

Abstract
A novel dissection and recording technique is described for optical monitoring staining and de-staining of lanceolate terminals surrounding hair follicles in the skin of the mouse pinna. The preparation is simple and relatively fast, reliably yielding extensive regions of multiple labeled units of living nerve terminals to study uptake and release of styryl pyridinium dyes extensively used in studies of vesicle recycling. Subdividing the preparations before labeling allows test vs. control comparisons in the same ear from a single individual. Helpful tips are given for improving the quality of the preparation, the labeling and the imaging parameters. This new system is suitable for assaying pharmacologically and mechanically-induced uptake and release of these vital dyes in lanceolate terminals in both wild-type and genetically modified animals. Examples of modulatory influences on labeling intensity are given.
Keywords: Neuroscience, Issue 110, Lanceolate ending, hair follicle, mechanosensation, ear, skin, mouse, electrophysiology
Introduction
Mechanosensory nerve endings are typically small, diffusely distributed and difficult to access in situ, as they are usually buried deep in the skin or other surrounding tissues. Visualizing them, therefore, usually requires sectioning followed by a prolonged immunolabeling/staining period, or the delay and expense of obtaining mouse lines with genetic expression of fluorescent labels such as green or yellow fluorescent protein (GFP/YFP)1. Here we show a convenient tissue and a quick and simple method to gain access to large numbers of hair follicle afferents for study, and how they can be quickly labeled for optical monitoring of terminal function2. With practice, the whole technique from dissection to imaging can be completed in as little as 2 hr.
The lanceolate terminals of sensory axons innervating hair follicles in mammals form palisades around the epithelium of the hair-follicle, with each terminal sandwiched between glial/Schwann cell processes2-4. The purpose of the terminals is to detect mechanical displacement of the hairs they surround. They are a mixture of rapidly and slowly adapting endings, but they predominantly produce short bursts of activity in response to hair movement. Typically, the firing stops very quickly when movement ceases, even in the presence of continued displacement.
This murine pinna model for studying lanceolate terminals by optical methods has many advantageous features for studying the structure and function of these endings. The pinna is predominantly two layers of skin apposed back-to-back, with only a small amount of cartilage tissue between. The skin is very thin and easily dissected due to minimal amounts of weakly adhesive connective tissue relative to other regions of the body. The easily separated skin layers, therefore, give good access to the follicles and terminals. The innervation is easily accessible and identifiable. The hair follicles are more sparsely distributed than in other skin regions, facilitating the imaging of individual or small groups of follicles. The thin underlying dermal layer gives good accessibility to dyes and pharmacological drugs, so is ideal for imaging by fluorescence microscopy without further processing. The imaging of labeled terminals can either be of living terminals or, if using a fixable dye analogue, after fixation and further histological processing.
We have used this preparation to show that membrane recycling occurs in the lanceolate endings, evidenced by uptake (endocytosis) and release (exocytosis) of a styryl pyridinium dye (FM1-43; N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino) styryl) pyridinium dibromide). The dye does not, however, seem to significantly label the investing Schwann cell processes2. We also showed that this dye uptake/release, and hence membrane recycling, is subject to glutamatergic modulation through an atypical (phospholipase D-coupled) metabotropic glutamate receptor. The outcomes of simple stimulation and analysis protocols are illustrated, and common potential analysis issues are also highlighted.
Protocol
These methods were used in the research reported in Banks, R.W. et al.2 Mice were humanely euthanised by cervical dislocation. In the UK, this is a legally approved Schedule 1 method listed in the Animals (Scientific Procedures) Act, 1986 and European Directive 2010/63/EU. This federal legislation is enforced locally at the University of Aberdeen by the Animal Welfare and Ethical Review Board who have approved all procedures.
1. Preparing Ears for Labeling
Prepare a standard physiological saline, such as Liley's (1956)5 and saturate it with 90% O2/5% CO2 (carbogen). Liley's saline is (mM): NaHCO3 (12), KCl (4), KH2PO4 (1), NaCl (138.8), MgCl2 (1), CaCl2 (2) and glucose (11). NOTE: Throughout the remainder of this manuscript, 'saline' will refer to saline that is fully saturated with carbogen. During dissection, refresh the saline covering the preparation every 15 - 20 min.
