

Abstract
In order to understand organogenesis, the spatial and temporal alterations that occur during development of tissues need to be recorded. The method described here allows time-lapse analysis of normal and impaired kidney development in zebrafish embryos by using a fluorescence dissecting microscope equipped for structured illumination and z-stack acquisition. To visualize nephrogenesis, transgenic zebrafish (Tg(wt1b:GFP)) with fluorescently labeled kidney structures were used. Renal defects were triggered by injection of an antisense morpholino oligonucleotide against the Wilms tumor gene wt1a, a factor known to be crucial for kidney development.
The advantage of the experimental setup is the combination of a zoom microscope with simple strategies for re-adjusting movements in x, y or z direction without additional equipment. To circumvent focal drift that is induced by temperature variations and mechanical vibrations, an autofocus strategy was applied instead of utilizing a usually required environmental chamber. In order to re-adjust the positional changes due to a xy-drift, imaging chambers with imprinted relocation grids were employed.
In comparison to more complex setups for time-lapse recording with optical sectioning such as confocal laser scanning or light sheet microscopes, a zoom microscope is easy to handle. Besides, it offers dissecting microscope-specific benefits such as high depth of field and an extended working distance.
The method to study organogenesis presented here can also be used with fluorescence stereo microscopes not capable of optical sectioning. Although limited for high-throughput, this technique offers an alternative to more complex equipment that is normally used for time-lapse recording of developing tissues and organ dynamics.
Keywords: Developmental Biology, Issue 110, Zebrafish, time-lapse, fluorescence dissecting microscope, autofocus strategy, relocation grid, kidney development
Introduction
Following gastrulation, organogenesis is the next stage of an individual's life cycle. It involves the rearrangement, interaction and very often migration of cells to produce tissues and organs. Inaccuracy in the processes underlying the development of organs often leads to diseases that can manifest themselves either immediately or also later in life. Thus, understanding organogenesis has been a major effort in developmental biology and biomedical research. In order to be able to investigate the development of a particular organ, it has to be visible even through the body wall of the embryo. This enables the recording of the spatial and temporal alterations that occur during development. Also in order to analyze the importance of factors involved in organogenesis, it needs to be susceptible to manipulation.
One organism that is exceedingly suitable for the investigation of in vivo organogenesis is zebrafish. Its transparency during embryonic development in combination with the use of fluorescent transgenic lines allows observation of protein localization and expression dynamics, as well as the visualization of the inner organs 1. This provides a unique advantage concerning the investigation of organ development in real time compared to mammalian models whose organs are inaccessible for microscopic analysis. Moreover, different tools are available to manipulate embryonic development of zebrafish. Beside the generation of mutant lines, antisense morpholino oligonucleotides (MO) can be used to knockdown the activity of particular genes that are involved in the development of certain organs. MOs either target a splice site or the translational start codon (AUG), and thereby interfere with splicing of pre-mRNA or translation.
The zebrafish embryonic kidney, the pronephros, is an anatomically simple but valuable model to study kidney development and the function of genes related to kidney diseases 2. It consists of only two functional units called nephrons. Each nephron consists of a glomerulus where blood filtration takes place 3. Further components are the short neck region as well as the segmented tubule for secretion and reabsorption of solutes and the duct, which ends in the cloaca 4. Regardless of its simple composition, the organization and the different cell types of the zebrafish pronephros are very similar to the mammalian kidney 5,6.
One factor, which is critically involved in kidney development, is encoded by the Wilms Tumor suppressor gene Wt17. Zebrafish possess two paralogs called wt1a and wt1b being expressed in an overlapping but not identical pattern during development of the pronephros 8. By using transgenic zebrafish with GFP-labeled pronephros structures, it has been shown that complete knock-down of wt1a or of a particular splice form leads to severe or mild malformations of the embryonic kidney, respectively 9,10.
The method described here allows time-lapse analysis of normal and impaired nephrogenesis in zebrafish embryos by employing a fluorescence dissecting microscope equipped for optical sectioning via structured illumination. Optical sections in general allow the acquisition of images which only contain in-focus information. Out-of-focus information can be avoided by various approaches such as mathematical algorithms (e.g. deconvolution), optical design (e.g. confocal laser scanning microscopy) or a combination of both (e.g. structured illumination).
