

Abstract
FBXL4 deficiency is a recently described disorder of mitochondrial maintenance associated with a loss of mitochondrial DNA in cells. To date, the genetic diagnosis of FBXL4 deficiency has been established in 28 individuals. This paper retrospectively reviews proxy-reported clinical and biochemical findings and evaluates brain imaging, morphological and genetic data in 21 of those patients. Neonatal/early-onset severe lactic acidosis, muscular hypotonia, feeding problems and failure to thrive is the characteristic pattern at first presentation. Facial dysmorphic features are present in 67 % of cases. Seven children died (mean age 37 months); 11 children were alive (mean age at follow-up 46 months), three children were lost to follow-up. All survivors developed severe psychomotor retardation. Brain imaging was non-specific in neonates but a later-onset, rapidly progressive brain atrophy was noted. Elevated blood lactate and metabolic acidosis were observed in all individuals; creatine kinase was elevated in 45 % of measurements. Diagnostic workup in patient tissues and cells revealed a severe combined respiratory chain defect with a general decrease of enzymes associated with mitochondrial energy metabolism and a relative depletion of mitochondrial DNA content. Mutations were detected throughout the FBXL4 gene albeit with no clear delineation of a genotype- phenotype correlation. Treatment with Bmitochondrial medicationsˆ did not prove effective. In conclusion, a clinical pattern of early-onset encephalopathy, persistent lactic acidosis, profound muscular hypotonia and typical facial dysmorphism should prompt initiation of molecular genetic analysis of FBXL4. Establishment of the diagnosis permits genetic counselling, prevents patients undergoing unhelpful diagnostic procedures and allows for accurate prognosis.
Introduction
FBXL4 is a protein involved in phosphorylation-directed ubiquitination (Cenciarelli et al 1999; Winston et al 1999) but recently shown to contain a mitochondrial localization signal and found within mitochondria (Bonnen et al 2013; Gai et al 2013). In fibroblasts from patients with biallelic FBXL4 mutations, a disturbance of the mitochondrial network, decreased activity of enzymes associated with mitochondrial energy metabolism and depletion of mitochondrial DNA (mtDNA) content in relation to nuclear DNA has been shown (Bonnen et al 2013; Gai et al 2013). Thus, although the precise function of FBXL4 remains undetermined, there is strong evidence to support an important role in mtDNA maintenance or mitochondrial maintenance per se (Bonnen et al 2013; Gai et al 2013). In contrast to other disorders of mtDNA maintenance, FBXL4 deficiency (MIM 615471) leads to a decrease in mitochondrial mass within the cell although the mechanism of this presumed pathologically-relevant process remains unclear.
To date, FBXL4 mutations have been mainly identified through whole exome sequencing initiated in children with encephalopathy, muscular hypotonia, persistent lactic acidosis and combined mitochondrial respiratory chain deficiencies in skeletal muscle tissue (Bonnen et al 2013; Gai et al 2013). The aim of the present study was to analyse whether individuals with proven FBXL4 mutations share clinical, neuroradiological or biochemical features which might facilitate earlier diagnosis. In addition, by collating the extensive clinical information and DNA sequencing data available from 21 patients we have explored the possibility of genotype-phenotype associations.
Methods
Data on patients’ genotype, phenotype, clinical course, biochemical and neuroradiological findings were obtained by means of a questionnaire from physicians involved in the clinical care and follow-up of individuals with FBXL4 mutations. Contact addresses of the physicians were retrieved from the databases of referrers of diagnostic material to specialized laboratories and from specialist networks. Caregivers gave their consent to submit the retrospective data in pseudonymized form to their physicians.
A Survey Monkey questionnaire was constructed (provided as supplementary material) based on published features encountered in individuals with FBXL4 mutations and in addition, included open questions addressing yet unpublished symptoms and organ involvement. Furthermore, physicians were asked to send their patient’s brain MRI images to have them assessed by an experienced child neurologist.
The activity of respiratory chain enzymes, pyruvate dehydrogenase and citrate synthase was determined in different diagnostic laboratories according to published standard methods (Berger et al 2003; Meierhofer et al 2004; Kirby et al 2007; Feichtinger et al 2014). The relative content of mtDNA per nuclear genome was determined by quantitative real-time PCR (Acham-Roschitz et al 2009). Immunohistochemical staining was performed with formalin-fixed muscle tissue as reported previously (Feichtinger et al 2010).