Humanely euthanize an adult mouse without damaging the skull. NOTE: We have used C57/Bl6J and MF1 mice but any mouse strain can be used. The most important aspect is not to use large adults (>25 g). Labeling in larger mice is less reliable, often being confined to small and scattered groups of follicles.
Remove the external ears (pinnae) near the base with ~25 cm scissors, just above the dense hair line, and submerge in saline in a silicone rubber-lined culture/petri dish (50 mm is usually convenient) .
Position the pinna anterior (concave) side down and pin the margins to the silicone at regular intervals with fine insect pins (~6 - 8 per ear, ~0.2 mm diameter pins). Refresh saline every 15 - 20 min, or use a constantly perfused bath.
- Carefully peel away the posterior skin from the anterior skin by blunt dissection, initially accessing from the cut base of the pinna over the thick central cartilage and systematically working towards the thin, cartilage-free external margins.
- To do this, hold the center of the cut edge of the posterior skin with a pair of no. 3 forceps. Then, with the jaws closed, place the points of another set of no. 3 forceps in the gap between the skin and the underlying cartilage.
- Gently work the points of the closed forceps from side-to-side within the gap, using the minimum required force, carefully loosen adhesions while peeling back the posterior skin to separate the layers.
With the posterior skin removed, leave the anterior skin in position. Pin the posterior skin, dermal side up, beside the anterior skin (as per 1.5, above).
Next, carefully and completely remove the pinna cartilage that was sandwiched between the skin layers from the anterior skin by the same blunt dissection technique as in 1.6. Avoid making puncture holes with the forceps.
Then, for both pinna skin preparations gently peel, pluck and rub away the thin layer of connective tissue, resembling expanded polystyrene foam, that covers the hair follicle bases. NOTE: Obtaining optimal clearing can take a little practice; insufficient tissue removal obstructs access, both for dye solution and for imaging, while scraping too vigorously removes the neural networks which are at the base of the follicles.
Refresh the saline.
2. Lanceolate Terminal Labeling
NOTE: The following protocol is optimized for styryl pyridinium dye. Other, chemically similar, styryl pyridinium dyes should work, perhaps after some adjustment in concentration and incubation time. Wear gloves and a lab coat when handling dye solutions. Either buy 10 mM stock dye solution directly from the manufacturer or prepare stock by dissolving the dye powder in saline without glucose. Aliquot (10 µl is usually convenient, but adjust this for large scale experiments) and store frozen for up to 6 months at -20 °C or ~1 year at -80 °C. Powder will store for at least 2 years. Avoid repeated freeze/thaw cycles of dye solutions; this rapidly denatures the dye. Once defrosted, store at 4 °C and use within 1 week.
Pre-heat a water bath to 30 °C, with a platform ~3 - 5 mm below the surface of the water.
Freshly prepare 10 µM styryl pyridinium dye solution (10 µl of freshly defrosted 10 mM styryl pyridinium dye in 10 ml saline) and equilibrate in water bath.
Pin each preparation in a silicone-rubber lined 35 mm culture dishes submerged in 1 - 2 mm of saline (typical volume, ~2 ml). Each pinna skin preparation will yield two preparations if cut in half from apex to base with small scissors (10 - 15 cm length), to minimize animal use.
Place the 35 mm dishes containing the saline-covered pinna preparations on the submerged platform in the 30 °C waterbath. Ensure the water depth is sufficient for adequate warming but does not contaminate the saline in the open dish.
Pin fine (0.5 - 1 mm outer diameter) tubing with flowing O2/CO2 into the silicone base of each dish to continuously gas the preparations. Also place into the water bath 100 ml of dye-free saline for washing/dish refreshing. Use a tightly stoppered-bottle to maintain carbogen saturation and prevent water contamination.
Equilibrate everything at 30 °C for 30 min then replace the saline over the pinna preparations from the 100 ml stoppered bottle. Incubate a further 30 min. NOTE: This replacement compensates for saline evaporation and any bulk volume losses resulting from gas-line bubbling.
Pour away dye-free saline into a 100 ml waste beaker, then quickly and carefully blot away any liquid remaining around the preparation with tissue paper to minimize dilution effects on the dye solution to be added next. Take care not to touch (i.e., damage) the exposed deep dermal surfaces directly.
Incubate pinna preparations in 2 - 3 ml of 10 µM styryl pyridinium dye for 40 min at 30 °C then return to R/T for the rest of the procedure.