To induce defects in kidney development we used an antisense MO against wt1a that was injected into a transgenic zebrafish line (Tg(wt1b:GFP)). This line shows GFP-fluorescence in the glomerulus, neck and the anterior part of the tubule 9,10. In comparison to more complex setups for time-lapse recordings with optical sectioning such as laser scanning or light sheet microscopes, a zoom microscope is easy to handle and less expensive. Moreover, equipment for structured illumination can easily be combined with conventional fluorescence equipment (e.g. fluorescence lamp, filters) and offers dissecting microscope-specific advantages such as an extended working distance and large field of view. Problems with drift were solved without using additional equipment (environmental chamber, anti-vibration table) usually required for stable results. To correct for focal drift an autofocus strategy was established and imaging chambers with imprinted relocation grids were utilized to re-adjust the positioning following a drift in x or y direction.
The presented method can also be applied to microscopes without optical sectioning options, such as fluorescence stereo microscopes and offers an alternative to more complex equipment that is normally used for time-lapse recording of developing tissues and organ dynamics.
Protocol
All animal experiments were performed according to the 'Principles of laboratory animal care' as well as to the current version of the German Law on the Protection of Animals.
1. Preparation of Antisense-Morpholino (MO)
To prepare a 3 mM MO stock solution, add 100 µl of sterile, ultrapure water to the lyophilized MO (300 nmol in glass vial). For complete dissolution, heat the stock solution to 65 °C for 5 min. Seal the vial with Parafilm and store the MO stock solution at room temperature (RT). Do not chill the MO stock solution because that could cause an association of the MO with the vial walls.
- Prepare a 1 mM MO working solution by diluting the MO stock solution with sterile, ultrapure water and add 0.5% phenol red solution to a final concentration of 0.05%.
- To get 10 µl of a MO working solution, add 3.3 µl MO stock solution and 1 µl of 0.5% phenol-red solution to 5.7 µl water. Before preparing a new batch of working solution heat the MO stock solution to 65 °C for 5 min.
Prior to use, centrifuge the MO working solution to prevent needle clogging. For example, spin 10 µl of MO stock solution at 15,000 x g for 5 min (RT) and remove 8 µl of the supernatant for injection. NOTE: Loss of activity may occur in morpholino solutions over time 11. To exclude this, one can test the solutions by UV spectrometry following the protocol supplied by the manufacturer. In case of the used wt1a morpholino a reduced effectiveness has not been observed over a long period of storage (3 mM working solution was stored longer than one year).
2. Setting Up Mating Pairs and Collection of Fertilized Eggs
The afternoon prior to microinjection, set up five pairs of the Tg (wt1b: GFP) line, each in a breeding container equipped with a removable sieve to separate males and females during night. NOTE: To ensure that enough eggs are available for a successful injection experiment, fish used as mating pairs should have been pre-screened and evaluated as good "producers".
The next morning, after the lights turn on, put the male and female together above the sieve that prevents the fish from eating up their eggs.
Watch the spawning. Once an amount of eggs are laid that can be injected before the 4-cell stage is reached (approximately 50), separate male from female again. Transfer the eggs with a Pasteur pipette into a petri dish filled with embryo water for a first round of injection. To get additional rounds of eggs, place a mating pair together and separate several times.
3. Preparatory Work for Microinjection
NOTE: Perform steps 3.1 and 3.2 in advance.
To prepare a dish that holds the embryos in position during the injection (microinjection dish) insert a glass slide at a 40-45° angle into a 94 mm Petri dish filled with liquid, transparent, pure, food grade silicon. Remove the slide when the silicon is hardened. That produces a groove in the silicon layer. NOTE: Alternatively, use 1.5-3% agarose instead of silicon and store the dish at 4 °C.
- Generate injection needles from glass capillaries with a needle puller.
- Use capillaries that contain internal filaments. NOTE: The filament will help to transport the MO solution toward the tip of the needle after backfilling (see 3.3).
- Establish appropriate settings on the needle puller. Good needles for injection into zebrafish should have long and thin tips 12. Generate tips with a length of approximately 10 mm and a diameter of 1.5-2 µm at their very end. For example, use a horizontal needle puller with following settings: Heat = 330, Pull = 0, Velocity = 40, Time = 100.
- Examine the quality of the needles under a dissecting microscope and sort out those with short or too long tips. NOTE: Needles with short tips tend to damage the embryo while such with very long and thin tips bend when touching the chorion and do not penetrate it.
Backfill the needle with 2 µl of MO working solution by using a microloader tip and insert it into the needle holder of the microinjection apparatus.