All patients were genetically diagnosed by whole exome sequencing (Haack et al 2014) or Sanger sequencing (primer and PCR conditions are available on request). Splice site mutations were predicted by the mutation taster (Schwarz et al 2010) and post-translational modification sites by PTMcode. The alignment (ClustalW2; embl-ebi) consists of the following species (with corresponding uniprot identifiers), Homo sapiens (Q9UKA2), Pan troglodytes (H2QTF8), Macaca mulatta (F7G6N0), Mus musculus (V9GXH8), Bos taurus (F1N1M5), Gallus gallus (E1C3J3), Xenopus tropicalis (K9J7L5), Danio rerio (Q5U388).
Results
Fourteen physicians reported data on 21 affected children (13 males, eight females) from 19 families (two pairs of siblings). Three children (two males, one female) have been published before by Gai et al 2013, one male has been published by Bonnen et al 2013 (see Supplementary Tables 1 and 2). Probably affected, but genetically unconfirmed, siblings were encountered in four additional families. Parental consanguinity was present in 58 % of families. Eight children were of Arabian (two siblings), four of Middle and South European, three of Turkish, two (siblings) of Mexican (Zacatecas/Guanajuato), and one each of Palestinian, Pakistani and Portuguese/African American descent. Ethnic background was unknown for one case.
General paediatricians and specialists in paediatric metabolic medicine or paediatric neurology performed clinical assessment of all patients. The majority of patients were born at term (mean and median gestational week 38; range 35–41; n= 19). Birth weight was below the 3rd centile in six individuals and normal, but predominantly in the low centile range (n=10 between 3rd and 50th, n = 2 > 50th percentile) (Barbier et al 2013) for the others. Data on head circumference at birth were available for only five individuals and below the 3rd centile in two/five children.
Clinical symptoms at initial workup and during the disease course
Age at onset of first symptoms and at follow-up
First symptoms occurred very early in life (mean 115 days, median 2 days, range 1d-24 months) with 14 patients presenting symptoms in the neonatal period while another five patients first exhibited disease between 3 and 12 months of age. A further two patients showed a somewhat later onset at the age of 17 and 24 months, respectively. At the time of the study, 11 children are alive (mean age at follow-up 46 months; median 41 months, range 2–139 months) while seven individuals had died (mean age at death 37 months; median 26 months, range 24–75 months). No information was available for three subjects.
Typical pattern of symptoms at first presentation
Main clinical features for the cohort over time are depicted in Fig. 1. More than 80 % of patients presented with failure to thrive/feeding problems, muscular hypotonia and neurodevelopmental impairment at initial workup combined with elevated lactate and metabolic acidosis.
Fig. 1.

Main clinical features (columns) and number of available ratings for each parameter (lines) at first presentation and ever during the course in 21 patients with FBXL4 mutations
Typical pattern of symptoms during the disease course
As the disease evolved, neurocognitive impairment, commonly severe psychomotor retardation and developmental delay, became readily apparent, but perhaps surprisingly, there were few individuals with features often associated with central nervous system involvement of mitochondrial disease such as seizures, hearing impairment and movement disorders (Fig. 1).
Cardiac disease
Cardiac disease was noticed at first presentation in seven subjects and evolved in another patient during the course. Non- progressive cardiomyopathy was present in two individuals at presentation. Left ventricular non-compaction, borderline left ventricular hypertrophy and hyperdynamic left ventricular systolic function and small combined atrial and ventricular septum defects were each present in a single child. No detailed information was available for four patients, including the single patient developing cardiac involvement during the course of the disease. Other visceral organs were very rarely affected.
Rarely reported symptoms: eye disease
Congenital cataracts were present in two children and optic nerve atrophy with significant visual impairment evolved during the course of the illness in one female.
Rarely reported symptoms: neutropenia
One child presented with episodic neutropenia associated with recurrent infections. In another child, persistent neutropenia was accompanied by recurrent infections and wound healing deficit. The patient died from fatal gastrointestinal bleeding 1 year after placement of a percutaneous endoscopic gastrostomy (PEG) tube.
Late-onset symptoms
A female patient aged 11 years 7 months at last follow-up and thus representing by far the oldest individual in the cohort (mean 46 months) developed ptosis, sensorineural hearing loss, optic nerve atrophy with significant visual impairment and stroke (at age 10 years) causing left sided spastic hemiparesis besides the above mentioned episodic neutropenia. Individual courses are described in Supplementary Table 1.
