). These were most commonly observed in patients who had received lenvatinib at dose levels >10 mg. Antitumor activity was observed in patients with various solid tumor types, including melanoma, medullary thyroid, non–small cell lung, endometrial, renal, pancreatic, and ovarian. Responses to lenvatinib in patients with medullary thyroid cancer is of special interest, since several multikinase inhibitors have since been granted FDA approval for this indication (17, 18).
Hypertension, fatigue, and proteinuria have been seen with other VEGF inhibitors, including bevacizumab and cediranib (19-21), and are a common side effect of antiangiogenic therapy (22). The incidence of hypertension AEs in the present study (43%) was similar to that in a lenvatinib phase I study of once-daily continuous dosing (40%) (11), but lower than that in a lenvatinib phase I study of bid dosing on an interrupted schedule of 2 weeks on and 1 week off (67%) that explored higher doses (up to 20 mg bid) (23). Results from a population PK/PD analysis suggest that active management of side effects, including treatment with antihypertensives for hypertension in association with AE-guided dose de-escalations, may be effective in allowing 80% of patients to continue treatment with lenvatinib for 16 weeks (24).
The incidences of proteinuria (39%) and fatigue (42%) in the present study are largely consistent with those of other studies of lenvatinib administered to patients with solid malignancies (11, 15, 23). Differences observed in a once-daily dosing study (proteinuria, 26%; fatigue, 18%) (11) and in the bid dosing study with higher doses of lenvatinib (proteinuria, 63%; fatigue, 70%) (23) may be attributable, in part, to differences in doses, schedules, and patient populations.
In the current study, 9 patients had confirmed PRs; when patients with uPRs were included as a best response, the proportion was 15.6% (n=12). The SD rate was 51.9% (40/77 patients), with 19 (24.7%) patients having durable SD for ≥23 weeks. The combined PR/uPR/durable SD rate was 40.3% (31/77 patients). The overall anti-tumor activity observed in the present study was comparable to anti-tumor activities observed in other phase 1 studies of lenvatinib (11, 23). The combined PR/uPR/durable SD rate (40.3%) is also broadly comparable to that observed with other tyrosine kinase inhibitors in the treatment of advanced solid tumors. For those agents, the rates of PR and sustained SD ranged from 20% to 50%, with the values varying depending on the definition of durable SD (25-29).
The activity of lenvatinib as a single agent in melanoma is encouraging. Three (10.3%) of the 29 patients with melanoma had a confirmed PR, and 7 (24.1%) had durable SD. Two melanoma patients had uPRs; therefore, 17.2% of patients with melanoma had at least 30% regression as their best response. Among these responders was a patient with mutant NRAS. The 3 PR/uPR and 8 SD among the BRAFWT patients are of special interest because currently there are no targeted therapies approved for this patient population.
Melanoma is a highly vascular tumor (30), and angiogenesis plays a crucial role in malignant melanoma (31). Ugurel et al (32) found that serum concentrations of angiogenic factors, such as angiogenin, basic FGF, VEGF, and IL-8, were higher in patients with more advanced stages of melanoma, and that higher serum concentrations of these pro-angiogenic molecules were associated with diminished OS and PFS. Mehnert et al (33) reported higher expression of VEGFR2 in metastases relative to primary tumors, and suggested that angiogenesis is critical to melanoma metastasis. Lenvatinib may exert a greater effect on angiogenesis by targeting multiple signaling pathways, including VEGFR, FGFR, PDGF, RET, and KIT.