As the needles are closed at the tip end, generate an opening by pinching back the very end of the needle with sharp tweezers under the dissection microscope. Alternatively use the microinjection apparatus to gently push the tip to a rigid surface (e.g. small metal block or back of tweezers).
Calibrate the injection volume by injecting the MO working solution into a drop of mineral oil placed on a graticule. Adjust the duration of injection for producing a drop diameter of 75 µm to obtain an injection volume of approximately 200 pl.
4. Microinjection Procedure
Arrange the 1-cell stage embryos along the groove of the microinjection dish with the vegetal pole toward the direction from where the needle comes (toward the 45° angle of the groove).
Perforate the chorion and the yolk with the needle and inject 200 pl of the 1 mM MO working solution (0.2 pmol) into the yolk of 1- to 2-cell embryos.
Using a Pasteur pipette with a wide opening, collect all of the injected embryos in a Petri dish filled with embryo water and incubate them at 28.5 °C.
In the afternoon, remove dead embryos by using a Pasteur pipette with a wide opening and replace the embryo water.
5. Preparation of Embryos for In Vivo Imaging
The next morning, replace the embryo water with embryo water containing 0.003% N-phenylthiourea (PTU) to inhibit melanization. Do not apply PTU before the end of gastrulation because it interferes with early embryonic development. CAUTION: PTU is toxic and teratogenic. Wear gloves at all times and avoid inhalation and contact with skin.
Dechorionate the embryos with sharp tweezers under a dissection microscope at the desired developmental stage. Grab the chorion with one pair of tweezers and try to grab the other side of the chorion with a second pair of tweezers. Carefully pull the tweezers in opposite directions without disrupting the embryo.
Anesthetize the embryo by replacing the embryo water containing PTU (0.003%) with embryo water containing PTU (0.003%) and 0.02% Ethyl 3-aminobenzoate methanesulfonate (Tricaine).
- Embed the embryo in low melting agarose on a coverglass-bottomed µ-dish with a 50 µm relocation grid. Proper embedding requires some practice.
- Prepare 1 ml aliquots of 0.7% agarose in 1.5 ml reaction tubes and put them on a heat block set to 36 °C. Use a Pasteur pipette with wide opening to transfer the embryo to the µ-dish. Remove excess embryo water using a Pasteur pipette with ultra-thin tip and overlay the embryo with tempered agarose.
- While the agarose is still liquid, orient the embryo with two gel loader tips in the desired position, with regard to the structure of interest as well as the distance to the relocation grid. Place the structure of interest (here the pronephros) as close as possible to the glass bottom (here: dorsal down) and in close vicinity to the grid.
- For optimal image quality minimize the agarose layer between the embryo and the glass bottom by placing the structure of interest of the embryo as close as possible to the bottom. Besides that, locate the embryo in close vicinity to the grid but take care that the grid will not overlay with the structure of interest. Embed 3-4 embryos in different µ-dishes each and use the one that is positioned best.
- When the agarose is hard enough to hold the embryo wait for 2-3 min and overlay the embedded embryo with embryo water containing 0.003% PTU and 0.02% Tricaine. Decant the excess embryo water or remove it by using a Pasteur pipette with ultra-thin tip. Close the lid of the µ-dish at the lock position to prevent evaporation. NOTE: Avoid the occurrence of air bubbles in the µ-dish, as they would impair image recording.
6. Microscopy
NOTE: Use a microscope equipped for optical sectioning via structured illumination. Record images with a software containing the following modules: Multichannel, Z-Stack, Time Series, Software Autofocus and Extended Focus.
- Acquisition of Z-stacks
- Invert the µ-dish. The glass bottom now faces toward the objective and the µ-dish serves as an imaging chamber.
- Toggle the slider into the grid position and enable the structured illumination mode in the software controls.
- Calibrate the grid focus for the channel(s) of choice with the specimen to be examined (embedded embryo) by following the instructions of the Focus Calibration Wizard.
- Activate the z-Stack acquisition mode and set it to center mode. Focus into the middle of the specimen and set it as the center plane of the z- stack by clicking Center. Set the dimension of the z-stack by defining the range around the center position. NOTE: Object structures should not be seen sharply outside the first and the last plane of the z-stack.
