

ABSTRACT
The 5S rDNA gene is a non-coding RNA that can be found in 2 copies (type I and type II) in bony and cartilaginous fish. Previous studies have pointed out that type II gene is a paralog derived from type I. We analyzed the molecular organization of 5S rDNA type II in elasmobranchs. Although the structure of the 5S rDNA is supposed to be highly conserved, our results show that the secondary structure in this group possesses some variability and is different than the consensus secondary structure. One of these differences in Selachii is an internal loop at nucleotides 7 and 112. These mutations observed in the transcribed region suggest an independent origin of the gene among Batoids and Selachii. All promoters were highly conserved with the exception of BoxA, possibly due to its affinity to polymerase III. This latter enzyme recognizes a dT4 sequence as stop signal, however in Rajiformes this signal was doubled in length to dT8. This could be an adaptation toward a higher efficiency in the termination process. Our results suggest that there is no TATA box in elasmobranchs in the NTS region. We also provide some evidence suggesting that the complexity of the microsatellites present in the NTS region play an important role in the 5S rRNA gene since it is significantly correlated with the length of the NTS.
KEYWORDS: Internal Control Region, Microsatellites, Non-transcribed Spacer, RNA secondary structure, TATA-box
Abbreviations
- TFIIIA
Transcription factor IIIA
- ICR
Internal control region
- NTS
Non-transcribed Spacer
Introduction
The organization of ribosomal genes includes a transcribed followed by a non-transcribed region arranged in tandem within the nucleolar organizing region (NOR)1 whose numbers vary among organisms.2,3 In most eukaryotes, these genes are classified in 2 different groups: 45S and 5S rDNA. Following a typical organization, the 5S rDNA contains a 120 bp transcribed region that is highly conserved due to its essential function in stabilizing the ribosomal structure and enhancing the peptidyl transferase activity.4 This transcribed region is adjacent to a Non-transcribed Spacer (NTS) that varies in length among species.1,5 This NTS region has a higher mutation rate than the transcribed region.1 Insertions and deletions, rather than nucleotide substitutions, is believed to be the main cause of the observed variation.6
The 5S rDNA codes for a small structural RNA and is classified as a class III gene. This implies that it is transcribed by RNA polymerase III. RNA Polymerase III promoters are divided into 3 types according to their organization. The promoter for 5S rDNA is of type 1 and consists of BoxA, an Internal Element (IE), and BoxC; that is found within the transcribed region.7 These three elements are grouped in the Internal Control Region (ICR)8 and in elasmobranchs (shark, rays, and skates) have been reported to be respectively at positions 50 – 64, 67 – 72, and 80 – 97 bp of the transcript.9,10 The transcribed region also contains a termination signal consisting of a poly-T relic, that is still recognized by RNA polymerases III during transcription. Another feature of 5S rDNA is that a TATA box is not strictly required for transcription. However, in mammals, it has been shown that there is a TATA sequence in the NTS region that may play an important role in regulating 5S rDNA expression.5,12 In some fish species and in elasmobranchs, 5S rDNA has a TATA-like sequence, suggesting that this sequence influences gene expression.9,10,13,14,15,16 This TATA-like element in fish has not been experimentally investigated yet. In Xenopus borealis a deletion of 55 nucleotides in the upstream 5′ end of the 5S rRNA only expressed a part of the gene, showing that this region influences in the transcription process.17
Like many ribosomal genes, 5S rDNA has been shown to follow the model of concerted evolution.18 This model proposes that paralogous genes in a species are more similar among themselves than with orthologous genes across species as a result of homogenization.19,20,21 This homogenization has been attributed to the replicative transposition, gene amplification, and gene conversion.22,23 It has also been proposed that the unequal crossing over among the repetition units play an important role in this process.24 Walsh,25 using computer simulations and taking into account more parameters, showed that this process deleted tandem arrays. Later, this was supported by Stephan26 who evaluated the structural patterns of tandem repetitions with the simulations of different recombination rates. Their results demonstrated that at high recombination rates, the repetitive structures are distributed in a regular pattern.
In elasmobranchs (as well as in several bony fishes27) 5S rDNA occurs frequently in 2 forms: type I and II. Both types are differentiated by the length and the sequence of its NTS region. The longest of the 2 is designated as type II. The evolutionary history of type II gene suggests that it might have originated in the elasmobranch lineage by genome duplication. This further implies that it is paralogous to 5S rRNA type II in bony fish.28 The aim of this study is to compile and analyze the molecular organization and the secondary structure of 5S rDNA previously as type II in elasmobranchs.
Results and discussion
Transcribed region: mutations in the ICR components and termination signal of the
RNA polymerase III
PCR amplification of the 5S rDNA type II in Urotrygon with the primers 5SRajid amplified a unique band of 1,5 Kbp (supplementary Fig. 1) and its partial sequencing revealed that this band is composed of the 5S rRNA gene as well as its associated NTS. The 5S rRNA segment contains all the elements of the ICR and the terminal signal of the polymerase III. Since the transcribed region has a functional role within the organism, it is expected to be strictly conserved. However, there are point mutations in the transcribed region covering both the inside and outside of the ICR. The BoxA has 7 positions with mutations (positions 51, 52, 56, 59, 60, 61, and 64). From these positions the nucleotide 60 is the one with highest inferred information (∼1 bit). The BoxC only shows 2 mutations (positions 84 and 92), while the IE has a single mutation with high information (∼1.8 bit) corresponding to the nucleotide 68. Outside the ICR 15 mutations were found, with the nucleotides 18, 24, 35, and 112 showing low information (≤ 1 bit) (Fig. 1).
Figure 1.
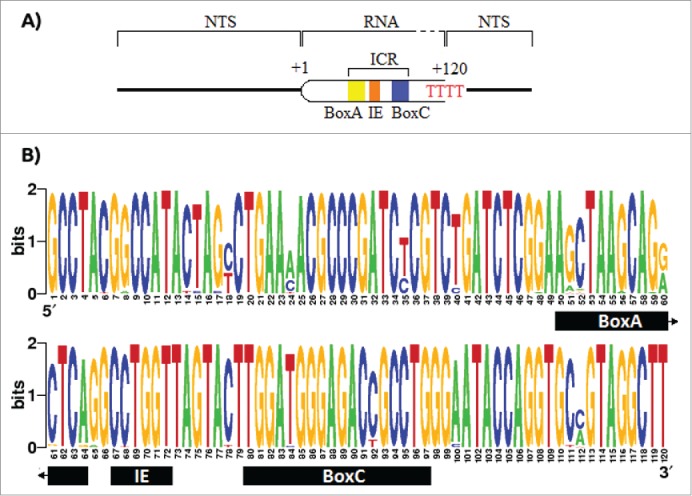
Promoter and terminator elements description and entropy index for the transcribed region of the 5S rDNA. A) The promoter of the 5S rDNA contains the elements BoxA, the Internal Element, and the BoxC, altogether called Internal Control Region (ICR). The RNA polymerase recognizes a poly dT as stop signal. B) The entropy index for the 5S rRNA using all the 91 sequences retrieved from the GenBank. However, due the fact that the first 43 nucleotides in Potamotrygon motoro, Potamotrygon falkneri and Paratrygon aiereba transcript region sequences were missing, these were assumed to be equal to those in Urotrygon.