- Remove non-internalized dye in three stages, using solutions at R/T. NOTE: These steps are best performed in minimal ambient light (blackout blinds, red/orange low intensity lighting) to begin eye dark-adaption for imaging.
- Pour away the labeling solution from the dishes into a waste beaker and quickly rinse in three changes of dye-free saline in rapid succession. This removes the residual styryl pyridinium dye solution adhering to the preparations and any dye that easily departitions from exposed membranes.
- Incubate in a final change of dye-free saline for a further 30 min to departition most remaining dye from surface membranes, i.e., dye not internalized by the terminals.
- Remove persistent dye stuck to the external leaflet of exposed membranes by chelating with sulfonated b-cyclodextrin derivative (Advasep-7, 1 mM, 5 min - see Table 1). Thus, all remaining dye will be within the tissues.
Place in fresh dye-free saline for imaging and dry the outside of the dish thoroughly with tissue paper. NOTE: The terminals are now ready for imaging. It is important to be aware that the lanceolate endings are alive and, therefore, slowly releasing the endocytosed styryl pyridinium dye again. Although this will be slower at R/T, it is important to minimize delays before/during imaging. If the experiment requires labeling of multiple preparations, initially maintain them all unlabeled at 30 °C. They will be viable for several hours. Then label sequentially at intervals, by labeling the second preparation (steps 2.7 - 2.8.3) while imaging the first.
3. Imaging Labeled Lanceolate Endings.
NOTE: The observer should be dark-adapted for the following imaging steps. The increased visual acuity this affords means lower excitation light levels are required to find the follicles for imaging. This is most important for minimising phototoxicity rather than reducing the inconvenience of photobleaching. Free radicals generated by full intensity illumination can rapidly (within 60 sec) kill living nerve terminals (G.S. Bewick, S. Fadul and W.J. Betz, unpublished observations).
- Before imaging, check the preparation under a dissection stereomicroscope and carefully remove any surface debris. This prevents auto-fluorescence contaminating the images.
- Mount the dish on the stage of an upright epifluorescence microscope with a standard fluorescein filter set (480 - 488 nm excitation, 500 - 520 nm emission for styryl pyridinium dye).
Attenuate the excitation light intensity with neutral density filters until just sufficient to comfortably locate the terminals.
Locate labeled follicles by observing with low (3.5x - 4x dry objective), then progressively higher (10x and 20x water immersion objective) magnification objectives.
Capture images of labeled lanceolate terminals through 10x dry objective for low power and 20x water immersion objectives for higher magnification. Minimize the light intensity and exposure time (a camera integration time of 0.5 - 1 sec is suggested).
Capture images on a standard digital camera and save images on computer hard drive using proprietary software. NOTE: A typical example is shown in Figure 1.
Image ~20 lanceolate endings from each preparation. Start at the margin furthest from the cut edge at the base of the pinna skin and work along this margin imaging each follicle in turn. Work across in slightly overlapping fields parallel to the cut edge, taking care not to image the same follicle twice and any that are obviously damaged. NOTE: There are typically too many lanceolate terminals to image them all before significant spontaneous de-staining occurs. Thus, we suggest systematically sampling the population using the following protocol.
After completing the marginal strip, move one field-width towards the base of the pinna, and repeat the process in the reverse direction.
Image all terminals encountered, except occasional ones clearly oriented very much more obliquely to the optical plane and unlikely to fit entirely within the standard annular analysis ROI (see section 4). Do not exclude unusually brighter or dimmer lanceolates compared to the surrounding terminals, unless they are obviously damaged or background intensity is particularly high, indicating localized tissue damage.
Discard the preparation if more than 10% of the skin area/terminals are damaged/unusable. NOTE: As lanceolates destain with time, complete imaging each preparation within 20 min of the end of the wash.
If using more than one preparation, ensure intensity comparisons are valid by time-matching preparations. To do this, offset timings of staining each preparation by ~30 min, to ensure comparability of de-stain times.
To monitor de-stain (exocytosis), image the same terminal at 5 or 10 min intervals. With practice, it is possible to image up to 3 separate terminals at each time point in any one preparation.
4. Analyzing Labeled Lanceolate Endings.
Note: The following procedure is a rapid method for analyzing terminal intensity.