The utility of pharmacodynamic biomarkers evaluated in the present study was 2-fold: first, to demonstrate that lenvatinib was engaging the molecular target for which it was designed; and second, to attempt to identify subjects who could maximally benefit from lenvatinib treatment. Pretreatment expression levels of cytochrome C correlated favorably with maximum tumor shrinkage. Cytochrome C induces caspase activation and apoptosis in tumor cells (34). Higher baseline levels of serum cytochrome C may sensitize tumor cells to the apoptotic pathway upon lenvatinib treatment, resulting in greater tumor reduction. Tumor shrinkage was also associated with elevated levels of M30, an antibody that recognizes caspase-cleaved cytokeratin-18, on day 8 relative to day 1 of cycle 1 (35). Cytokeratins are intermediate filament proteins found primarily in epithelial cells (35). Cytokeratin-18 is cleaved by caspases during apoptosis, and thus, elevated caspase-cleaved cytokeratin-18 levels (as indicated by M30) is a measure of increased apoptosis (35). In the present study, the association between an increase in M30 levels between day 8 of cycle 1 (vs baseline) and greater tumor shrinkage suggests that apoptosis may serve as an indicator of, and potential mechanism for, response with lenvatinib in patients with melanoma.
In this study, decreases in sTie-2 levels at cycle 1 day 8 correlated with lenvatinib exposure. In addition, rapid decreases in serum levels of angiopoietin-1 at 2 hours post-lenvatinib treatment relative to baseline were associated with shorter PFS. Although lenvatinib does not directly target the Tie-2 receptor tyrosine kinase, the signaling pathways of both VEGF/VEGFR and angiopoietin/Tie-2 have been shown to be instrumental for tumor angiogenesis, and the angiopoietin-1/Tie-2 signaling network may be associated with antitumor activity of VEGFR signaling inhibitors (36). The relatively small sample size of the current pharmacodynamic analyses limits the ability to draw any conclusions; larger cohorts will be needed to further validate these findings. Importantly, in multivariate Cox proportional hazard model analyses, increased baseline sBP (P=0.04) and a decreased ratio of angiopoietin-1 at 2 hours versus baseline (P=0.03) were associated with longer PFS. Other studies have also shown a correlation between BP and response to antiangiogenic agents (37, 38). This preliminary result suggests that BP changes may serve as a marker for activity, and additional investigations are warranted.
In conclusion, in the present study of patients with advanced solid tumors, the MTD of lenvatinib was 10 mg bid. Lenvatinib was generally well tolerated, with the majority of AEs related to VEGFR inhibition. Antitumor activity was observed in several tumor types, particularly melanoma and medullary thyroid cancer, adding to the body of evidence for lenvatinib antitumor activity (4, 5, 11, 23, 39). The emerging evidence of antitumor activity across multiple tumor types has prompted the initiation of several phase II and phase III studies of lenvatinib in melanoma, advanced radioiodine-refractory differentiated thyroid cancer, medullary thyroid cancer, hepatocellular carcinoma, endometrial cancer, renal cell carcinoma, and glioblastoma. Biomarker analyses suggest that baseline levels or changes in angiogenesis-related and apoptosis-related markers are associated with PFS in lenvatinib trials. Ongoing phase II and phase III trials will further explore the prognostic and/or predictive utility of these biomarkers in lenvatinib treatment.
Supplementary Material
Statement of Translational Relevance.
Angiogenesis impacts the proliferation, growth, progression, migration, and metastasis of tumors and surrounding stromal cells. Lenvatinib is an antiangiogenic agent that acts specifically against tyrosine kinase receptors implicated in angiogenesis and related pathways (VEGFR 1–3, FGFR 1–3, and PDGFRα), as well as RET and KIT. This phase I study evaluated the maximum tolerated dose (MTD), dose-limiting toxicities, safety, pharmacokinetics, pharmacodynamics, and efficacy of lenvatinib given twice daily in patients with diverse tumor types, and in an expansion cohort of patients with melanoma. The MTD for lenvatinib was established at 10 mg twice daily. Encouraging antitumor activity was observed in patients with thyroid cancer and melanoma. Pharmacodynamic analyses suggested that several serum biomarkers of apoptosis and angiogenesis may potentially be predictive of clinical outcome in patients with advanced melanoma treated with lenvatinib.
Acknowledgments
We would like to thank all patients, as well as investigators and their teams, who participated and are participating in this study. We thank Joann Aaron, MA, for scientific editing of the manuscript. Editorial support was provided by Phase Five Communications and Oxford PharmaGenesis Inc., and was funded by Eisai Inc.