Unlike BoxC, the IE, and BoxA show a higher number of mutations and ratio between the number of mutations over length (BoxA = 7/14, IE = 1/5, and BoxC = 2/17). This might be due to the effects of the IE and BoxA in the affinity to the Transcription Factor IIIA (TFIIIA) compared to the BoxC.29,30,31 It has been shown that BoxC interacts with the first 3 amino-terminal fingers of the TFIIIA, and that these are required for affinity to the 5S rDNA.32,33 This may be the reason why BoxC is highly conserved among individuals. In Rajiformes the termination signal of the RNA polymerase III (poly-T tail) is longer (up to 4 dTs longer) than in other clades (up to 4 nucleotides as the expected range for eukaryotes34). Some studies have shown gene transcripts (like the tRNALys in rabbits and X. laevis, and the VA RNA-II in adenovirus) that contain extensive dT tail that are more efficient at terminating transcription.35 Similar cases have been reported in Saccharomyces pombe and S. cerevisiae, where effective transcription required 5 and 6 thymidine fragments, respectively.36 Apparently, a higher content of dT nucleotides in the termination signal make the transcription more robust limiting the influence of surrounding sequences. This allows that adjacent dC and dG nucleotides improve the effectiveness of the termination while the dA nucleotide decreases it.37 However, there are some exceptions like the tRNALys of X. laevis where an extension of residues dA downstream to the termination signal enhances its effectiveness.38 In Rajiformes, both upstream and downstream sequences in the termination signal are similar to that of other species. This makes it difficult to conclude that the long tail of poly-T in Rajiformes is due to surrounding sequences that interfere with the effectiveness in the termination signal of the RNA polymerase III. Also, the poly-T tail has several adjacent nucleotides dC and dG that increase the effectiveness of the transcription termination.37 Considering that RNA polymerase III often ignores the original termination signal and recognizes the posterior poly-dT cluster,39 it is possible that the significant excess of dT residues (p < 0.01; Table 2) in the original termination signal in Rajiformes, with respect to other clades, is an adaptation to a better efficiency of transcription termination.
Table 2.
Contingency table of Chi-square to test the equality of the stop signal in all the elasmobranchii individuals studied here.
| Observed |
Expected |
Chi Squared |
||||||
|---|---|---|---|---|---|---|---|---|
| Present | Absent | Present | Absent | Present | Absent | Total | p-Value | |
| dT7 | 26 | 0 | 7,9 | 19,1 | 39,6 | 17,2 | 56,7 | |
| dT4 | 0 | 60 | 18,1 | 41,9 | 17,2 | 7,4 | 24,6 | |
| Total | 26 | 60 | 26,0 | 60,0 | 56,7 | 24,6 | 81,3 | <0,01 |
Changes in the transcribed region and secondary structure through elasmobranch
Evolution
The low information inferred for the positions 18, 24, 35, 60, and 112 is mainly due to variability in some taxonomic units. Mutations C18T, G60A, and T112M (where M = A or C) are characteristic of a division at a higher taxonomic level: Batoidea (Rajiformes and Myliobatiformes, in this study) with respect to Selachii (Carchariniformes and Lamniformes, in this study). However, the synapomorphic mutation of the position 112 is absent in the genus Galeocerdo. This might be due to the under-representation of this genus in the sampling (1 individual). Mutations A24C and T35C are synapomorphic in Myliobatiformes with respect to Rajiformes (Fig. 2). The synapomorphic mutations C18T, G60A, and T112M agree with the hypothesis of an independent evolution of Batoideans and Selachians based on the fossil record41 and also congruent with several molecular phylogenetic studies.42,43,44,45,46 However, Compagno's hypothesis,47 that batoids are derived from Pristiophoriformes, cannot be tested with our data since the sharks of the superorder Squalimorphi are not represented in our sampling.
Figure 2.
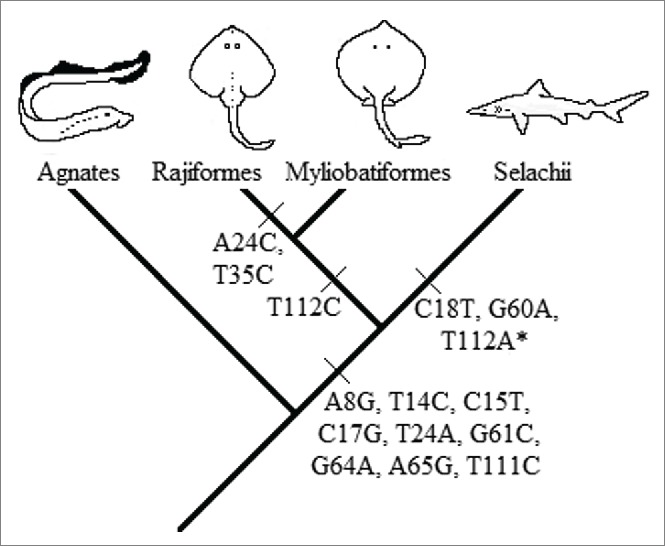
Evolutionary interpretation of the synapomorphic mutation events. (*)The synapomorphic mutation of the nucleotide 112 is absent in Galeocerdo cuvier. This might be due to the underrepresentation of this genus or a back mutation in that position.
The mutations at positions 18, 60 and 7, 112 form pairs in the secondary structure proposed by Barciszewska et al.48 (Fig. 3A), and is similar to our secondary structure consensus (Fig. 3B) with the following exceptions: a) mismatch in the basepair (7,112) in sharks except in G. cuvier, b) within the loop C the basepairs (33,42) and (34,41) are joint, c) the bulge located at position 84 in Barciszewska et al. is now located at position 83, and d) basepair (80,96) mismatches in all the structures obtained. The aforementioned 7–112 mismatch (feature a, Fig. 3B) is located in helix I. Therefore, a large effect is expected on the transcription level since the TFIIIA binds to this site.50,51 In addition, this feature is highly conserved in Selachii. An experimental validation of this claim is needed to assert this effect.
Figure 3.
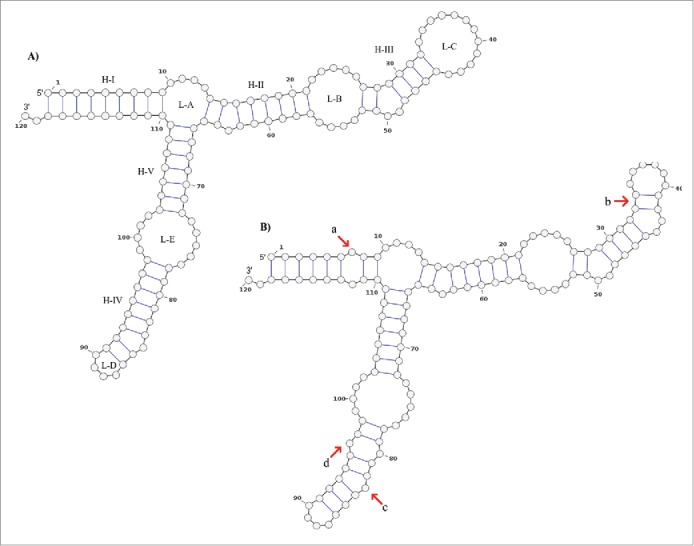
Secondary structure consensus of the 5S rRNA type II predicted by RNAalifold. The structure A) is proposed by Barciszewska et al.48 which corresponds to the general model of 5S rRNA for eukaryotes. The structure B) is the consensus structure predicted for the 5S rRNA type II by RNAalifold. The main differences are those marked with red arrows: a) positions 7 and 112 with no pair in all sharks but G. cuvier; b) all the sequences fit well with the basepair 34C:41G and 33U:42A which reduce the loop C size; c) the bulge at the position 84 (with base dU in this study) proposed by Barciszewska et al.48 is now located at the position 83 (dA). According to these authors the bulge located in the helix IV must be a purine; d) positions 80 and 96 are not joined in all the sequences.