Physicians’ overall assessment of the disease course
The course of the disease was considered by the physicians to improve over time in one case; remain stable in seven and deteriorate in ten individuals (n=18). Seven children died at a mean age of 37 months, mostly from multiorgan failure associated with overwhelming metabolic acidosis, which in some cases was triggered by infections. All survivors developed severe psychomotor retardation. Only one child was able to walk with assistance while 14 of 15 were completely unable to do so. Fifteen of 16 children had no or severely impaired functional speech and communicative abilities.
Treatment and physicians’ overall assessment of outcome
Treatment approaches included coenzyme Q10 in six, riboflavin in three, long-term sodium bicarbonate in four, sodium citrate in two, carnitine in four, thiamine, biotin, vitamin D and folate in two individuals each. Bezafibrate, high fat diet or low protein diet were applied in a single case each. One female with neutropenia required treatment with granulocyte colony stimulating factor. Fifty-seven percent of 21 patients received more than one medication, 38 % never had specific Bmitochondrialˆ treatment. Physician’s overall assessments underpin the severity of handicap and the limited biochemical and clinical response to treatment. Physicians agreed or strongly agreed to the statement Bmy patient shows no biochemical response to treatmentˆ in 61 %; to the statement Bmy patient shows no clinical response to treatmentˆ in 72 % and to the statement Bmy patient is severely handicappedˆ in 93 % of cases. In addition, physicians stated that both patients’ and parental quality of life was severely impaired (93 % of cases).
Dysmorphic features
Dysmorphic features or malformations were reported in 67 % of the affected individuals (supplementary table 1). Figure 2 illustrates the typical facial appearance of the patients such as prominent forehead; thick, diffuse eyebrows and synophris; short, upslanting palpebral fissures; broad nasal root with depressed nasal bridge; smooth philtrum, and low set, dysplastic ears.
Fig. 2. Patients with FBXL4 deficiency share typical facial characteristics.
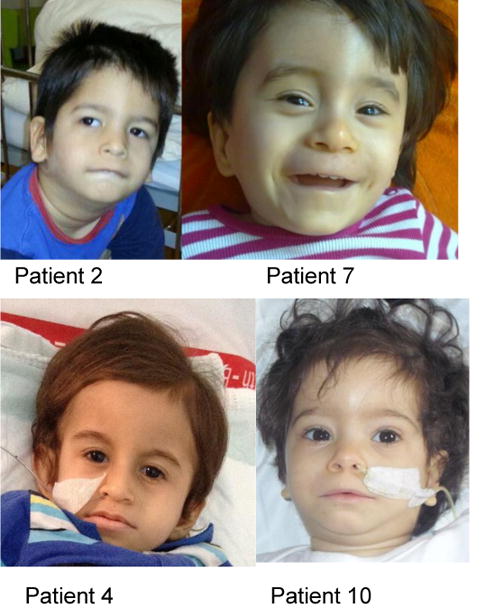
Patient 2: Prominent, narrow forehead with intertemporal narrowing, thick, medial diffuse eyebrows with slight synophris, bilateral epicanthus, periorbital fullness, broad nasal root and broad nasal ridge, fullness of paranasal tissue, flat malar bones, long philtrum, large, low set, protruding ears with overfolded helices.
Patient 7: Prominent forehead, thick, diffuse eyebrows, short, upslanting palpebral fissures, broad nasal root, depressed nasal bridge, wide nasal base, smooth philtrum, thin vermillion of the upper lip, low set ears.
Patient 4: Broad, prominent forehead, thick eyebrows, short palpebral fissures, broad nasal root, wide nasal base, low set ears.
Patient 10: Prominent forehead, medial diffuse eyebrows with slight synophris, short, upslanting palpebral fissures, broad nasal root, depressed nasal bridge, wide nasal base, smooth philtrum, thin vermillion of the upper lip, low set, protruding ears.
Brain imaging
Data on brain magnetic resonance imaging were available for 14 cases; serial imaging had been performed in six children. Central assessment of the imaging series by an experienced child neurologist was possible for 11 cases. Cerebellar hypoplasia was present in two patients. Five children were imaged during the neonatal period, between 4 and 10 days of age. All had increased white matter signal on the T2-weighted and correspondingly reduced signal on the T1-weighted images. In addition, the white matter appeared swollen, indicating increased water content (Fig. 3a–f). In some children, diffusion- weighted imaging was available with increased diffusivity in the supratentorial white matter and diffusion restriction in the dorsal tegmental tracts (n = 4) and posterior limb of the internal capsule (n = 1). Two children had already small defects in the basal ganglia, four had large cysts, adjacent to the anterior horns or body of the lateral ventricles, in some of them with evidence of an earlier bleeding, three had increased pericerebellar CSF spaces. One neonate had two small areas of diffusion-restriction bilaterally in the parietooccipital cortex with subsequent (1 month later) development of cortical defects.