Grant Support: This work was supported by Eisai Inc. and by the NIH Clinical and Translational Science Award UL1 RR024148 and NIH Cancer Center Support Grant (CCSG) award CA016672 to MD Anderson Cancer Center.
Footnotes
Conflict of Interest Statement: David S. Hong: Research funding, Eisai Inc.
Razelle Kurzrock: Nothing to disclose.
Jennifer J. Wheler: Nothing to disclose.
Aung Naing: Nothing to disclose.
Gerald S. Falchook: Nothing to disclose
Siqing Fu: Nothing to disclose.
Kevin B. Kim: Honorarium and research funding, Eisai Inc.
Michael A. Davies: Nothing to disclose.
Ly M. Nguyen: Nothing to disclose.
Goldy C. George: Nothing to disclose.
Lucy Xu: Employee of Eisai Inc.
Robert Shumaker: Employee of Eisai Inc.
Min Ren: Employee of Eisai Inc.
Jennifer Mink: Nothing to disclose.
Cynthia Bedell: Nothing to disclose.
Corina Andresen: Nothing to disclose.
Pallavi Sachdev: Employee of Eisai Inc.
James P. O'Brien: Employee of Eisai Inc.
John Nemunaitis: Research funding from Eisai Inc.
References
- 1.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 2.Korc M, Friesel RE. The role of fibroblast growth factors in tumor growth. Curr Cancer Drug Targets. 2009;9:639–51. doi: 10.2174/156800909789057006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matsui J, Funahashi Y, Uenaka T, Watanabe T, Tsuruoka A, Asada M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin Cancer Res. 2008;14:5459–65. doi: 10.1158/1078-0432.CCR-07-5270. [DOI] [PubMed] [Google Scholar]
- 5.Matsui J, Yamamoto Y, Funahashi Y, Tsuruoka A, Watanabe T, Wakabayashi T, et al. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer. 2008;122:664–71. doi: 10.1002/ijc.23131. [DOI] [PubMed] [Google Scholar]
- 6.Okamoto K, Kodama K, Takase K, Sugi NH, Yamamoto Y, Iwata M, et al. Antitumor activities of the targeted multi-tyrosine kinase inhibitor lenvatinib (E7080) against RET gene fusion-driven tumor models. Cancer Lett. 2013;340:97–103. doi: 10.1016/j.canlet.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 7.Shumaker R, Aluri J, Fan J, Martinez G, Ren M, Chen K. Evaluation of the effects of formulation and food on the pharmacokinetics of lenvatinib (E7080) in healthy volunteers. Int J Clin Pharmacol Ther. 2014;52:284–91. doi: 10.5414/CP201937. [DOI] [PubMed] [Google Scholar]
- 8.Shumaker R, Aluri J, Fran J, Martinez G, Thompson GA, Ren M. Effects of ketoconazole on the pharmacokinetics of lenvatinib (E7080) in healthy participants. Clin Pharmacol Drug Devel. 2015;4:155–60. doi: 10.1002/cpdd.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shumaker R, Aluri J, Fan J, Martinez G, Thompson GA, Ren M. Effect of rifampicin in the pharmacokinetics of lenvatinib in health adults. Clin Drug Investig. 2014;4:651–59. doi: 10.1007/s40261-014-0217-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dubbelman AC, Rosing H, Nijenhuis C, Huitema AD, Mergui-Roelvink M, Gupta A, et al. Pharmacokinetics and excretion of (14)C-lenvatinib in patients with advanced solid tumors or lymphomas. Invest New Drugs. 2015;33:233–40. doi: 10.1007/s10637-014-0181-7. [DOI] [PubMed] [Google Scholar]
- 11.Boss DS, Glen H, Beijnen JH, Keesen M, Morrison R, Tait B, et al. A phase I study of E7080, a multitargeted tyrosine kinase inhibitor, in patients with advanced solid tumours. Br J Cancer. 2012;106:1598–604. doi: 10.1038/bjc.2012.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simon R, Freidlin B, Rubinstein L, Arbuck SG, Collins J, Christian MC. Accelerated titration designs for phase I clinical trials in oncology. J Natl Cancer Inst. 1997;89:1138–47. doi: 10.1093/jnci/89.15.1138. [DOI] [PubMed] [Google Scholar]