The results obtained by mutual information show 5 pairs of positions with scores above the critical value (Mijc ≈0.212), these positions are (60,18), (112,18), (92,60), (112,60), (60,24) with Mij equal to 0.99, 0.51, 0.24, 0.45, and 0.23, respectively. Moreover, the covariance-like measure shows 3 pairs of positions with values above its critical value (Cijc ≈0.172), these positions are (18,51), (18,60), and (35,60) with Cij equal to 0.31, 0.49, and 0.44, respectively. Although there is some evidences of the formation of base triples in RNA structures,82 it is well known that the 5S rRNA structure form only base pairs.83 Given that one nucleotide can join only to another one nucleotide, we opted to choose only the pair position (18,60), since this is the only pair common to the 2 analyses and has the highest score. We used this information to constrain the secondary structure prediction for each haplotype. This resulted in 25 different secondary structures which can be classified into 3 groups (Fig. 4). Group α represents 84% of all the sequences studied and have a structure similar to the consensus structure, with 3 arms. The structure of the group β have 4 arms and represents 5% of all sequences. The group γ comprises 11% of all the sequences and has a structure with 4 arms arranged differently from those of group β. Despite the amount of different predicted secondary structures, the RNA sequence fit well to the consensus secondary structure (Fig. 3B) with exceptions in some positions due to SNP's. This lower accuracy in the 5S rRNA secondary structure prediction with respect to other RNA molecules has been reported previously56 (and references therein). This could be due to possible interactions that are needed for the proper folding of the 5S rRNA. For example, within the group γ (Fig. 4), the sequence FJ517193.1 has a structure with a free energy of -37.41 kcal/mol. This is notably different from the others due to the mutation G116A which seems to destabilize helix I. When this mutation is reverted, the resulting secondary structure is similar to those of the group α and its free energy is reduced to -43.60 kcal/mol. However, experimental validation of this hypothesis is needed. In addition, we also marked those sequences whose promoter elements are different to the consensus sequence of metazoan 5S rRNA in the tree of structures (supplementary Fig. 2). We found not only that these sequences are interspersed in the tree (among the 3 groups), but also those sequences with promoter elements that fit the consensus sequences. These results suggest that SNP's both inside and outside the ICR can change the pattern of folding into another conformation with lower free energy.
Figure 4.
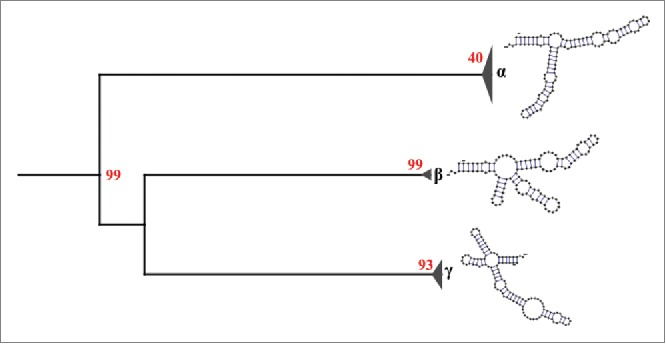
Secondary structural groups of the 5S rRNA type II defined by neighbor joining. The results show 3 different structural groups: α, β and γ. The group α is composed of structures that are similar to our consensus secondary structure of the 5S rRNA type II, while the groups β and γ are composed of structures different to our consensus. Red numbers represent the bootstrap values.
The compensatory mutation between nucleotides 18 and 60 indicates that helix II in the secondary structure is important for transcription. This may be because it is the region where TFIIIA's 7 zinc fingers attach.49,50 In fact, it has been proven that a minimum fragment of 5S RNA that includes the helices I, II, and V, and the loops A and E, is sufficient to bind to TFIIIA.50,51 This is an essential component of the initiation complex in the transcription of RNA polymerase III.52,53,54 Nucleotides 24, 35 and 40 are not tightly bound to the secondary structure and can mutate more freely. Also the loops B and C containing these mutations don't have an important role for the union of the TFIIIA's zinc fingers to the 5S rRNA molecule.55
NTS region
TATA Box - The NTS region of the 5S rDNA type II is more conserved within species than expected considering the overall substitution rate over the wider phylogenetic range.57,58 This can be explained either by concerted evolution or because of strong selective pressures due to the regulation of the gene.5,59 In bony fishes, a TATA-like box was found between position -20 and -30 upstream from the transcription start site.13,14,60 This suggests a possible influence on the level of transcription. However, despite the presence of a TATA-like box in Rhizoprionodon (with an AATT motif starting at nucleotide -27)10 and in Rajiformes (as TACA at nucleotide -25),9 in both Urotrygon species there is not a TATA-like element within the positions −30 and −20 (5′-TGTGAAGAGGC-3') similar to those reported previously in Rhizoprionodon and Rajiformes. This discrepancy in both sequence and location with respect to a TATA-like box among the 3 orders may indicate that these TATA-like boxes are not really essential factors. Furthermore, since the promoter of 5S rDNA is of type 1, all of its elements are internal and therefore not necessary for transcription.11 However, more studies are needed to determinate whether these TATA boxes have some level of influence on the transcription process. There are hybrid promoters like the U6 snRNA gene in S. cerevisiae that contain both internal promoters and a TATA box61,62 which contributes to better transcription.63,64,65
Microsatellites - The different microsatellites arrangements between genera at the NTS region are the main contributors to the variation in the NTS length. It has been shown that there is a correlation between the length of the NTS region and the number of motif repetitions within each region of microsatellites in Rajiformes.9 But in addition, it is possible that there exists a correlation among the number of microsatellite cluster regions and the length of the NTS region. This process is possible because the NTS shows several clusters of repetitions such as (dNdN)2 (see supplementary Fig. 3). To confirm this correlation, a linear regression was performed. However, missing data in Urotrygon, Potamotrygon and Paratrygon might bias the analyses. To test whether the missing region could be safely ignored, we performed the Clark-Evans and the Diggle-Cressie-Loosmore-Ford (DCLF) test in Rhizoprionodon, Raja and Dipturus to check if the microsatellites were distributed randomly, regularly or aggregatively. If the distribution is random or regular, and assuming that the NTS of Urotrygon, Potamotrygon and Paratrygon behaves similarly, the regression won't be affected by the absence of the fragments. Our results show that indeed the distribution of the microsatellite is random in some species, regular in others, but never aggregated (Table 3). Therefore, the partial sequence of the NTS of Urotrygon, Potamotrygon and Paratrygon species can be used for the linear regression.
Table 3.