- Adjust the spacing between optical sections. For best z-resolution click the Optimal button. Based on the objectives depth of field the software will set a recommended distance following the Nyquist criterium (50% overlap). NOTE: If aiming for smaller file sizes and the structure permits, set a larger spacing size manually. Setting smaller steps than recommended will merely result in larger file sizes without increasing lateral information.
- Time-lapse Imaging
- Open the Time Series tool and set the interval of the time series experiment. For example record z-stack images every 30 min over a period of 5 hr.
- To correct for focal drift over time, set up an autofocus strategy. Check the Focus Strategy dialogue box, select Software Autofocus from the drop-down list and choose the TL-Brightfield channel as reference channel.
- Start each time point with the autofocus. In order to assure a reliable search range within an optimized time frame, set a fixed 200 µm relative search range. This range is fully scanned (Range Coverage parameter) with basic quality (Quality parameter) in a maximum step size (Sampling parameter).
- To increase precision of the autofocus, activate the metering within a focus region of interest (focus ROI) and position said focus ROI in the live view window around the structure of interest. NOTE: If the system lacks the Software Autofocus module which updates the center z-position automatically during time-lapse recording, pause the experiment at a given time and adjust the focus manually and continue the experiment.
- To compensate for a potential xy-drift during time series recording, position a particular point of the imprinted grid (of the imaging chamber) into the crosshairs of the software and save the positioning by making a screenshot.
- Start the time series experiment. Before the time series interval is over, pause the experiment and, if necessary, re-adjust positioning according to the screenshot. Continue with the experiment.
7. Image Processing
Switch to the processing tab and select the Extended Depth of Focus (EDF) in the Method dialogue box. Since optical sections are acquired, apply the Maximum Projection algorithm to the image series. NOTE: Here the algorithm will project the brightest or the darkest pixel from stack to a composite 2D image in a first step and eventually use the one with the highest variance as a result image for each time point.
Use the Movie Export method to create a time lapse movie (e.g. avi or move file) out of the EDF image series.
Representative Results
Time-lapse microscopy is a powerful technique to watch dynamic biological processes over a longer period of time. An often occurring problem during time-lapse imaging is a movement in x, y or z direction, called drift, which is induced by temperature variations and mechanical vibrations. The method presented here enables time-lapse recording of fluorescently labeled structures in zebrafish embryos or larvae on a dissecting microscope without environmental chamber and anti-vibration table.
For compensation of xy-drift, the specimen was embedded in an imaging chamber with an imprinted relocation grid (Figure 1). The location of a particular point of the grid related to the crosshairs of the software tool was used to return the specimen to the pre-adjusted position (Figure 2A). The presented results were obtained by using a zoom microscope with an integrated slider for optical sectioning via structured illumination (Figure 2B). For continuous focal correction, an autofocus strategy was established. In this strategy, the software autofocus is used to find the focus based on maximum contrast before every time point. This so defined focal plane is subsequently set as center plan for z-stack acquisition. In order to minimize phototoxicity in the sample, the transmitted light brightfield channel is used as reference channel as it allows sufficiently short exposure times during the autofocus step (Figure 2C).
The method was applied for comparative analysis of kidney development in control- and wt1a morphant embryos of a transgenic zebrafish line (Tg(wt1b:GFP)). During early nephrogenesis this line shows green fluorescence in the intermediate mesoderm, were the kidney progenitors arise from 9,10 and later in the forming tubules and nephron primordia (Figure 3).
Time-lapse image recording reveals that nephrogenesis in control morpholino injected embryos is unaltered in comparison to uninjected ones (results not shown) and follows the described steps of early zebrafish kidney development 3. At 20 hpf the developing pronephric tubules are visible and at their anterior tips, spherical accumulations of cells, representing the forming nephron primordia can be detected. During the next hours, tubules and nephron primordia grow and later on the primordia start to fuse at the midline (Figure 4, video 2). In contrast, nephrogenesis is severely disrupted in wt1a morphant embryos. Although GFP-positive tubular structures are visible at 20 hpf, they appear to be more diffuse and less developed. Furthermore, no proper nephron primordia have been formed. The most striking difference to the control embryos at this time point, however, is the appearance of a large number of fluorescent cells outside the developing pronephros (Figure 4, video 2). Subsequently, these cells leave the pronephric field and migrate ventrally (video 2).
Kidney development in control embryos and wt1a morphants of the wt1b transgenic line have been previously compared by taking images at different fixed time points 9,10. In contrast to this static method, time-lapse recording allows to follow dynamics of normal nephrogenesis and misrouting of kidney progenitor cells caused by wt1a depletion.