Make an annular region of interest (ROI) centered on the tip of the hair shaft at the base of the follicle (Figure 2). Set the inner circumference of the annular ROI large enough to avoid the hair shaft auto-fluorescence in any follicle to be analyzed but still encompass all the terminal fluorescence. Ensure that the outer circumference is big enough to enclose the largest terminal likely to be encountered that is oriented close to the main optical/follicle axis.
Make a background ROI approximately equal in area to the target annulus (square or circle, typically ~400 µm2) to determine the local staining intensity in the vicinity of the terminal under analysis. Note: The sizes of these two ROIs are set at the beginning of the analysis and used to analyze all subsequent follicles/lanceolate terminals across all the preparations in any one experiment. So, it is important to set their parameters with care at the beginning.
Place the background ROI close enough to the follicle to represent local background in the sebaceous glands that underlie the terminals, not the lower intensity skin between the follicles, but be careful not to include any terminal regions.
Record the mean intensity of the two ROIs using the 'measure' or 'analyze ROI' function of the software and transfer data to a spreadsheet.
In the spreadsheet, for each individual follicle subtract the mean local background intensity from that in the annulus to give a net intensity. This adjustment for localized background greatly reduces between-follicle variability.
Representative Results
The hair follicles in this pinna preparation are easily seen under transillumination without fluorescence (Figure 1A), illustrating the wafer-thin nature of the preparation and the relative ease of accessibility it affords to these mechanosensory terminals. Each is typified by the prominent hair shaft base piercing the dark crescent of the sebaceous gland. Under epifluorescence, the labeled lanceolate terminals surrounding each hair follicle are clearly seen and show the typically robust spontaneous styryl dye uptake (Figure 1B). This occurs without imposed movement. The lanceolate terminals are seen to encircle the hair shaft, although the extent of encirclement is variable6. Much of the labeling is punctate (spots) rather than the linear arrangement that might be expected. The punctate pattern reflects terminals being observed in an optical plane along the length of the terminal. This preparation received no further processing for visualization after labeling, it was simply transferred to a microscope stage and imaged in situ, which again illustrates the simplicity of this preparation.
Example experiments to which this novel preparation is suited are shown in Figure 3. It can be used to study both endocytosis and exocytosis in living terminals, as well as their modulation. Thus, for endocytosis, glutamate receptor agonists can increase the labeling intensity, while blocking Ca2+ channels with ligands or cations such as Mg2+, Ni2+ or Cd2+ decreases it2. Alternatively, this preparation can also be used to study the kinetics of exocytosis, as shown in Figure 3C. First, a labeled terminal is seen de-staining spontaneously. However, this destain is accelerated by the exocytosis stimulant, latrotoxin. More details of the experimental outcomes can be seen in Banks, R.W. et al.2.
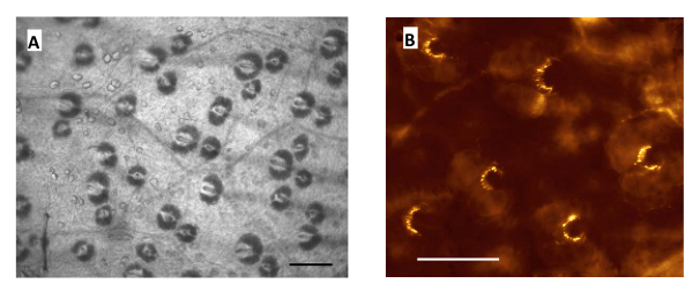 Figure 1. Robust Styryl Dye Labeling Recorded in the Mouse Pinna. (A) Part of an unlabeled pinna preparation in bright field illumination, showing how clearly hair follicles appear in areas cleared of overlying areolar/adipose tissues. The dark, usually bilobed, crescent shapes are sebaceous glands surrounding the central hair shaft. (B) A similar area, at a slightly higher magnification and imaged with epifluorescence, showing lanceolate endings labeled with styryl dye. Scale bars - 40 µm. Please click here to view a larger version of this figure.
Figure 1. Robust Styryl Dye Labeling Recorded in the Mouse Pinna. (A) Part of an unlabeled pinna preparation in bright field illumination, showing how clearly hair follicles appear in areas cleared of overlying areolar/adipose tissues. The dark, usually bilobed, crescent shapes are sebaceous glands surrounding the central hair shaft. (B) A similar area, at a slightly higher magnification and imaged with epifluorescence, showing lanceolate endings labeled with styryl dye. Scale bars - 40 µm. Please click here to view a larger version of this figure.