Statistical results for the distribution of the microsallite region inside the NTS. Significant p-values for R. montagui and R. miraletus indicate that its distribution is regular. For other species the distribution is random.
| Species | Clarc-Evans testtwo tail (p-value) | DCLF(p-value) |
|---|---|---|
| R. lalandii | 0.67 | 0.87 |
| R. porosus | 0.22 | 0.60 |
| R. polystigma | 0.28 | 0.73 |
| R. montagui | 0.03 | 0.65 |
| R. miraletus | 0.03 | 0.87 |
| R. clavata | 0.55 | 0.30 |
| D. oxyrinchus | 0.29 | 0.80 |
| R. asterias | 0.26 | 0.18 |
The linear regression analysis shows a strong relationship between these 2 variables (r = 0.95, p − value < 0.01; Fig. 5), even if both species of Urotrygon are excluded from the analysis (r = 0.75, p − value < 0.01). With this, we speculate that the microsatellite clusters play a functional role in transcription where a mechanism of unwinding is providing and used during the transcription process.66 However, according to Pinhal et al.10 this is not possible because the NTS region of the Rhizoprionodon genus does not present a high microsatellite arrangement complexity like in Rajidae. This low complexity could be due to the short length of the NTS region in Rhizoprionodon where the amount of microsatellites needed to maintain the arrangement and other functions is lower (directly proportional). Therefore it is possible to propose that fewer microsatellites are needed to maintain the arrangement and other functions, but this is a speculative claim.
Figure 5.
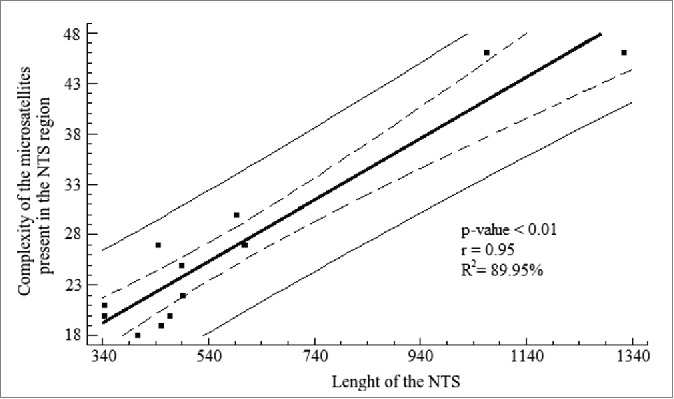
Linear regression analysis for the length of the NTS and the complexity of the microsatellites present in the NTS region. The analysis shows a high coefficient determination, which was significant. Solid line represents the linear regression according to the equation Y = 10.78 + 0.03(X) + e . Dashed lines represent the 95% confidence interval for the median value of Yi in any Xi . Thin lines are the prediction limits of 95% for new observations.
Species belonging to Rajiformes have a highly similar NTS which Pasolini et al.9 interpret as evidence of the existence of a similar NTS organization in the 5S rRNA genes of the common ancestor of the Rajiformes families from Upper Cretaceous (97 Mya). However, this high level of similarity may be the product of a more recent event of speciation. Valsecchi et al.67 by molecular studies of the control region and the 16S rRNA estimated that the divergence of the Rajini tribe could be less than 7 Mya. This estimate is also supported by the paleogeography and paleoclimate events that gave rise to the Mediterranean fauna where the samples of the genus Raja and Dipturus were obtained.9,68
Conclusions
In this study we show that mutations in the primary structure and the conformation of the secondary structure of the 5S rRNA type II can be tracked in their evolution among the elasmobranchii group. Despite the fact that the secondary structure is highly conserved, there are several mutations present. We advise that future studies take into account the tertiary structure of the 5S rRNA since it might be more informative with respect to the possible changes in the spatial conformation. Moreover, mutations that maintain the secondary structure and others characteristics (e.g. terminal signal for RNA polymerase III and the low variation in the promoters), indicates that this non-coding gene is under strong purifying selection. Also, our study shows that the complexity of the microsatellite present throughout the NTS region is positively correlated with its length, which supports the idea that microsatellite, and therefore the NTS regions, play an important role in the function and evolution of the gene.
Materials and methods
Sample collection and storage
Samples from Urotrygon rogersi and Urotrygon aspidura were collected by artisanal shrimp trawling in Juanchaco and La Bocana localities in the Colombian Pacific Ocean (3 o 52 N − 77 o 18 W ). From each individual approximately 1 cm3 of muscle was extracted and preserved in Longmire's buffer69 as suggested by Hleap et al.70 until DNA extraction.
DNA extraction, PCR amplification, and DNA sequencing
The DNA extraction was performed with proteinase-K digestion following a phenol/chloroform purification with the conditions reported by Hleap et al.70. The PCR amplification of the 5S rDNA type II was performed using the primer set 5SRajid designed to amplify specifically the 5S rDNA type II in the genera Raja and Dipturus.9 The primer 5SRajid-F (5′-GGAATACCAGGTGCCGTAGG-3') binds between the positions 55 - 74 of the positive strand, whereas the primer 5SRajid-R (5′-GGTATTCCCAGGCGGTCT-3') binds between the positions 88 – 105 in the negative strand. The PCR reaction contained 1X Taq Buffer, 1 mM Mg, 200 pmol dNTP's, 10 μM of each primer and 1 U of Taq polymerase. Thermal conditions were the following: an initial denaturation step at 95 ° C for 3 min, 30 cycles of 95 ° C for 30 s, 56.5 ° C for 30 s and 72 o C for 1 min. Finally an extension at 72 o C for 7 min was made. A negative control (all above except DNA, the volume of which was replaced by water) was used to test for any contamination. The PCR products were assessed by electrophoresis in 1.0% agarose gels and visualized by ethidium bromide staining under ultraviolet illumination. The PCR purification was done with the polyethylene/glycol (PEG) protocol.71 Nine sequences were gathered (5 samples of U. aspidura and 4 of U. rogersi) through the standard Sanger sequencing service of Macrogen Inc.. in Korea and were submitted to GenBank (www.ncbi.nlh.nih.gov) under the accession numbers JQ670255 – JQ670263.
In silico data collection, sequence alignment and RNA secondary structure prediction
A total of 82 sequences are reported for the 5S rDNA type II among 13 species of elasmobranchs and 3 Petromyzontiformes were obtained from GenBank (Table 1). Sequences from Galeocerdo cuvier, Scyliorhinus canicula and Alopias superciliosus were assigned to belong to the type II 5S rRNA by BLAST (nucleotide blast).72 The BLAST search was optimized for highly similar sequences (megablast) showing higher similarity with the 5S rRNA type II among all elasmobranchii to the exclusion of type I. Similarly, the 5S rDNA sequences of Raja montagui was assigned to belong to type II since it showed higher similarity to the type II sequence of Raja and Dipturus genus instead of type I. The authors68 also reported that this sequence corresponded to the heaviest band of 2 bands obtained. Therefore, the study included 91 specimens (82 retrieve from NCBI database, plus 5 of U. aspidura and 4 of U. rogersi described in this study). All sequences were aligned with MUSCLE73 implemented in MEGA V5.074 with default options. In order to ensure the alignment of homologous positions, low complexity regions were manually removed. However, the microsatellite often present in such regions were taken into account to discuss the NTS sequence.
Table 1.