 Figure 1: Schematic Illustration of an Embedded Embryo in a Commercially Available Chamber with Relocation Grids. The dish has four grids, each subdivided into a repeat distance of 50 µm, imprinted into a glass coverslip bottom. The embryo to be imaged is embedded upside down, with kidney structure in close proximity to an observation square but without overlaying with the grid. Please click here to view a larger version of this figure.
Figure 1: Schematic Illustration of an Embedded Embryo in a Commercially Available Chamber with Relocation Grids. The dish has four grids, each subdivided into a repeat distance of 50 µm, imprinted into a glass coverslip bottom. The embryo to be imaged is embedded upside down, with kidney structure in close proximity to an observation square but without overlaying with the grid. Please click here to view a larger version of this figure.
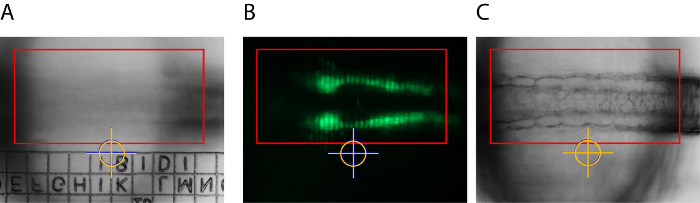 Figure 2: Adjustments for Compensation of Drift in Time-lapse Imaging and Grid Projection of Structured Illumination. A distinct position of the relocation grid was brought into the image center (marked by yellow crosshairs) and serves as a reference point for later xy-alignment in case of small shifts. The red rectangle represents the region of interest (ROI) used in the autofocus strategy (A). Optical sectioning was obtained by structured illumination. The image shows a grid structure projected in the focal plane after correct calibration (B). The ROI for the autofocus strategy (red rectangle) is set to cover distinctive embryonic structures high in contrast (here somites) that allow reliable autofocusing into the structure of interest (C). Please click here to view a larger version of this figure.
Figure 2: Adjustments for Compensation of Drift in Time-lapse Imaging and Grid Projection of Structured Illumination. A distinct position of the relocation grid was brought into the image center (marked by yellow crosshairs) and serves as a reference point for later xy-alignment in case of small shifts. The red rectangle represents the region of interest (ROI) used in the autofocus strategy (A). Optical sectioning was obtained by structured illumination. The image shows a grid structure projected in the focal plane after correct calibration (B). The ROI for the autofocus strategy (red rectangle) is set to cover distinctive embryonic structures high in contrast (here somites) that allow reliable autofocusing into the structure of interest (C). Please click here to view a larger version of this figure.
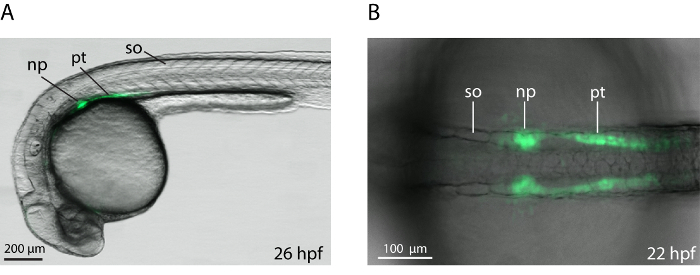 Figure 3: Transgenic wt1b:GFP Embryos with Green Fluorescence in the Developing Kidney. Overlays of dorsal (A) or lateral (B) transmission and fluorescence images are shown with anterior to the left. np, nephron primordium; pt, pronephric tubule; so, somite. Please click here to view a larger version of this figure.
Figure 3: Transgenic wt1b:GFP Embryos with Green Fluorescence in the Developing Kidney. Overlays of dorsal (A) or lateral (B) transmission and fluorescence images are shown with anterior to the left. np, nephron primordium; pt, pronephric tubule; so, somite. Please click here to view a larger version of this figure.