- 13.Trotti A, Colevas AD, Setser A, Rusch V, Jaques D, Budach V, et al. CTCAE v3.0: development of a comprehensive grading system for the adverse effects of cancer treatment. Semin Radiat Oncol. 2003;13:176–81. doi: 10.1016/S1053-4296(03)00031-6. [DOI] [PubMed] [Google Scholar]
- 14.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 15.Keizer RJ, Gupta A, Mac Gillavry MR, Jansen M, Wanders J, Beijnen JH, et al. A model of hypertension and proteinuria in cancer patients treated with the anti-angiogenic drug E7080. J Pharmacokinet Pharmacodyn. 2010;37:347–63. doi: 10.1007/s10928-010-9164-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eskens FA, Verweij J. The clinical toxicity profile of vascular endothelial growth factor (VEGF) and vascular endothelial growth factor receptor (VEGFR) targeting angiogenesis inhibitors; a review. Eur J Cancer. 2006;42:3127–39. doi: 10.1016/j.ejca.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 17.Elisei R, Schlumberger MJ, Müller SP, Schöffski P, Brose MS, Shah MH, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31:3639–46. doi: 10.1200/JCO.2012.48.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wells SA, Jr, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134–41. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Izzedine H, Massard C, Spano JP, Goldwasser F, Khayat D, Soria JC. VEGF signalling inhibition-induced proteinuria: Mechanisms, significance and management. Eur J Cancer. 2010;46:439–48. doi: 10.1016/j.ejca.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Robinson ES, Matulonis UA, Ivy P, Berlin ST, Tyburski K, Penson RT, et al. Rapid development of hypertension and proteinuria with cediranib, an oral vascular endothelial growth factor receptor inhibitor. Clin J Am Soc Nephrol. 2010;5:477–83. doi: 10.2215/CJN.08111109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu X, Wu S, Dahut WL, Parikh CR. Risks of proteinuria and hypertension with bevacizumab, an antibody against vascular endothelial growth factor: systematic review and meta-analysis. Am J Kidney Dis. 2007;49:186–93. doi: 10.1053/j.ajkd.2006.11.039. [DOI] [PubMed] [Google Scholar]
- 22.van den Meiracker AH, Lankhorst S, van Esch Joep HM, Jan Danser AH, Kappers Mariëtte HW. Hypertension induced by antiangiogenic therapy: clinical and pathophysiological aspects. Eur J Hosp Pharm. 2012;19:327–9. [Google Scholar]
- 23.Yamada K, Yamamoto N, Yamada Y, Nokihara H, Fujiwara Y, Hirata T, et al. Phase I dose-escalation study and biomarker analysis of E7080 in patients with advanced solid tumors. Clin Cancer Res. 2011;17:2528–37. doi: 10.1158/1078-0432.CCR-10-2638. [DOI] [PubMed] [Google Scholar]
- 24.Keizer RJ, Gupta A, Shumaker R, Beijnen JH, Schellens JH, Huitema AD. Model-based treatment optimization of a novel VEGFR inhibitor. Br J Clin Pharmacol. 2012;74:315–26. doi: 10.1111/j.1365-2125.2012.04197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hurwitz HI, Dowlati A, Saini S, Savage S, Suttle AB, Gibson DM, et al. Phase I trial of pazopanib in patients with advanced cancer. Clin Cancer Res. 2009;15:4220–7. doi: 10.1158/1078-0432.CCR-08-2740. [DOI] [PubMed] [Google Scholar]