List of the 5S rDNA type II gene sequences obtained from 14 elasmobranchs and 3 Petromyzontiformes species retrieve from the NCBI database.
| Species | Number of sequences | NCBI Accession code | Order |
|---|---|---|---|
| Entosphenus japonicus | 2 | X04308-X04309 | Petromyzontiformes |
| Lampetra reissneri | 1 | X13038 | Petromyzontiformes |
| Lethenteron camtshaticum | 1 | D00076 | Petromyzontiformes |
| Rhizoprionodon lalandii | 19 | FJ517239-FJ517257 | Carcharhiniformes |
| Rhizoprionodon porosus | 16 | FJ517223-FJ517238 | Carcharhiniformes |
| Galeocerdo cuvier | 1 | FJ539131 | Carcharhiniformes |
| Scyliorhinus caniculus | 1 | M24954 | Carcharhiniformes |
| Alopias superciliosus | 1 | FJ539130 | Lamniformes |
| Potamotrygon motoro | 6 | JF792331-JF792336 | Myliobatiformes |
| Potamotrygon falkneri | 3 | JF92328-JF92330 | Myliobatiformes |
| Paratrygon aiereba | 5 | JF92323-JF92327 | Myliobatiformes |
| Raja asterias | 5 | DQ020522-DQ020526 | Rajiformes |
| Raja clavata | 6 | DQ020527-DQ020532 | Rajiformes |
| Dipturus oxyrinchus | 6 | DQ020541-DQ020546 | Rajiformes |
| Raja miraletus | 4 | DQ020537-DQ020540 | Rajiformes |
| Raja polystigma | 4 | DQ020533-DQ020536 | Rajiformes |
| Raja montagui | 1 | AY278250 | Rajiformes |
The level of conservation of each nucleotide position of 5S rRNA was assessed with WebLogo75 which uses information gained at each position.
The consensus secondary structure of the 5S rRNA type II was obtained through RNAalifold76 using the matrix of sequences. However, since a consensus structure can overshadow the structure variability of the molecule, we also made the secondary structure prediction for each haplotype sequence. The haplotype sequences were retrieved with a python script that only keeps unique sequences. Then we use the covariation among positions with the mutual information (Mij) using:
| (Eq. 1) |
where fi(X) is the frequency of base X at aligned position i and fij(XY) is the frequency of finding X in i and Y in j and (XY) is the set of allowed basepair B = {GC,CG,AU,UA,GU,UG} so that (XY) ∈ B. Mutual information is stringent respect to the information contained in consistent mutations such as GC↔GU. When one position is conserved the result of Mij is equal to zero. Therefore, we use also the covariation-like measure proposed by Hofacker et al.77 divided by 2 since Cij make use of a symmetric matrix:
| (Eq. 2) |
where D(XY;X'Y') is the Hamming distance of (XY) and (X'Y') and both (XY) and (X'Y') ∈ B77. Null models were fitted to test for the statistical significance of the results of both Mij and Cij . The null models were computed by 1) making 2 vectors with a length equal to the number of sequences studied, where each vector has a composition of 25% for each possible base, 2) estimating Mij or Cij between these 2 vectors and storing the results, 3) repeating steps 1 and 2 for 1.0 × 105 iterations, 4) making a cumulative distribution function with the results and finding the value Xijc such as the result of the integral from Xijc to Xij max is equal to 0.01, where Xij represent both Mij or Cij and Xij max represents the maximum possible value of Mij or Cij (2 and 0.75, respectively), and finally 5) using the pair of columns from the real dataset whose values of Mij or Cij were higher than its respective Xijc to constrain the secondary structure prediction of each haplotype in RNAalifold. To define the groups of structures obtained, we built a matrix using the dot-bracket notation of each structure and then we applied the distance analysis of Neighbor Joining with 1000 replicates in SeaView v4.4.78
Analysis of the molecular structure of 5S rDNA type II
Analysis was performed by direct comparison of gene sequences between species, focused in the 5S rRNA, internal control region (ICR), microsatellites, TATA box sequences and termination signal of the RNA polymerase III. The microsatellite distribution along the NTS region was analyzed with the Clark-Evans test79 and the Diggle-Cressie-Loosmore-Ford (DCLF) test80 with 100000 iterations of a Monte Carlo simulation implemented in R v3.0.1 with the module Spatstat.81 The spatial reference for each microsatellite was its midpoint. In order to prove that a relationship between the complexity of microsatellite regions and the length of the NTS exist, we performed a linear regression analysis. For those samples of Urotrygon only the sequenced regions were taken into account. Also, a chi-square test with Yates's correction was performed to prove statistically that the stop signal in 5S rDNA is equal in all the individuals (Table 2).
Author's contributions
JSH and HCH coordinated the study. JSH collected the samples used in this study. SIC completed the laboratory work, the in silico analysis, the results interpretation and wrote the manuscript. JSH, HCH and CB helped in the analysis, the results interpretation and revised the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Hernán Paredes and Florentino Cuero and sons for the support in fieldwork and Agustin Martínez, owner of the motorboat “Arraiján” to furnish his ship for some of the sampling. To Jitka M. Krejci, Conor Mehan, and Johanna Zaglauer for the review of the English version, as well as the critical review of this manuscript. The SQUALUS Foundation logistically supported this work through contributions of PADI Foundation and IEA scholarships for the project “Biodiversidad e historia de vida de rayas del Pacífico vallecaucano.” The Universidad del Valle supported this work through research grant CI #7809 for the project entitled “Efecto de la selección sobre la heredabilidad estimada en algunos caracteres morfométricos de historia de vida en dos especies del género Urotrygon del Pacífico vallecaucano” and the research grant CI #7922 for the project entitled “Determinación de la relación génica entre el gen 5S rDNA tipo I y tipo II entre peces óseos y cartilaginosos.” NSERC supported this work through the grant No. 120504858. The government of Canada partially sponsored this project through the DFATD scholarship.