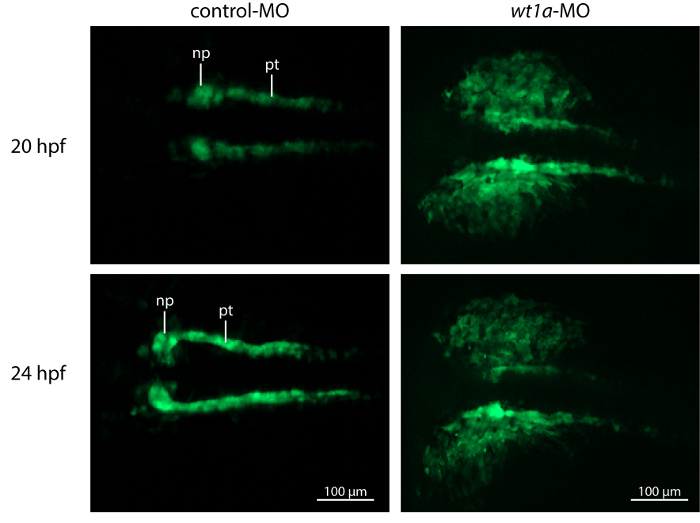 Figure 4: Knockdown of wt1a Disrupts Embryonic Kidney Development. Representative, extended depth of focus images from time-lapse recordings. In control morpholino injected embryos, kidney development shows normal progress with growing tubules and nephron primordia which start to fuse at the midline. In contrast, wt1a morphants fail to form proper nephron primordia and a massive amount of GFP positive cells are outside of the pronephric field. (np, nephron primordium; pt, pronephric tubule). Please click here to view a larger version of this figure.
Figure 4: Knockdown of wt1a Disrupts Embryonic Kidney Development. Representative, extended depth of focus images from time-lapse recordings. In control morpholino injected embryos, kidney development shows normal progress with growing tubules and nephron primordia which start to fuse at the midline. In contrast, wt1a morphants fail to form proper nephron primordia and a massive amount of GFP positive cells are outside of the pronephric field. (np, nephron primordium; pt, pronephric tubule). Please click here to view a larger version of this figure.
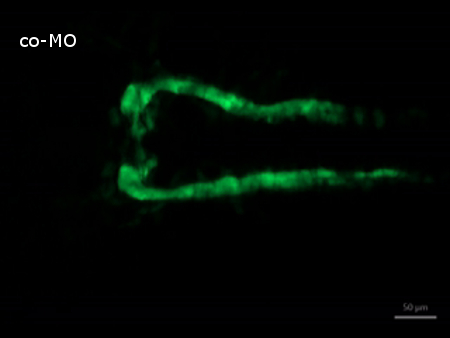 Supplemental Video 1. Time-lapse recording of normal kidney development in control morpholino injected embryos. (Right click to download). The video shows how tubules grow and nephron primordia begin to fuse. Starting at 20 hpf, images were taken in 30 min intervals over a period of 5 hr.
Supplemental Video 1. Time-lapse recording of normal kidney development in control morpholino injected embryos. (Right click to download). The video shows how tubules grow and nephron primordia begin to fuse. Starting at 20 hpf, images were taken in 30 min intervals over a period of 5 hr.
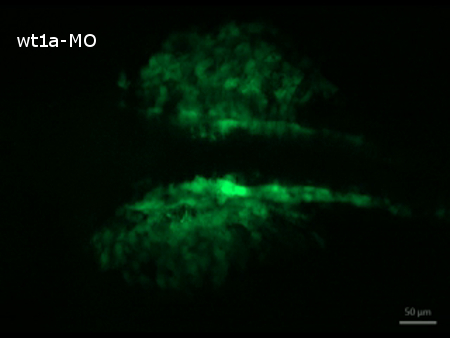 Supplemental Video 2. Time-lapse recording of disturbed nephrogenesis in wt1a morphant embryos. (Right click to download). The video shows the migration of GFP-positive cells out of the pronephric region. Starting at 20 hpf, images were taken in 30 min intervals over a period of 5 hr.
Supplemental Video 2. Time-lapse recording of disturbed nephrogenesis in wt1a morphant embryos. (Right click to download). The video shows the migration of GFP-positive cells out of the pronephric region. Starting at 20 hpf, images were taken in 30 min intervals over a period of 5 hr.