- 26.Kurzrock R, Sherman SI, Ball DW, Forastiere AA, Cohen RB, Mehra R, et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol. 2011;29:2660–6. doi: 10.1200/JCO.2010.32.4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strumberg D, Richly H, Hilger RA, Schleucher N, Korfee S, Tewes M, et al. Phase I clinical and pharmacokinetic study of the Novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43-9006 in patients with advanced refractory solid tumors. J Clin Oncol. 2005;23:965–72. doi: 10.1200/JCO.2005.06.124. [DOI] [PubMed] [Google Scholar]
- 28.Herbst RS, Maddox AM, L RM, Small EJ, Rubin EH, Baselga J, et al. Selective oral epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 is generally well-tolerated and has activity in non–small-cell lung cancer and other solid tumors: results of a phase I trial. J Clin Oncol. 2002;20:3815–25. doi: 10.1200/JCO.2002.03.038. [DOI] [PubMed] [Google Scholar]
- 29.Rosen LS, Kurzrock R, Mulay M, Van Vugt A, Purdom M, Ng C, et al. Safety, pharmacokinetics, and efficacy of AMG 706, an oral multikinase inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2007;25:2369–76. doi: 10.1200/JCO.2006.07.8170. [DOI] [PubMed] [Google Scholar]
- 30.Basu B, Biswas S, Wrigley J, Sirohi B, Corrie P. Angiogenesis in cutaneous malignant melanoma and potential therapeutic strategies. Expert Rev Anticancer Ther. 2009;9:1583–98. doi: 10.1586/era.09.135. [DOI] [PubMed] [Google Scholar]
- 31.Dewing D, Emmett M, Pritchard Jones R. The Roles of Angiogenesis in Malignant Melanoma: Trends in Basic Science Research over the Last 100 Years. ISRN Oncol. 2012;2012:546927. doi: 10.5402/2012/546927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ugurel S, Rappl G, Tilgen W, Reinhold U. Increased serum concentration of angiogenic factors in malignant melanoma patients correlates with tumor progression and survival. J Clin Oncol. 2001;19:577–83. doi: 10.1200/JCO.2001.19.2.577. [DOI] [PubMed] [Google Scholar]
- 33.Mehnert JM, McCarthy MM, Jilaveanu L, Flaherty KT, Aziz S, Camp RL, et al. Quantitative expression of VEGF, VEGF-R1, VEGF-R2, and VEGF-R3 in melanoma tissue microarrays. Hum Pathol. 2010;41:375–84. doi: 10.1016/j.humpath.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ledgerwood EC, Morison IM. Targeting the apoptosome for cancer therapy. Clin Cancer Res. 2009;15:420–4. doi: 10.1158/1078-0432.CCR-08-1172. [DOI] [PubMed] [Google Scholar]
- 35.Linder S, Olofsson MH, Herrmann R, Ulukaya E. Utilization of cytokeratin-based biomarkers for pharmacodynamic studies. Expert Rev Mol Diagn. 2010;10:353–9. doi: 10.1586/erm.10.14. [DOI] [PubMed] [Google Scholar]
- 36.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider BP, Wang M, Radovich M, Sledge GW, Badve S, Thor A, et al. Association of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 genetic polymorphisms with outcome in a trial of paclitaxel compared with paclitaxel plus bevacizumab in advanced breast cancer: ECOG 2100. J Clin Oncol. 2008;26:4672–8. doi: 10.1200/JCO.2008.16.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rini BI, Cohen DP, Lu DR, Chen I, Hariharan S, Gore ME, et al. Hypertension as a biomarker of efficacy in patients with metastatic renal cell carcinoma treated with sunitinib. J Natl Cancer Inst. 2011;103:763–73. doi: 10.1093/jnci/djr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glen H, Mason S, Patel H, Macleod K, Brunton VG. E7080, a multi-targeted tyrosine kinase inhibitor suppresses tumor cell migration and invasion. BMC Cancer. 2011;11:309. doi: 10.1186/1471-2407-11-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.