References
- 1.Long EO, David ID. Repeated genes in eukaryotes. Rev Biochem 1980; 49:727-64; http://dx.doi.org/ 10.1146/annurev.bi.49.070180.003455 [DOI] [PubMed] [Google Scholar]
- 2.Mindel DP, Honeycutt RL. Ribosomal RNA in vertebrates: evolution and phylogenetic applications. Annu Rev Ecol Syst 1990; 21:541-66; http://dx.doi.org/ 10.1146/annurev.es.21.110190.002545 [DOI] [Google Scholar]
- 3.Lewin B. (ed.): Genes VI. Oxford University Press, New York: 1997 [Google Scholar]
- 4.Barciszewska MZ, Szymanski M, Erdmann VA, Barciszewski J. Structure and function of 5S rDNA. Acta Biochim Pol 2001; 48:191-8; PMID:11440169 [PubMed] [Google Scholar]
- 5.Nederby NH, Hallenberg C, Frederiksen S, Sorensen PD, Lomholt B. Transcription of human 5S rRNA genes is influenced by an upstream DNA sequences. Nucleic Acids Res 1993; 26:3631-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campo D, Machado G, Horreo JL, García E. Molecular organization and evolution of 5S rDNA in the genus Merluccius and their phylogenetic implications. J Mol Evol 2009; 68:208-16; PMID:19247563; http://dx.doi.org/ 10.1007/s00239-009-9207-8 [DOI] [PubMed] [Google Scholar]
- 7.Pieler T, Oei S, Hamm J, Engelke U, Erdmann VA. Functional domains of the Xenopus laevis 5S gene promoter. EMBO J 1985; 4(13B):3751-6; PMID:3004969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hallenberg C, Nederby J, Frederiksen S. Characterization of 5S rRNA genes from mouse. Gene (Amst.) 1994; 142:291-5; http://dx.doi.org/ 10.1016/0378-1119(94)90277-1 [DOI] [PubMed] [Google Scholar]
- 9.Pasolini P, Costagliola D, Rocco L, Tinti F. Molecular organization of 5S rDNA in Rajidae (Chondrichthyes): Structural features and evolution of piscine 5S rRNA genes and Nontranscribed intergenic spacer. J Mol Evol 2006; 62:564-74; PMID:16612546; http://dx.doi.org/ 10.1007/s00239-005-0118-z [DOI] [PubMed] [Google Scholar]
- 10.Pinhal D, Yoshimura TS, Araki CS, Martins C. Molecular organization of 5S rDNA in sharks of the genus Rhizoprionodon: insights into the evolutionary dynamics of 5S rDNA in vertebrate genomes. Genet Res Camb 2009; 91:61-72; PMID:19220932; http://dx.doi.org/ 10.1017/S0016672308009993 [DOI] [PubMed] [Google Scholar]
- 11.Schramm L, Hernandez N. Recruitment of RNA polymerase III to its target promoters. Genes Dev 2002; 16:2593-620; PMID:12381659; http://dx.doi.org/ 10.1101/gad.1018902 [DOI] [PubMed] [Google Scholar]
- 12.Suzuki H, Sakurai S, Matsuda Y. Rat rDNA spacer sequences and chromosomal assignment of the genes to the extreme terminal region of chromosome 19. Cytogenetic Cell Genet 1996; 72:1-4; http://dx.doi.org/ 10.1159/000134149 [DOI] [PubMed] [Google Scholar]
- 13.Pendas AM, Moran P, Freije JP, Garcia-Vazquez E. Chromosomal mapping and nucleotide sequence of two tandem repeats of Atlantic salmon 5S rDNA. Cytogenetic Cell Genet 1994; 67:31-6; http://dx.doi.org/ 10.1159/000133792 [DOI] [PubMed] [Google Scholar]
- 14.Murakami M, Fujitani H. Characterization of repetitive DNA sequences carrying 5S rDNA of the triploid ginbuna (Japanese silver crucian carp, Carassius auratu langsdorfi). Genes Genet Syst 1998; 73:9-20; PMID:9546204; http://dx.doi.org/ 10.1266/ggs.73.9 [DOI] [PubMed] [Google Scholar]
- 15.Rocco L, Russo C, Stingo V, Aprea G, Odierna G. Characterization of 5S rDNA in Gasterosteus aculeatus (teleostei, Gasterosteidae). Ital J Zool 1999; 66:285-9; http://dx.doi.org/ 10.1080/11250009909356266 [DOI] [Google Scholar]
- 16.Martins C, Galetti PM. Organization of 5S rDNA in species of the fish Leporinus: two different genomic locations are characterized by distinct nontranscribed spacers. Genome 2001; 44:903-10; PMID:11681615; http://dx.doi.org/ 10.1139/g01-069 [DOI] [PubMed] [Google Scholar]
- 17.Sakonju S, Bogenhagen DF, Brown DD. A control region in the center of the 5S RNA gene directs specific initiation of transcription: I. The 5 border of the region. Cell 1980; 19:13-25; PMID:7357599; http://dx.doi.org/ 10.1016/0092-8674(80)90384-0 [DOI] [PubMed] [Google Scholar]
- 18.Ohta T, Dover G. The cohesive population genetics of molecular drive. Genetics 1984; 108:501-21; PMID:6500260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zimmer EA, Martin SL, Beverly SM, Kan YW, Wilson AC. Rapid duplication and loss of genes coding for the α chains of hemoglobin. Proc Natl Acad Sci USA 1980; 77:2158-62; PMID:6929543; http://dx.doi.org/ 10.1073/pnas.77.4.2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dover GA. Molecular drive: a cohesive mode of species evolution. Nature 1982; 299:111-6; PMID:7110332; http://dx.doi.org/ 10.1038/299111a0 [DOI] [PubMed] [Google Scholar]
- 21.Elder JF, Turner BJ. Concerted evolution of repetitive DNA sequences in eukaryotes. Q Rev Biol 1995; 70:297-320; PMID:7568673; http://dx.doi.org/ 10.1086/419073 [DOI] [PubMed] [Google Scholar]
- 22.Hillis DM, Moritz C, Porter CA, Barker RJ. Evidence for biased gene conversion in concerted evolution of ribosomal DNA. Science 1991; 251:308-10; PMID:1987647; http://dx.doi.org/ 10.1126/science.1987647 [DOI] [PubMed] [Google Scholar]
- 23.Liao D, Pavelitz T, Kidd J, Kidd K, Weiner A. Concerted evolution of the tandemly repeated genes encoding human U2 snRNA (the RNU2 locus) involves rapid intrachromosomal homogenization and rare interchromosomal gene conversion. EMBO J 1997; 16:588-98; PMID:9034341; http://dx.doi.org/ 10.1093/emboj/16.3.588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown DD, Wensink PC, Jordan E. Xenopus laevis and Xenopus mulleri: the evolution of tandem genes. J Mol Biol 1972; 63:57-73; PMID:5016971; http://dx.doi.org/ 10.1016/0022-2836(72)90521-9 [DOI] [PubMed] [Google Scholar]
- 25.Walsh JB. Persistence of tandem arrays: implication for satellite and simple sequence DNAs. Genetics 1987; 115:553-67; PMID:3569882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stephan W. Tandem-repetitive noncoding DNA: forms and forces. Mol Biol Evol 1989; 6:198-212; PMID:2716519 [DOI] [PubMed] [Google Scholar]
- 27.Martins C, Galetti PM. Two 5S rDNA arrays in neotropical fish species: is it a general rule for fishes? Genetica 2001; 111:439-46; PMID:11841188; http://dx.doi.org/ 10.1023/A:1013799516717 [DOI] [PubMed] [Google Scholar]
- 28.Pinhal D, Yoshimura TS, Araki CS, Martins C. The 5S rDNA family evolves through concerted and birth-and-death evolution in fish genome: an example from freshwater stingray. BMC Evol Biol 2011; 11:151; PMID:21627815; http://dx.doi.org/ 10.1186/1471-2148-11-151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pieler T, Hamm J, Roeder RG. The 5S gene internal control region is composed of three distinct sequences elements, organized as two functional domains with variable spacing. Cell 1987; 48:91-100; PMID:3791417; http://dx.doi.org/ 10.1016/0092-8674(87)90359-X [DOI] [PubMed] [Google Scholar]