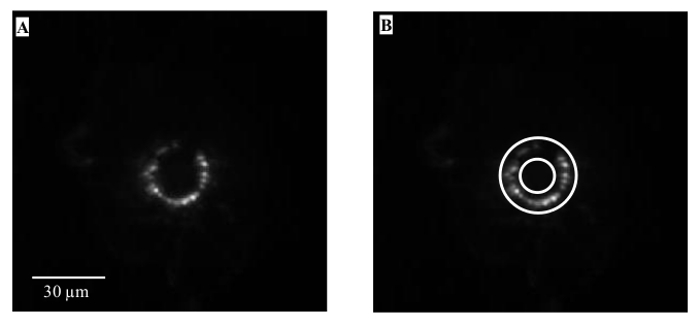 Figure 2. Analysis of Styryl Dye Labeling Intensity. (A) Typical circular pattern of hair follicle labeling with styryl dye, centered on the hair shaft (not visible in this image). (B) The main analysis 'region of interest' (ROI) is defined by a standard annulus superimposed onto the position of the palisade of lanceolate nerve endings encircling the follicle. This ROI is kept constant in shape and area for any single experiment to ensure labeling intensity can be fairly compared between preparations and across all treatments. Net intensity of each ROI is calculated by subtracting the intensity of an adjacent square or circular background region (~400 µm2). Again, this square background region is kept constant in shape and area throughout any given experiment. Please click here to view a larger version of this figure.
Figure 2. Analysis of Styryl Dye Labeling Intensity. (A) Typical circular pattern of hair follicle labeling with styryl dye, centered on the hair shaft (not visible in this image). (B) The main analysis 'region of interest' (ROI) is defined by a standard annulus superimposed onto the position of the palisade of lanceolate nerve endings encircling the follicle. This ROI is kept constant in shape and area for any single experiment to ensure labeling intensity can be fairly compared between preparations and across all treatments. Net intensity of each ROI is calculated by subtracting the intensity of an adjacent square or circular background region (~400 µm2). Again, this square background region is kept constant in shape and area throughout any given experiment. Please click here to view a larger version of this figure.
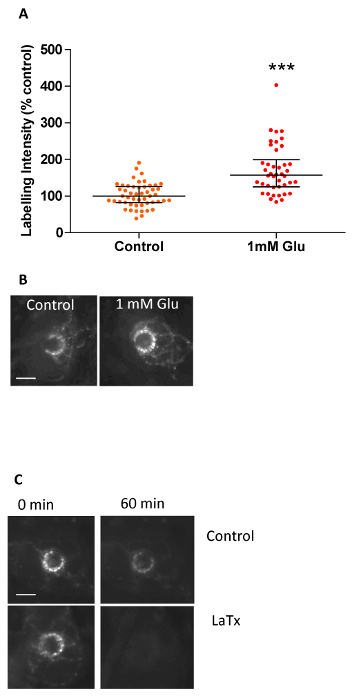 Figure 3. Labeling Intensity of Hair Follicle Afferent Lanceloate Endings is not Normally Distributed, and can be Used to Examine Both Endo- and Exocytosis. Typical data from styryl dye intensity measurements in spontaneously labeled lanceolate terminals using the analysis method described above. (A) Labeling intensity under control conditions (54 follicles, 4 ears) and 1 mM glutamate (42 follicles, 4 ears). The intensity values of individual endings are displayed as cluster dot plots. Each point represents the intensity of one terminal palisade around a follicle. Note that even under control conditions a few data points (follicles) are extremely intense, while most are clustered at lower intensities. The first dot at a particular intensity is plotted at the center and subsequent data points of the same intensity are plotted progressively more laterally on either side. Thus, lateral spread represents the frequency of follicles of any particular labeling intensity. The horizontal lines represent median ± 25% interquartile range. Incubation with 1 mM glutamate enhances the median intensity by ~50% (***P<0.001, Mann-Whitney), but labeling can be enhanced by 200 - 300% in some terminals. (B) Images demonstrating glutamate-mediated enhanced dye uptake (endocytosis). Incubation of the hair follicle preparation in glutamate (1 mM) markedly increases styryl dye uptake, as shown by the increased labeling intensity. Scale bar 20 µm. (C) Enhancing dye release (exocytosis). After labeling, styryl dye is spontaneously released again, as the labeling at 0 min is clearly brighter than at 60 min, even after background subtraction to control for photobleaching. This release is greatly potentiated by latrotoxin, a black widow spider venom constituent, which triggers uncontrolled vesicle exocytosis. After 60 min in toxin, terminal labeling is almost undetectable. This reversibility of styryl dye labeling supports the hypothesis that terminal fluorescence is due to internalization during local recycling of synaptic-like vesicles. Scale bar 20 µm. Please click here to view a larger version of this figure.