Fig. 3. Magnetic resonance imaging of the brain from patient 2 (4 days), patient 4 (9 months) and patient 19 (3 years).
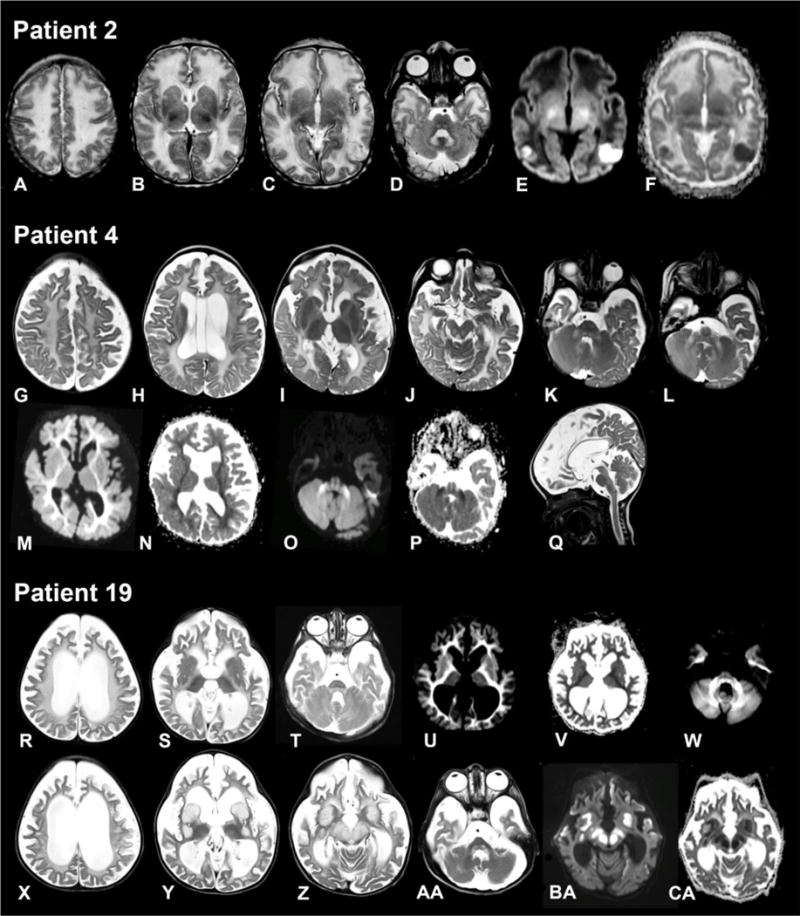
Patient 2 (age 4 days). Axial T2-weighted images (A–D) show swollen and slightly hyperintense aspect of supratentorial white matter. There are several defects in the basal ganglia (B). There are two small areas at the parietooccipital border on both sides where the cortex is less well defined (C) with corresponding impaired diffusion (E) and decreased apparent diffusion coefficient (ADC) (F). Signal of the dorsal tegmental tracts and hilus of the dentate nucleus is increased (D). In the supratentorial white matter, diffusivity is increased (E) with elevated ADC (F), with exception of the posterior limb of the internal capsule.
Patient 4 (age 9 months). Axial T2-weighted images (G–L) demonstrate global supratentorial atrophy, a cavum vergae and a left paraventricular cyst (H) and a mild diffuse increase of the supratentorial white matter signal (G–I). The tectum (J), pyramidal tracts, dorsal tegmentum (K) and middle cerebellar peduncles (K, L) have an elevated signal. Diffusion is impaired with decreased ADC in the supratentorial white matter (M, N) and the middle cerebellar peduncles, dorsal tegmental tracts and pyramidal tracts (O, P). The sagittal T2-weighted image (Q) shows normal volume of the cerebellum.
Patient 19 aged 3 years (R–W) and 2.5 months later (X-CA). Severe supratentorial atrophy is evident on the axial T2-weighted images (R–T), with now also thinning of the brain stem (T). White matter signal is diffusely elevated (R–T) with impaired diffusion (U, W) and a corresponding low ADC (V). 2.5 months later, basal ganglia (Y), substantia nigra and red nucleus (Z) are grossly swollen and hyperintense with corresponding impaired diffusion (BA) and ADC (CA). Also the dorsal tegmental tracts are swollen (AA).