- 30.Sands MS, Bogenhagen DF. TFIIIA binds to different domains of 5S RNA and the Xenopus borealis 5S RNA gene. Mol Cell Biol 1987; 7:3985-93; PMID:3431548; http://dx.doi.org/ 10.1128/MCB.7.11.3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.You Q, Veldhoen N, Baudin F, Romaniuk PJ. Mutations in 5S DNA and 5S RNA have different effects on the binding of Xenopus transcription factor IIIA. Biochemistry 1991; 30:2495-500; PMID:2001375; http://dx.doi.org/ 10.1021/bi00223a028 [DOI] [PubMed] [Google Scholar]
- 32.Christensen JH, Hansen PK, Lillelund O, Thogersen HC. Sequence-specific binding of the N-terminal three-finger fragment of Xenopus transcription factor IIIA to the interval control region of a 5S RNA gene. FEBS Lett 1991; 281:181-4; PMID:2015891; http://dx.doi.org/ 10.1016/0014-5793(91)80388-J [DOI] [PubMed] [Google Scholar]
- 33.Liao XB, Clements KR, Tannant L, Wright PG, Gottesfeld JM. Specific interaction of the first three zinc finger of TFIIIA with the internal control region of the Xenopus 5S RNA gene. J Mol Biol 1992; 223:857-71; PMID:1538401; http://dx.doi.org/ 10.1016/0022-2836(92)90248-I [DOI] [PubMed] [Google Scholar]
- 34.Allison DS, Hall BD. Effects of alterations in the 3' flanking sequence on in vivo and in vitro expression of the yeast SUP4-0 tRNA Tyr gene. EMBO J 1985; 4:2657-64; PMID:3902472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gunnery S, Ma Y, Mathews MB. Termination sequence requirements vary among genes transcribed by polymerase III. J Mol Biol 1999; 286:745-57; PMID:10024448; http://dx.doi.org/ 10.1006/jmbi.1998.2518 [DOI] [PubMed] [Google Scholar]
- 36.Hamada M, Sakulich AL, Koduru SB, Maraia RJ. Transcription termination by RNA polymerase III fission yeast. A genetic and biochemically tractable model system. J Biol Chem 2000; 275:29076-81; PMID:10843998; http://dx.doi.org/ 10.1074/jbc.M003980200 [DOI] [PubMed] [Google Scholar]
- 37.Bogenhagen DF, Brown DD. Nucleotide sequences in Xenopus 5S DNA required for transcription termination. Cell 1981; 24:261-70; PMID:6263489; http://dx.doi.org/ 10.1016/0092-8674(81)90522-5 [DOI] [PubMed] [Google Scholar]
- 38.Mazabraud A, Scherly D, Muller F, Rungger D, Clarkson SG. Structure and transcription termination of a lysine tRNA gene from Xenopus laevis. J Mol Biol 1987; 195:835-45; PMID:2443712; http://dx.doi.org/ 10.1016/0022-2836(87)90488-8 [DOI] [PubMed] [Google Scholar]
- 39.Akusjarvi G, Mathews MB, Andersson P, Vennstrom B, Petterson U. Structure of genes for virus-associated RNAI and RNAII of adenovirus type-2. Proc Natl Acad Sci USA 1980; 77:2424-8; PMID:6930642; http://dx.doi.org/ 10.1073/pnas.77.5.2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao JY, Ma LM, Guo YH, Zhang YC, Zhou H, Shao P, Chen YQ, Qu LH. Deep Sequencing of human nuclear and cytoplasmic small RNAs reveals an unexpectedly complex sub cellular distribution of miRNAs and tRNA 3' trailers. Plos One 2010; 5:e10563; PMID:20498841; http://dx.doi.org/ 10.1371/journal.pone.0010563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kriwet J, Kiessling W, Klug S. Diversification trajectories and evolutionary life-history in early sharks and batoids. Proc R Soc B 2009; 276:945-51; PMID:19129130; http://dx.doi.org/ 10.1098/rspb.2008.1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dunn KA, Morressey JF. Molecular phylogeny of elasmobranchs. Copeia 1995; 3:526-31; http://dx.doi.org/ 10.2307/1446750 [DOI] [Google Scholar]
- 43.Kitamura T, Takemura A, Watabe S, Taniuchi T, Shimizu M. Molecular phylogeny of the sharks and rays of superorder Squalea based on mitochondrial cytochrome b gene. Fisheries Science 1996; 62:340-3 [Google Scholar]
- 44.Douady CJ, Dosay M, Shivji MS, Stanhope MJ. Molecular phylogenetic evidence refuting the hypothesis of Batoidea (rays and skates) as derived sharks. Mol Phyl Evol 2003; 26:215-21; http://dx.doi.org/ 10.1016/S1055-7903(02)00333-0 [DOI] [PubMed] [Google Scholar]
- 45.Winchell CJ, Martin AP, Mallatt J. Phylogeny of elasmobranchs based on LSU and SSU ribosomal genes. Mol Phyl Evol 2004; 31:214-24; http://dx.doi.org/ 10.1016/j.ympev.2003.07.010 [DOI] [PubMed] [Google Scholar]
- 46.Human BA, Owen EP, Compagno LVJ, Harley EH. Testing morphologically based phylogenetic theories within the cartilaginous fishes with molecular data, with special reference to the catshark family (Chondrichthyes: Scyliorhinidae) and the interrelationships among them. Mol Phyl Evol 2006; 39:384-91; http://dx.doi.org/ 10.1016/j.ympev.2005.09.009 [DOI] [PubMed] [Google Scholar]
- 47.Compagno LVJ. Sharks of the World An annotated and illustrated catalogue of sharks species know to date. Volume 2. Bullhead, mackerel and carpet sharks (Heterodontiformes, Lamniformes and Orectolobiformes). FAO Species Catalogue for Fishery Purposes 2002; 2:1-3 [Google Scholar]
- 48.Barciszewska MZ, Szymaski M, Erdmann VA, Barciszewski J. 5S ribosomal RNA. Biomacromolecules 2000; 1:291-302; http://dx.doi.org/ 10.1021/bm000293o [DOI] [PubMed] [Google Scholar]
- 49.Clements KR, Wolf V, McBryant SJ, Zhang P, Liao XB, Wright PE, Romaniuk PJ, Gottesfeld JM. Molecular basis for specific recognition of both RNA and DNA by a zinc finger protein. Science 1993; 260:530-3; PMID:8475383; http://dx.doi.org/ 10.1126/science.8475383 [DOI] [PubMed] [Google Scholar]
- 50.Neely LS, Lee BM, Xu J, Wright PE, Gottesfeld JM. Identification of a minimal domain of 5S ribosomal RNA sufficient for high affinity interaction with the RNA-specific zinc finger of transcription factor IIIA. J Mol Biol 1999; 291:549-60; PMID:10448036; http://dx.doi.org/ 10.1006/jmbi.1999.2985 [DOI] [PubMed] [Google Scholar]
- 51.Theunissen O, Rudt F, Pieler T. Structural determinants in 5S RNA and TF IIIA for 7S RNP formation. Eur J Biochem 1998; 258:758-67; PMID:9874245; http://dx.doi.org/ 10.1046/j.1432-1327.1998.2580758.x [DOI] [PubMed] [Google Scholar]
- 52.Hanas JS, Gaskins CJ, Smith JF, Ogilvie MK. Structure, function, evolution of transcription factor IIIA. Prog Nucleic Acid Res Mol Biol 1992; 43:205-39; PMID:1410446; http://dx.doi.org/ 10.1016/S0079-6603(08)61048-X [DOI] [PubMed] [Google Scholar]
- 53.Pieler T, Theunissen O. Nine fingers - three hands? Trends Biochem Sci 1993; 18:226-30; PMID:7688487; http://dx.doi.org/ 10.1016/0968-0004(93)90194-R [DOI] [PubMed] [Google Scholar]
- 54.Shastry BS. Transcription factor IIIA (TFIIIA) in the second decade. Cell Science 1996; 109:535-9 [DOI] [PubMed] [Google Scholar]