Figure 3. Labeling Intensity of Hair Follicle Afferent Lanceloate Endings is not Normally Distributed, and can be Used to Examine Both Endo- and Exocytosis. Typical data from styryl dye intensity measurements in spontaneously labeled lanceolate terminals using the analysis method described above. (A) Labeling intensity under control conditions (54 follicles, 4 ears) and 1 mM glutamate (42 follicles, 4 ears). The intensity values of individual endings are displayed as cluster dot plots. Each point represents the intensity of one terminal palisade around a follicle. Note that even under control conditions a few data points (follicles) are extremely intense, while most are clustered at lower intensities. The first dot at a particular intensity is plotted at the center and subsequent data points of the same intensity are plotted progressively more laterally on either side. Thus, lateral spread represents the frequency of follicles of any particular labeling intensity. The horizontal lines represent median ± 25% interquartile range. Incubation with 1 mM glutamate enhances the median intensity by ~50% (***P<0.001, Mann-Whitney), but labeling can be enhanced by 200 - 300% in some terminals. (B) Images demonstrating glutamate-mediated enhanced dye uptake (endocytosis). Incubation of the hair follicle preparation in glutamate (1 mM) markedly increases styryl dye uptake, as shown by the increased labeling intensity. Scale bar 20 µm. (C) Enhancing dye release (exocytosis). After labeling, styryl dye is spontaneously released again, as the labeling at 0 min is clearly brighter than at 60 min, even after background subtraction to control for photobleaching. This release is greatly potentiated by latrotoxin, a black widow spider venom constituent, which triggers uncontrolled vesicle exocytosis. After 60 min in toxin, terminal labeling is almost undetectable. This reversibility of styryl dye labeling supports the hypothesis that terminal fluorescence is due to internalization during local recycling of synaptic-like vesicles. Scale bar 20 µm. Please click here to view a larger version of this figure.
Discussion
Bewick and Betz originally developed the N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino) styryl) pyridinium dibromide-related styryl pyridinium dyes7-10 to monitor local recycling of synaptic vesicle membrane. Dye uptake, and hence increased fluorescence, therefore reflects synaptic vesicle membrane internalization (endocytosis). Subsequently, this internalized dye can be re-released by further electrical activity, showing dye loss monitors vesicle membrane externalization (exocytosis). In synapses, this is a proxy for neurotransmitter secretion. At the same time, we also found these dyes label primary mechanosensory nerve endings7. More recently11, Bewick, Banks and colleagues then showed that this reflects a similar process in mature, differentiated mechanosensory terminals ex vivo. Here again, the dyes label 50 nm diameter 'synaptic-like' vesicles (SLVs) that recycle constitutively to release glutamate. In mechanosensory terminals, unlike their synaptic counterparts, this recycling is primarily spontaneous. It is also modulated by mechanical, rather than simply electrical, stimulation.
For sensory neurons in culture and in cochlea hair cells, styryl dyes in general and styryl pyridinium dye in particular have been shown to pass through the mechanosensory channels, blocking the mechanosensory channels and irreversibly labeling internal membranes12. However, at comparable styryl dye concentrations in differentiated mechanosensory terminals in situ, as here in lanceolate endings2, or in Ia endings in muscle spindles11, and in hair cells that are not mechanically stimulated13, 14, labeling is by membrane endocytosis. In mechanosensory nerve terminals, for instance, labeling is reversible and does not block the mechanosensory responses at the concentrations used here2, 11, 15. While some dye internalization by channel permeation in these endings cannot be completely ruled out, the near total destain with latrotoxin indicates the great majority of the labeling in mature terminals is by internalization with recycling vesicle membrane. We have, therefore, used this technique to study the activity dependence11 and, most recently, the pharmacology2 of SLV recycling in mechanosensory afferent terminals by examining drug effects on the uptake and release of the dyes.