Discussion
The zebrafish has become a popular model organism for studies of vertebrate development and modelling of human diseases. Because zebrafish embryos remain transparent, developing organs like brain and heart can be observed using a standard preparation microscope. Taking advantage of transgenic lines with organ specific fluorescence enables assessments of organogenesis throughout different stages of development within living embryos by fluorescence microscopy. One limitation for imaging structural details of whole organs with standard epifluorescence microscopy is the impact of signals from objects above and below the focal plane. This out-of-focus light not only results in decreased image contrast and resolution, it also may obscure important structures of interest 13,14. Several techniques such as laser scanning confocal-, spinning disk confocal- or multiphoton microscopy have been developed to minimize the out-of-focus information and thereby increase image contrast as well as axial resolution. An alternative method to obtain optical sections is the laser- and scanning free structured illumination microscopy, a wide field based illumination technique which is and simple to implement on a regular microscope 15. The results presented here illustrate that optical sectioning, achieved by structured illumination, implemented on a dissecting microscope and combined with reconstruction of extended focus images, enables visualization of structural details of normal and disturbed kidney development in zebrafish. In particular, the GFP-positive cells in wt1a morphants are dispersed at various focal depths, and images of only one plane with out-of- focus blur would underestimate the three dimensional complexity of the phenotype.
To follow the dynamics of organ organization, rearrangement or disruption, time-lapse fluorescence microscopy is a powerful albeit complex technique. During time-lapse imaging slight vibrations or minor temperature fluctuations cause drifts in x, y or z direction. This demands additional equipment (environmental chamber, anti-vibration table) in order to obtain stable results. The presented method uses the capability of a dissecting microscope with a motorized focus drive for recording time-lapse images without extra devices. To maintain a stable focus, an autofocus strategy was established and a relocation grid imprinted into the bottom of the imaging chamber was used for correction of x,y- instability.
Proper embedding of the embryo is a critical step within the protocol. Some practice is needed to place the structure of interest as close as possible to the glass bottom and in close vicinity to the grid without overlaying with it.
The main advantage of the method is that it is an affordable and simple tool for direct observation of developmental processes such as growth and migration in living embryos over several hours. Moreover, the dissection microscope-specific benefits such as large field of view and extended working distance facilitate examination of larger samples including whole organs. However, some limitations have to be kept in mind. Pausing of the experiment to check and readjust the positioning makes the procedure time-consuming and the absence of a robust temperature control changes the "standard" developmental time, which is defined as hr post fertilization at 28.5 °C for zebrafish 16. Another potential problem is the restricted depth of imaging into the animal, particularly when the structure of interest is located inside the embryo or larvae. Such a structure is the zebrafish pronephric glomerulus, which is situated between the somites and the yolk sac. The limiting depth for imaging the glomerulus was found to be about 200 µm, a distance that is reached after 5 days of development. In contrast, fluorescent structures that are located closer to the surface of the animal can be imaged for a longer time. For example liver cells are accessible for imaging at least until 9 dpf. A further limitation is that long term immobilization of the embryo or larvae in agarose and Tricaine treatment induce growth retardation and cardiac edema, respectively. Because this may interfere with normal development, it is recommended to restrict the duration of time-lapse recording to a certain period of interest. To ensure that during imaging structures of interest develop normally it is helpful to compare (at the end) the overall appearance with that of an animal kept under same experimental conditions but without embedding and anesthetization.
Recording a z-stack of optical sections every 30 minutes over a period of 5 hr and subsequent calculation of extended depth of focus images provided spatial and temporal information about early events of normal and disturbed kidney development which was induced by morpholino-mediated knockdown of wt1a. Previously performed morpholino knockdown experiments have already shown that Wt1a fulfills an early and essential role in pronephros formation. In situ hybridization with kidney marker 17,18 and employment of the transgenic wt1b:GFP line for imaging pronephros development at fixed time points 9,10 revealed that wt1a deficiency results in disrupted glomerular morphogenesis and failed podocyte specification. In contrast to these static approaches, time-lapse recordings directly visualize dynamics of early nephrogenesis in control embryos and the migration of GFP-positive cells away from the pronephric region in wt1a morphants. The possibility to track misrouted cells over time allows a more detailed phenotype analysis.
In general, the method that is presented here provides an inexpensive and easy to use alternative to the complex, and less accessible setups normally used for time-lapse imaging such as laser scanning microscope (equipped with an environmental chamber and anti-vibration table) or light sheet microscope. The described routine to fix drift problems can also be applied to perform time-lapse images on setups without optical sectioning options, such as fluorescence stereo microscopes.
The technique is not only suitable to investigate kidney development but can be also applied to monitor normal and defective morphogenesis of other organs such as heart or liver. Furthermore, the method can be used to observe various more dynamic processes in embryonic and adult model organisms such as wound healing or regeneration.
Disclosures
The author Michael Graf is an employee of Carl Zeiss Microscopy GmbH that produces instruments used in this Article.