In six children, imaging had been performed in the chronic phase of the disease, between age 3.5 months and 5 years. (Figure 3g–q). Supratentorial atrophy was prominent in four individuals and already present at age 3.5 months. The brain stem was hypotrophic in three patients. Pericerebellar spaces were prominent in five children. The white matter signal was diffusely elevated on the T2-weighted images with restricted diffusion in some patients. Myelination was severely delayed also on the T1-weighted images. Basal ganglia lesions were present in three cases.
In three children, imaging showed acute changes (Fig. 3x–ca). In two, basal ganglia and mesencephalon were swollen and their T2-signal of the basal ganglia was inhomogenously elevated with restricted diffusion. Signal abnormalities and diffusion restriction of the dorsal tegmental tracts were more prominent than in the chronic phase. One child had in addition increased T2 signal and impaired diffusion in both medial thalami and the cervical central spinal cord (not shown). In one child, the oldest of this series, at the age of 11 years there was evidence of an old stroke-like episode involving the right parietal and occipital lobes with cortical haemorrhagic necrosis and a new lesion with impaired diffusion of the left parietooccipital cortex. Clinically she presented with left- sided spastic hemiparesis. This child also had old defects in the basal ganglia and a partly destructed cerebellum.
Biochemical findings
Blood lactate was elevated in every reported case (mean 13.2; median 13.1; range 6.3 – 21; reference range 0.5 – 2.2 mmol/L) (Thomas 2007). Blood alanine concentrations (n = 13) exceeded the reference range in eight cases (mean 678, median 640; range 353–1580; reference range 120–600 μmol/L) (Blau et al 2003). Blood glucose concentrations were usually normal though hyperglycemia was recorded in one patient and hypoglycaemia in another two patients. Cerebrospinal fluid (CSF) glucose was normal (n = 2) but lactate was elevated (n = 4) in those children who had a lumbar puncture performed. Creatine kinase was elevated in four of 11 individuals at initial diagnostic work-up (mean 574, median 184, range 61 – 2000; reference range 25 – 172 U/l [females] and 27 – 242 U/l [males]) (Thomas 2007) and subsequently noted as abnormal (730 and 2263 U/L) in a further two patients. Blood ammonia testing was mildly elevated in seven of 13 patients at initial work-up (mean and median 99, range 36–166; reference range 15–70 μmol/L) (Thomas 2007). Lysine was elevated in two of nine patients (457 and 628 μmol/L; reference range 66–270 μmol/L) (Blau et al 2003). When performed, blood urea nitrogen, creatinine, uric acid, amylase and lipase were all reported normal, liver transaminases were generally normal or at maximum very mildly elevated. The interpretation of molecular and mitochondrial respiratory chain specific biochemical data is shown in Supplementary Table 2. All available measurements showed diminished citrate synthase (CS), respiratory chain complexes and mtDNA content. In contrast to patients with typical mtDNA depletion syndromes due to a disorder of mtDNA replication or nucleoside salvage/synthesis, the mtDNA/CS ratio was normal in patients with FBXL4 deficiency.
Muscle tissues (available from one patient included in this study) revealed reduced activity in COX-staining indicating a generally reduced number of mitochondria. However, intramitochondrial signals showed a normal pattern. Our findings are in line with the observation by Bonnen that mild subsarcolemmal accumulations were detected by modified Gomori Trichrome staining of muscle tissue (Bonnen et al 2013) in the absence of ragged red fibers (Gai et al 2013). Furthermore, immunohistochemical staining revealed a decreased number of positively staining mitochondria for all oxidative phosphorylation enzymes and porin (Supplementary Fig. 4). There was a decrease in the number of positive granule but the intensity was unaffected, which is comparable to COX activity staining.
Molecular genetic analyses
Figure 4 summarises all pathogenic mutations identified in FBXL4 to date (including mutations reported by Bonnen et al 2013; Gai et al 2013 depicted in grey). At present, 28 patients known to carry 23 distinct disease-associated FBXL4 mutations have been identified. The seven predicted loss-of-function mutations (five nonsense mutations, one frameshift, one 18 bp deletion) and two predicted splice site mutations (affecting exons 4 and 9) are equally distributed over all coding exons. In contrast, 13 of the 14 missense mutations, all affecting evolutionarily-conserved amino acids, ten cluster inside the leucine-rich repeat domains and three around a predicted phosphorylation site at position p.202 (p.Ile205Thr, Asp221Val, Asp221His). The clustering of mutations hints towards an interference of the mutations with the function of FBXL4 by impairing protein-protein interactions or regulation of FBXL4. We found no significant phenotypic differences between patients with biallelic missense or predicted loss of function mutations and as such no obvious genotype-phenotype correlation could be delineated.