As with most practical techniques, reproducibility requires repetition and practice. Some of the key points for ensuring reliability will now be discussed. Lanceolate labeling in younger animals is more reliable - the smallest animal we have used was 15 g body weight. It is not entirely clear why this is the case, but it may be because dissection of the younger connective tissue requires less mechanical trauma and leaves less remaining residue when removing the tissues overlying the innervation layer.
Overall, the tissue preparation is quite simple, with peeling the skin layers apart being the most technically challenging practical aspect and then only in older mice. In these, the skin layers adhere strongly together at the base where the dissection starts, but even here separation of the layers becomes much easier towards the margins. Thankfully, or perhaps consequently, these thinner marginal skin areas usually give the most satisfactory labeling, as does the anterior skin generally. Therefore, particularly avoid grasping the very thin skin at the margins. To minimize the risk of damage, manipulate preparations indirectly by pushing with closed forceps rather than grasping directly, or if essential grasp only tougher tissues unlikely to be labeled. Be thorough in removing the 'sheet polystyrene' layer immediately overlying the follicles, as this is the main mechanical obstruction to imaging and drug access. However, it is essential to minimize physical contact with underlying structures as this damages (i.e., strips away) the neural networks and terminals just below. If the predominant fluorescence is yellow/white in the sebaceous glands, prominent auto-fluorescence of the hair shaft bases and few crescents of orange/yellow lanceolate endings, this indicates the nerve plexus layer and associated lanceolate terminals have been removed during clearance. This appears to be the main problem with older (>30 g) mice. Throughout the staining procedures, ensure everything is at 30 °C and well-oxygenated. Be aware that labeled terminals are still alive. Consequently, dye will be secreted by ongoing spontaneous (constitutive) exocytic release. This will be slower at R/T, but it is good practice to minimize the delay between the removal of chelator solution and imaging, to minimize dye loss. Moreover, for between-groups comparative studies of intensity, it is essential to ensure imaging of all groups is time-matched. Use of a dye-chelating agent before imaging greatly improves image contrast. So, while relatively expensive, the chelation step is quite important for ensuring good image quality. Chelator use can be minimized by reusing the solution multiple times, and even on 2 - 3 consecutive days, if stored at 4 °C between uses. Discard the chelator solution once it goes noticeably pink, showing its dye sequestration is approaching saturation.
During imaging, be aware of the potential for phototoxic damage to these living terminals, which is a cumulative result of both the intensity and the exposure time of the excitation light. This consideration is less important for single timepoint images, where image quality is the primary concern. However, it is crucial during repeated imaging in kinetics studies to reduce excitation light exposure to a minimum. While developing these dyes to label synapses, we found excitation for >1 min at full power excitation illumination will cause dramatic and irreparable damage to nerve terminals. They first subsequently expand hugely, then collapse completely over a period of ~15 min. We did not systematically test the relative contributions of exposure time and intensity, but these observations suggest the guiding principle should be to minimize both. So, it is recommended that excitation light intensity and duration are the minimum commensurate with getting good images, and appropriate control images are taken in time-course studies. To test that the data are not affected by phototoxicity under repeated imaging conditions, at each time point take an additional image of a previously unviewed terminal. This is to ensure the behavior of the labeling in naive terminals resembles that in follicles being imaged repeatedly.
A number of factors can reduce the within-group variability of the net intensity data. Accurate placement of the ROIs, particularly the background ROI, is important, as even small areas of inappropriate staining can greatly affect between-follicle data variability. The variability of net intensity is also influenced by how completely each hair follicle is encircled by the palisade of endings. Compare for example, the crescent shaped labeling distribution in Figure 1B with the almost completely circular pattern seen in Figure 2. More complex analysis, perhaps using automated software detection and thresholding, are necessary if the degree of encirclement is a relevant parameter. Please note that the intensity distributions for even the most accurately analyzed follicle groups are not normally distributed (see Figure 3), necessitating the use of non-parametric statistics for between-treatment comparisons of population medians rather than means. Normalization techniques, such as expressing intensities as a percentage of contralateral control, or of a control group made up of several preparations, can be applied to further reduce variability. The final technical item to be considered is the experimental design for pharmacological testing. During pharmacological investigations, pre-incubate the preparation in drug solution for 30 min before dye application. Then, maintain the drug concentration in the dye solution. In controls, use either saline or, if the drug is dissolved in a vehicle, saline plus vehicle.