- 55.McBryant SJ, Veldhoen N, Gedulin B, Leresche A, Foster MP, Wright PE, Ramaniuk PJ, Gottesfeld JM. Interaction of the RNA binding fingers of Xenopus transcription factor IIIA with specific regions of 5S ribosomal RNA. J Mol Biol 1995; 248:44-57; PMID:7731045; http://dx.doi.org/ 10.1006/jmbi.1995.0201 [DOI] [PubMed] [Google Scholar]
- 56.Sun FJ, Caetano-Anollés G. The evolutionary history of the structure of 5S ribosomal RNA. J Mol Evol 2009; 69:430-543; PMID:19639237; http://dx.doi.org/ 10.1007/s00239-009-9264-z [DOI] [PubMed] [Google Scholar]
- 57.Hosikawa Y, Iida Y, Iwabuchi M. Nucleotide sequence of the transcriptional initiation region of Dictyostelium discoideum rRNA gene and comparison of the initiation regions of three lower eukaryotes genes. Nucl Acids Res 1983; 11:1725-34; PMID:6300775; http://dx.doi.org/ 10.1093/nar/11.6.1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suzuki H, Moriwaki K, Sakurai S. Sequences and evolutionary analysis of mouse 5S rDNA. Mol Biol Evol 1994; 11:704-10; PMID:8078409 [DOI] [PubMed] [Google Scholar]
- 59.Hallenberg C, Frederiksen S. Effects of mutations in the upstream promoter on the transcription of human 5S rRNA genes. Biochim Biophys Acta 2001; 1520:169-73; PMID:11513959; http://dx.doi.org/ 10.1016/S0167-4781(01)00264-0 [DOI] [PubMed] [Google Scholar]
- 60.Inafaku J, Nabeyama M, Kikuma Y, Saitoh J, Kubota S, Kohono S. Chromosomal location and nucleotide sequence of 5S ribosomal DNA of two cyprinid species (Osteichthyes, Pisces). Chromosome Res 2000; 8:193-9; PMID:10841046; http://dx.doi.org/ 10.1023/A:1009292610618 [DOI] [PubMed] [Google Scholar]
- 61.Brown DA, Guthrie C. Transcription of a yeast U6 snRNA gene requires a polymerase III promoter element in a novel position. Genes Dev 1990; 4:1345-56; PMID:2227412; http://dx.doi.org/ 10.1101/gad.4.8.1345 [DOI] [PubMed] [Google Scholar]
- 62.Eschenlauer JB, Kaiser MW, Gerlach VL, Brown DA. Architecture of a yeast U6 RNA gene promoter. Mol Cell Biol 1993; 13:3015-26; PMID:8474459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yukawa Y, Sugita M, Choisne N, Small I, Sugiura M. The TATA motif, the CAA motif and the poly(T) transcription termination motif are important for transcription re-initiation on plant tRNA. Plant J 2000; 22:439-47; PMID:10849359; http://dx.doi.org/ 10.1046/j.1365-313X.2000.00752.x [DOI] [PubMed] [Google Scholar]
- 64.Dieci G, Percudani R, Giuliodori S, Bottarelli L, Ottonello S. TFIIIC-independent in vitro transcription of yeast tRNA genes. J Mol Biol 2000; 299:601-13; PMID:10835271; http://dx.doi.org/ 10.1006/jmbi.2000.3783 [DOI] [PubMed] [Google Scholar]
- 65.Ouyang C, Martinez MJ, Young LS, Sprague KU. TATA-binding protein-TATA interaction is a key determinant of differential transcription of skillworn constitutive and silk gland-specific tRNA Ala genes. Mol Cell Biol 2000; 20:1329-43; PMID:10648619; http://dx.doi.org/ 10.1128/MCB.20.4.1329-1343.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Catasti P, Chen X, Mariappan SV, Bradbury EM, Gupta G. DNA repeats in the human genome. Genetica 1999; 106:15-36; PMID:10710707; http://dx.doi.org/ 10.1023/A:1003716509180 [DOI] [PubMed] [Google Scholar]
- 67.Valsecchi E, Pasolini P, Bertozzi M, Garoia F, Ungaro N, Vacchi M, Sabelli B, Tinti F. Rapid Miocene-Pliocene dispersal and evolution of Mediterranean rajid fauna as inferred by mitochondrial gene variation. J Evol Biol 2005; 18:436-46; PMID:15715849; http://dx.doi.org/ 10.1111/j.1420-9101.2004.00829.x [DOI] [PubMed] [Google Scholar]
- 68.Rocco L, Costagliola D, Fiorillo M, Tinti F, Stingo V. Molecular and chromosomal analysis of ribosomal cistrons in two cartilaginous fish, Taeniura lymma and Raja montagui (Chondricthyes, Batoidea). Genetica 2005; 123:245-53; PMID:15954495; http://dx.doi.org/ 10.1007/s10709-004-2451-3 [DOI] [PubMed] [Google Scholar]
- 69.Longmire JL, Maltbie M, Baker RJ. Use of “lysis buffer” in DNA isolation and its implications for museum collection. Occasional Papers, The Museum of Texas Tech University 1997; 163:1-3 [Google Scholar]
- 70.Hleap JS, Cárdenas H, García-Vallejo F. Preservación no criogénica de tejido y extracción de ADN: Una aplicación para peces cartilaginosos. PanamJAS 2009; 4:283-93 [Google Scholar]
- 71.Lis JT. Fractionation of DNA fragments by polyethylene glycol induced precipitation. Methods Enzymol 1980; 65:347-53; PMID:6246357; http://dx.doi.org/ 10.1016/S0076-6879(80)65044-7 [DOI] [PubMed] [Google Scholar]
- 72.Morgulis A, Coulouris G, Raytselis Y, Madden TL, Agarwala R, Schaffer AA. Database indexing for production MegaBLAST searches. Bioinformatics 2008; 24:1757-64; PMID:18567917; http://dx.doi.org/ 10.1093/bioinformatics/btn322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acid Res 2004; 32:1792-7; PMID:15034147; http://dx.doi.org/ 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using Maximum Likelihood, Evolutionary distances, and Maximum parsimony methods. Mol Biol Evol 2011; 282731-2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: A sequence logo generator. Genome Res 2004; 14:1188-90; PMID:15173120; http://dx.doi.org/ 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics 2008; 9:474; PMID:19014431; http://dx.doi.org/ 10.1186/1471-2105-9-474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hofacker IL, Fekete M, Stadler PF. Secondary Structure Prediction for Aligned RNA Sequences. J Mol Biol 2002; 319:1059-66; PMID:12079347; http://dx.doi.org/ 10.1016/S0022-2836(02)00308-X [DOI] [PubMed] [Google Scholar]
- 78.Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplataform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 2010; 27:221-4; PMID:19854763; http://dx.doi.org/ 10.1093/molbev/msp259 [DOI] [PubMed] [Google Scholar]
- 79.Clarck PJ, Evans FC. Distance to nearest neighbor as a measure of spatial relationships in populations. Ecology 1954; 35:445-53; http://dx.doi.org/ 10.2307/1931034 [DOI] [Google Scholar]
- 80.Loosmore N, Ford ED. Statistical inference using the G or K point pattern spatial statistics. Ecology 2006; 87:1925-31; PMID:16937629; http://dx.doi.org/ 10.1890/0012-9658(2006)87%5b1925:SIUTGO%5d2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- 81.Baddeley A, Turner R. Spatstat: an R package for analyzing spatial point patterns. J Stat Soft 2005; 12:1-42; http://dx.doi.org/ 10.18637/jss.v012.i06 [DOI] [Google Scholar]
- 82.Huthoff H, Girard F, Wijmenga SS, Berkhout B.: Evidence for a base triple in the free HIV-1 TAR RNA. RNA 2004; 10:412-23; PMID:14970387; http://dx.doi.org/ 10.1261/rna.5161304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ben-Shem A, Jenner L, Yusupova G, Yusupov M. Crystal Structure of the Eukaryotic Ribosome. Science 2010; 330:1203-9; PMID:21109664; http://dx.doi.org/ 10.1126/science.1194294 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.