Fig. 4. FBXL4 Mutation Status and Gene Structure.

(A) Gene structure of FBXL4 with localization of mutations in 28 patients. Known functional domains are colored in green and red, regions of unknown function in grey. Red and blue asterisks indicate post-translational modifications. Mutations labeled in bold origin from patients reported in this study. (B) Conservation of amino acid residues affected by mutations. Numbering and coloring correspond to NP_001265645 alignment.
Discussion
We describe and review the clinical course in a cohort of 21 patients with biallelic FBXL4 mutations highlighting the severity of this recently described pan-ethnic mitochondrial disease. Our study has several limitations. This cohort represents the first group of patients with the diagnosis of an FBXL4-associated mitochondrial disease and late-onset, milder and atypical presentations of the disease will probably be described in the future, as it becomes routine to include FBXL4 mutations in the standard diagnostic workup of mitochondrial disease. The retrospective design and the variable follow-up strategies applied resulted in heterogeneous and partially incomplete data regarding biochemical parameters and clinical investigations. Furthermore, both the selection of physicians from the referrers of diagnostic material to specialized laboratories or specialist networks and physicians’ reports and assessments as data source carry a high risk of bias. Proxy-reported data may not or only in part reflect patients’ and caregivers’ perspectives.
In the reported cohort, seven of 21 patients died and survivors are severely handicapped. Most patients were young at follow-up and their symptoms may thus not represent the complete clinical spectrum of the disease. In the oldest patient, a female, aged 11 years and 7 months at last follow-up, stroke- like episodes and neutropenia evolved as additional clinical features. In 11 of 19 families, parental consanguinity (mostly first-cousin marriages) was reported. The cohort had a preponderance of males (13), but given the small size of the group and the manner in which individuals were recruited for investigation, we believe there may be significant bias.
Based on the observed pattern of clinical features in this cohort, we suggest that mutations in the FBXL4 gene are considered when children exhibit very early (neonatal in some cases) onset of lactic acidosis, profound muscular hypotonia, failure to thrive often (67 %) associated with characteristic facial features. While multiorgan involvement is a common finding in mitochondrial disease, the presence of a distinct dysmorphic phenotype is rare (Skladal et al 2003; Thorburn 2004; DiMauro and Gurgel-Giannetti 2005; Böhm et al 2006; Debray et al 2007; Cizkova et al 2008; Chinnery 2010; Emmanuele et al 2012; Desguerre et al 2014; Mayr 2014).
Many patients are critically ill from first presentation and may die at a very young age. In survivors, the initial pattern persists and development, motor and cognitive abilities become most severely impaired when patients grow older. Cardiac involvement in the form of non-progressive cardiomyopathy may be evident at initial presentation or evolve through the course of the disease. Other organs are rarely affected. The clinical course is generally progressive or at best stable and quality of life for patients and caregivers is considerably impaired.
There are no specific routine or basic laboratory parameters other than lactic acidosis and, in some cases, elevated creatine kinase, which may facilitate diagnosis. Where tested, skeletal muscle biochemical analysis reveals a severe, combined respiratory chain defect. Thus, loss of FBXL4 function results in a severe deficiency of mitochondrial energy metabolism, but rather unusually, this is characterised by a loss of mitochondrial number within cells. Diagnostic workup reveals a generally decreased amount of enzymes of the mitochondrial energy metabolism and mtDNA; the mtDNA/CS ratio remains normal (Supplementary Table 2) (Bonnen et al 2013; Gai et al 2013).
FBXL marks proteins containing an F-box and leucine-rich repeats. Both domains mediate protein-protein interactions. F- box proteins were first characterised as components of SCF ubiquitin-ligase complexes (named after their main components, Skp I, Cullin and an F-box protein), in which they bind substrates for ubiquitin-mediated proteolysis (Kipreos and Pagano 2000). Van Rechem et al (2011) identified FBXL4 in a screen for proteins interacting with the histone lysine demethylase JMJD2A. These experiments suggest that a SCF ubiquitin ligase complex, that contains cullin 1 and FBXL4, coordinates the turnover of JMJD2A and thereby regulates cell cycle progression. Experiments by Bonnen et al (2013) and Gai et al (2013) however, consistently provide a mitochondrial localisation of FBXL4, which suggests a different mode of action for FBXL4, in concordance with the severe mitochondrial dysfunction in patients harbouring recessive FBXL4 mutations. The precise function of FBXL4 and the mechanism by which FBXL4 mutations exert their effect is still unclear and warrants further work. The pattern of missense mutations in domains of protein-protein interaction and no obvious differences between these, as well as predicted biallelic loss of function mutations support an important scaffold function of FBXL4.