The present article demonstrates how this new pinna preparation offers high quality images of mechanosensory endings in skin with minimal preparation. We also show it can be used to examine the pharmacology of two critical aspects of vesicle recycling. We show first that a glutamate receptor agonist can increase dye internalization (a marker of endocytosis), and second, that dye is lost again with latrotoxin (a stimulant of exocytosis). The significance of this preparation and technique is that it offers excellent access to living skin mechanosenory terminals in situ for imaging experiments, affording unique opportunities for optical monitoring of terminal function, structure and their inter-relationships. To this latter end, we have also developed it further for use in combined optical/electrophysiological studies (see sister JoVE article for details).
Disclosures
The authors have nothing to disclose.
Acknowledgments
The work was funded by UK Medical Research Council project grant G0601253 to G.S.B. and R.W.B. and a SULSA Bioskape grant to G.S.B.
References
- Li L, et al. The functional organization of cutaneous low-threshold mechanosensory neurons. Cell. 2011;147(7):1615–1627. doi: 10.1016/j.cell.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks RW, et al. Glutamatergic modulation of synaptic-like vesicle recycling in mechanosensory lanceolate nerve terminals of mammalian hair follicles. J. Physiol. 2013;591(10):2523–2540. doi: 10.1113/jphysiol.2012.243659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres KH. On the microstructure of receptors on sinus hair. Z. Zellforsch. Mikrosk. Anat. 1966;75:339–365. [PubMed] [Google Scholar]
- Shenton F, Bewick GS, Banks RW. A study of the expression of small conductance calcium-activated potassium channels (SK1-3) in sensory endings of muscle spindles and lanceolate endings of hair follicles in the rat. PLoS One. 2014;9:e107073. doi: 10.1371/journal.pone.0107073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liley AW. An investigation of spontaneous activity at the neuromuscular junction of the rat. J. Physiol. 1956;132(3):650–666. doi: 10.1113/jphysiol.1956.sp005555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Ebara S, Koike T, Tonomura S, Kumamoto K. How many hair follicles are innervated by one afferent axon? A confocal microscopic analysis of palisade endings in the auricular skin of thy1-YFP transgenic mouse. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci. 2012;88(10):583–595. doi: 10.2183/pjab.88.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, Mao F, Bewick GS. Activity-dependent fluorescent staining and destaining of living vertebrate motor nerve terminals. J. Neurosci. 1992;12(2):363–375. doi: 10.1523/JNEUROSCI.12-02-00363.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS, Ridge RM. Intracellular movements of fluorescently labeled synaptic vesicles in frog motor nerve terminals during nerve stimulation. Neuron. 1992;9(5):805–813. doi: 10.1016/0896-6273(92)90235-6. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science. 1992;255(5041):200–203. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS. Optical monitoring of transmitter release and synaptic vesicle recycling at the frog neuromuscular junction. J. Physiol. 1993;460(1):287–309. doi: 10.1113/jphysiol.1993.sp019472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewick GS, Reid B, Richardson C, Banks RW. Autogenic modulation of mechanoreceptor excitability by glutamate release from synaptic-like vesicles: evidence from the rat muscle spindle primary sensory ending. J. Physiol. 2005;562(2):381–394. doi: 10.1113/jphysiol.2004.074799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew L, Wood J. FM1-43 is a permeant blocker of mechanosensitive ion channels in sensory neurons and inhibits behavioural responses to mechanical stimuli. Molecular Pain. 2007;3 doi: 10.1186/1744-8069-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesinger CB, Richards CD, Ashmore JF. FM1-43 reveals membrane recycling in adult inner hair cells of the mammalian cochlea. J. Neurosci. 2002;22(10):3939–3952. doi: 10.1523/JNEUROSCI.22-10-03939.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesinger CB, Richards CD, Ashmore JF. Apical endocytosis in outer hair cells of the mammalian cochlea. Eur. J. Neurosci. 2004;20(1):41–50. doi: 10.1111/j.0953-816X.2004.03452.x. [DOI] [PubMed] [Google Scholar]
- Watson S, Aryiku C, Banks RW, Bewick GS. Comparison of gadolinium and FM1-43 as blockers of stretch-evoked firing of rat muscle spindle afferents. Proc. Physiol. Soc. 2010;21:P22. [Google Scholar]