Acknowledgments
We thank Thomas Bates for critically reading and improving the manuscript. We also thank Christina Ebert and Sabrina Stötzer for fish maintenance.
References
- Lieschke GJ. Animal models of human disease: zebrafish swim into view. Nat Rev Genet. 2007;8(5):353–367. doi: 10.1038/nrg2091. [DOI] [PubMed] [Google Scholar]
- Drummond IA. Kidney development and disease in the zebrafish. J Am Soc Nephrol. 2005;16(2):299–304. doi: 10.1681/ASN.2004090754. [DOI] [PubMed] [Google Scholar]
- Drummond IA, et al. Early development of the zebrafish pronephros and analysis of mutations affecting pronephric function. Development. 1998;125(23):4655–4667. doi: 10.1242/dev.125.23.4655. [DOI] [PubMed] [Google Scholar]
- Wingert RA, et al. The cdx genes and retinoic acid control the positioning and segmentation of the zebrafish pronephros. PLoS Genet. 2007;3(10):1922–1938. doi: 10.1371/journal.pgen.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer-Zucker AG, Wiessner S, Jensen AM, Drummond IA. Organization of the pronephric filtration apparatus in zebrafish requires Nephrin, Podocin and the FERM domain protein Mosaic eyes. Dev Biol. 2005;285(2):316–329. doi: 10.1016/j.ydbio.2005.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingert RA, Davidson AJ. The zebrafish pronephros: a model to study nephron segmentation. Kidney Int. 2008;73(10):1120–1127. doi: 10.1038/ki.2008.37. [DOI] [PubMed] [Google Scholar]
- Kreidberg JA, et al. Wt-1 Is Required for Early Kidney Development. Cell. 1993;74(4):679–691. doi: 10.1016/0092-8674(93)90515-r. [DOI] [PubMed] [Google Scholar]
- Bollig F, et al. Identification and comparative expression analysis of a second wt1 gene in zebrafish. Dev Dyn. 2006;235(2):554–561. doi: 10.1002/dvdy.20645. [DOI] [PubMed] [Google Scholar]
- Perner B, Englert C, Bollig F. The Wilms tumor genes wt1a and wt1b control different steps during formation of the zebrafish pronephros. Dev Biol. 2007;309(1):87–96. doi: 10.1016/j.ydbio.2007.06.022. [DOI] [PubMed] [Google Scholar]
- Schnerwitzki D, et al. Alternative splicing of Wilms tumor suppressor 1 (Wt1) exon 4 results in protein isoforms with different functions. Dev Biol. 2014;393(1):24–32. doi: 10.1016/j.ydbio.2014.06.026. [DOI] [PubMed] [Google Scholar]
- Bedell VM, Westcot SE, Ekker SC. Lessons from morpholino-based screening in zebrafish. Brief Funct Genomics. 2011;10(4):181–188. doi: 10.1093/bfgp/elr021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rembold M, Lahiri K, Foulkes NS, Wittbrodt J. Transgenesis in fish: efficient selection of transgenic fish by co-injection with a fluorescent reporter construct. Nat Protoc. 2006;1(3):1133–1139. doi: 10.1038/nprot.2006.165. [DOI] [PubMed] [Google Scholar]
- Conchello JA, Lichtman JW. Optical sectioning microscopy. Nat Methods. 2005;2(12):920–931. doi: 10.1038/nmeth815. [DOI] [PubMed] [Google Scholar]
- Jensen EC. Types of imaging, Part 2: an overview of fluorescence microscopy. Anat Rec (Hoboken) 2012;295(10):1621–1627. doi: 10.1002/ar.22548. [DOI] [PubMed] [Google Scholar]
- Chasles F, Dubertret B, Boccara AC. Optimization and characterization of a structured illumination microscope. Opt Express. 2007;15(24):16130–16140. doi: 10.1364/oe.15.016130. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203(3):253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Hsu HJ, Lin G, Chung BC. Parallel early development of zebrafish interrenal glands and pronephros: differential control by wt1 and ff1b. Development. 2003;130(10):2107–2116. doi: 10.1242/dev.00427. [DOI] [PubMed] [Google Scholar]
- O'Brien LL, et al. Wt1a, Foxc1a, and the Notch mediator Rbpj physically interact and regulate the formation of podocytes in zebrafish. Dev Biol. 2011;358(2):318–330. doi: 10.1016/j.ydbio.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]