Brain imaging shows non-specific findings with increased water content of the white matter and rather large but non- specific germinolytic periventricular cysts in neonates. Severe atrophy ensues quickly, and decreased apparent diffusion coefficient (ADC) in the affected white matter suggests intramyelinic oedema. In some patients, central grey matter structures including the mesencephalon show increased volume and T2 signal and decreased ADC, as in other mitochondrial disorders, indicating acute energy failure.
In conclusion, the clinical pattern of early-onset encephalopathy, congenital muscular hypotonia, persistent lactic acidosis associated with the typical facial dysmorphism should prompt direct initiation of molecular genetic analysis, which seems the clearest way to diagnosis. Even though treatment approaches with coenzyme Q10, carnitine and other Bmitochondrial medications did not prove effective, establishment of the diagnosis of FBXL4 deficiency permits accurate genetic counselling and discussion of future reproductive options, prevents patients undergoing further unhelpful but burdensome diagnostic procedures and allows for accurate prognosis with appropriate end of life planning.
Supplementary Material
Acknowledgments
We gratefully acknowledge the contribution of Georg F. Hoffmann, Franz A. Zimmermann and Richard J. Rodenburg. We thank C. Terrile for technical support.
This project was supported by grants from the BMBF funded German Network for Mitochondrial Disorders (mitoNET #01GM1113C) and by the E-Rare project GENOMIT (01GM1207) and GENOMIT FWF I 920B13 to WS and 01GM1207 to HP.
RWT and RM are supported by The Wellcome Trust Centre for Mitochondrial Research (906919 Z/11/Z), the Lily Foundation and the UK NHS Highly Specialised Commissioners which funds the BRare Mitochondrial Disorders of Adults and Childrenˆ Diagnostic Service in Newcastle upon Tyne (http://www.mitoresearch.org.uk/).
D.E.-F. acknowledges support from the Graduate Academy of the University of Heidelberg, the Young Investigator Award Program at Ruprecht-Karls-University Heidelberg Faculty of Medicine, the Daimler and Benz Foundation (Daimler und Benz Stiftung, Ladenburg, Germany) and the Reinhard-Frank Foundation (Reinhard-Frank-Stiftung, Hamburg, Germany).
Footnotes
Compliance with ethics guidelines Conflict of interest None.
References
- Acham-Roschitz B, Plecko B, Lindbichler F, Bittner R, Mache CJ, Sperl W, Mayr JA. A novel mutation of the RRM2B gene in an infant with early fatal encephalomyopathy, central hypomyelination, and tubulopathy. Mol Genet Metab. 2009;98:300–304. doi: 10.1016/j.ymgme.2009.06.012. [DOI] [PubMed] [Google Scholar]
- Barbier A, Boivin A, Yoon W, et al. New reference curves for head circumference at birth, by gestational age. New reference curves for head circumference at birth, by gestational age. Pediatrics. 2013;131:1158–1167. doi: 10.1542/peds.2011-3846. [DOI] [PubMed] [Google Scholar]
- Berger A, Mayr JA, Meierhofer D, et al. Severe depletion of mitochondrial DNA in spinal muscular atrophy. Acta Neuropathol. 2003;105:245–251. doi: 10.1007/s00401-002-0638-1. [DOI] [PubMed] [Google Scholar]
- Blau N, Duran M, Blaskovic ME, Gibson KM, editors. Physician’s guide to the laboratory diagnosis of metabolic diseases. 2nd. Springer; Berlin: 2003. [Google Scholar]
- Böhm M, Pronicka E, Karcmarewicz E, et al. Retrospective, multicentric study of 180 children with cytochrome c oxidase deficiency. Pediatr Res. 2006;59:21–26. doi: 10.1203/01.pdr.0000190572.68191.13. [DOI] [PubMed] [Google Scholar]
- Bonnen PE, Yarham JW, Besse A, et al. Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am J Hum Genet. 2013;93:471–481. doi: 10.1016/j.ajhg.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenciarelli C, Chiaur DS, Guardavaccaro D, Parks W, Vidal M, Pagano M. Identification of a family of human F-box proteins. Curr Biol. 1999;9:1177–1179. doi: 10.1016/S0960-9822(00)80020-2. [DOI] [PubMed] [Google Scholar]
- Chinnery PF. Defining neurogenetic phenotypes (or how to compare needles in haystacks) Brain. 2010;133:649–654. doi: 10.1093/brain/awq027. [DOI] [PubMed] [Google Scholar]
- Cizkova A, Stranecky V, Mayr JA, et al. TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat Genet. 2008;40:1288–1290. doi: 10.1038/ng.246. [DOI] [PubMed] [Google Scholar]
- Debray FG, Lambert M, Chevalier I, et al. Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics. 2007;119:722–733. doi: 10.1542/peds.2006-1866. [DOI] [PubMed] [Google Scholar]
- Desguerre I, Hully M, Rio M, Nabbout R. Mitochondrial disorders and epilepsy. Revue neurologique. 2014;170:375–380. doi: 10.1016/j.neurol.2014.03.010. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Gurgel-Giannetti J. The expanding phenotype of mitochondrial myopathy. Curr Opin Neurol. 2005;18:538–542. doi: 10.1097/01.wco.0000179761.63486.1a. [DOI] [PubMed] [Google Scholar]
- Emmanuele V, Lopez LC, Berardo A, et al. Heterogeneity of coenzyme Q10 deficiency. Arch Neurol. 2012;69:978–983. doi: 10.1001/archneurol.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feichtinger RG, Zimmermann F, Mayr JA, Neureiter D, Hauser- Kronberger C, Schilling FH, Jones N, Sperl W, Kofler B. Low aerobic mitochondrial energy metabolism in poorly or undifferentiated neuroblastoma. BMC Cancer. 2010;19:149. doi: 10.1186/1471-2407-10-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feichtinger RG, Weis S, Mayr JA, Zimmermann F, Geilberger R, Sperl W, Kofler B. Alterations of oxidative phosphorylation complexes in astrocytomas. Glia. 2014;62:514–525. doi: 10.1002/glia.22621. [DOI] [PubMed] [Google Scholar]
- Gai X, Ghezzi D, Johnson MA, et al. Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am J Hum Genet. 2013;93:482–495. doi: 10.1016/j.ajhg.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack TB, Gorza M, Danhauser K, et al. Phenotypic spectrum of eleven patients and five novel MTFMT mutations identified by exome sequencing and candidate gene screening. Mol Genet Metab. 2014;111:342–352. doi: 10.1016/j.ymgme.2013.12.010. [DOI] [PubMed] [Google Scholar]
- Kipreos ET, Pagano M. The F-box protein family. Genome Biol. 2000;1:reviews3002.1–reviews3002.7. doi: 10.1186/gb-2000-1-5-reviews3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby DM, Thorburn DR, Turnbull DM, Taylor RW. Biochemical assays of respiratory chain complex activity. Methods Cell Biol. 2007;80:93–119. doi: 10.1016/S0091-679X(06)80004-X. [DOI] [PubMed] [Google Scholar]
- Mayr JA. Lipid metabolism in mitochondrial membranes. J Inherit Metab Dis. 2014;38:137–144. doi: 10.1007/s10545-014-9748-x. [DOI] [PubMed] [Google Scholar]
- Meierhofer D, Mayr JA, Foetschl U, et al. Decrease of mitochondrial DNA content and energy metabolism in renal cell carcinoma. Carcinogenesis. 2004;25:1005–1010. doi: 10.1093/carcin/bgh104. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. Mutation taster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- Skladal D, Sudmeier C, Konstantopoulou V, et al. The clinical spectrum of mitochondrial disease in 75 pediatric patients. Clin Pediatr. 2003;42:703–710. doi: 10.1177/000992280304200806. [DOI] [PubMed] [Google Scholar]
- Thomas L. Labor und Diagnose. 7th. Stuttgart: Thieme; 2007. [Google Scholar]
- Thorburn DR. Mitochondrial disorders: prevalence, myths and advances. J Inherit Metab Dis. 2004;27:349–362. doi: 10.1023/B:BOLI.0000031098.41409.55. [DOI] [PubMed] [Google Scholar]
- Van Rechem C, Black JC, Abbas T, et al. The SKP1-Cul1-F-box and leucine-rich repeat protein 4 (SCF-FbxL4) ubiquitin ligase regulates lysine demethylase 4A (KDM4A)/Jumonji domain- containing 2A (JMJD2A) protein. J Biol Chem. 2011;286:30462–30470. doi: 10.1074/jbc.M111.273508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston JT, Koepp DM, Zhu C, Elledge SJ, Harper JW. A family of mammalian F-box proteins. Curr Biol. 1999;9:1180–1182. doi: 10.1016/S0960-9822(00)80021-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.