

ABSTRACT
Co-transcriptional splicing takes place in the context of a highly dynamic chromatin architecture, yet the role of chromatin restructuring in coordinating transcription with RNA splicing has not been fully resolved. To further define the contribution of histone modifications to pre-mRNA splicing in Saccharomyces cerevisiae, we probed a library of histone point mutants using a reporter to monitor pre-mRNA splicing. We found that mutation of H3 lysine 36 (H3K36) – a residue methylated by Set2 during transcription elongation – exhibited phenotypes similar to those of pre-mRNA splicing mutants. We identified genetic interactions between genes encoding RNA splicing factors and genes encoding the H3K36 methyltransferase Set2 and the demethylase Jhd1 as well as point mutations of H3K36 that block methylation. Consistent with the genetic interactions, deletion of SET2, mutations modifying the catalytic activity of Set2 or H3K36 point mutations significantly altered expression of our reporter and reduced splicing of endogenous introns. These effects were dependent on the association of Set2 with RNA polymerase II and H3K36 dimethylation. Additionally, we found that deletion of SET2 reduces the association of the U2 and U5 snRNPs with chromatin. Thus, our study provides the first evidence that H3K36 methylation plays a role in co-transcriptional RNA splicing in yeast.
KEYWORDS: Chromatin, coupling, co-transcriptional pre-mRNA splicing, histones, methylation, snRNPs, transcription
Abbreviations
- (H3K36)
histone H3 lysine 36
- (ChIP)
chromatin immunoprecipitations
- (RT-qPCR)
reverse transcription PCR
- (snRNP)
small nuclear ribonucleprotein
- (RNA Pol II)
RNA polymerase II
Introduction
The process of pre-mRNA splicing is carried out within the spliceosome, a large and dynamic ribonucleoprotein. The spliceosome is comprised of small nuclear ribonucleoprotein complexes (snRNPs) that assemble on a pre-mRNA molecule in a precise order through interactions with sequences in the introns.1 The U1 snRNP binds to the 5′ splice site, followed by ATP-dependent loading of the U2 snRNP onto the branchpoint sequence. The tri-snRNP (U4/U6•U5) joins next, and subsequent ATP-dependent RNA helicase-mediated rearrangements remove the U1 and U4 snRNPs and reorganize the remaining snRNPs to promote the 2 catalytic steps of splicing. In Saccharomyces cerevisiae, splicing takes place primarily during RNA Polymerase II (RNA Pol II) transcription2-4 and studies have revealed that snRNPs assemble onto the nascent pre-mRNA co-transcriptionally.5-9
Mounting evidence supports a model in which pre-mRNA splicing is tightly coordinated with transcription to ensure precise and efficient gene expression.10,11 The fact that pre-mRNA splicing occurs cotemporally with RNA Pol II-dependent gene transcription indicates that spliceosome assembly and subsequent catalytic steps take place in the context of a dynamic chromatin environment. There are currently 2 non-mutually exclusive proposed mechanisms for coupling transcription and RNA splicing. The first is a recruitment model in which RNA Pol II and/or transcription-related proteins, either directly or indirectly, physically recruit splicing factors to the nascent transcript leading to specific splicing outcomes.10,11 The second is a kinetic coupling model that explains how splicing can be impacted by transcription elongation rates. The splicing of a given intron is enhanced when the rate of elongation is slow because the splicing machinery has more time to access the splice sites. Exon skipping can occur when elongation is fast, as there is less time for the splicing machinery to assemble on the pre-mRNA.10,11
Most S. cerevisiae genes do not contain more than one intron, and thus extensive alternative splicing does not occur. However, several recent reports show that the splicing efficiency of specific subsets of transcripts changes in different environmental conditions12 or when splicing-related proteins are mutated.13,14 Mutations that alter transcription elongation rates or treatment with drugs that affect transcription can change alternative splicing outcomes in metazoa15-20 and splicing efficiency in yeast.8,21,22 For example, splicing of the DYN2 alternative splicing reporter pre-mRNA in S. cerevisiae changes when transcription elongation is slowed using the small molecules 6-Azauracil or mycophenolic acid, or by mutating the RNA Pol II subunit Rpb2.21 A recent point mutation epistatic miniarray profile (pE-MAP) paired with genome-wide splicing microarray analysis of 53 RNA polymerase mutants in S. cerevisiae revealed that altering the rate of elongation can change the efficiency of splicing; slow elongation enhances splicing, while fast elongation reduces splicing efficiency.22 Thus any protein that can alter RNA Pol II elongation rate has the potential to regulate RNA splicing.
In the context of chromatin, histone tails undergo extensive posttranslational modifications, such as lysine acetylation and methylation, altering the structure of chromatin23,24 and hence access of RNA Pol II to the DNA template. Recent genome-wide analysis in both metazoa25 and in yeast26 reveal that the presence of certain histone modifications differs between DNA sequences encoding exons and those encoding introns, leading to the emerging paradigm that histone modification can modulate RNA splicing.11 This paradigm is supported by several recent studies showing that both histone H3 acetylation27,28 and histone H2B-K123 ubiquitination29,30 enhance splicing efficiency in yeast. Furthermore, several histone modifications have recently been implicated in co-transcriptional recruitment of splicing factors, providing evidence for the recruitment model of coupling transcription with RNA splicing.10,11 For example, histone H2B ubiquitination by the Bre1 E3 ubiquitin ligase29 and Gcn5 histone acetyltransferase activity27,28 facilitate splicing by recruiting splicing factors to splicing substrates in yeast. In metazoa, depletion of SETD2, the chromatin modification enzyme that tri- methylates H3K36 (see below), changes alternative splicing patterns and both tri-methylated H3K4 and tri-methylated H3K36 interact with splicing proteins to recruit them during transcription.31-35 Thus, histone modifications and the changing chromatin landscape constitute an exciting frontier for splicing regulation that has yet to be fully explored.
Recently, large-scale studies have identified a potential role for the Set2 methyltransferase in yeast RNA splicing.29,30 Set2 methylates nucleosomal H3K36, and generates mono-, di-, and tri-methylated forms.36 Studies show that Set2 is associated with the elongating form of RNA Pol II and mediates H3K36me2/me3 to recruit a number of chromatin-modifying complexes (Rpd3S and Isw1b) that maintain a repressive chromatin environment that is resistant to pervasive transcription in the coding regions of genes.37-42 Although a number of studies have shown that the human homolog of Set2, SETD2, is important for alternative splicing 31,33 and that H3K36 is essential for viability in drosophila,43 the direct role of H3K36me3 and other methylation states (particularly H3K36me2) in both canonical and alternative splicing has not been clearly elucidated.
To identify novel regulators of RNA splicing in yeast, we recently carried out a genome-wide screen using a fluorescent reporter to monitor gene expression in a library of 4967 S. cerevisiae deletion mutants. These studies suggested that deletion of several transcription factors and histone modifiers may cause a pre-mRNA splicing defect.44 Here, we sought to further characterize the role of histone modification in RNA splicing. Utilizing the reporter to probe for splicing defects in a library consisting of hundreds of synthetic histone point mutants,45 we identified several histone point mutations displaying splicing-like defects. These defects also mimic those seen in deletion mutants of specific histone modification- and chromatin remodeling-enzymes, including set2Δ. We found that mutations altering H3K36me have genetic interactions with genes encoding RNA splicing factors and alter splicing efficiency of a set of endogenous transcripts. Further work demonstrated that changes in transcription elongation alone do not fully account for changes in RNA splicing efficiency, and showed that deletion of SET2 significantly reduces the association of snRNPs with chromatin, supporting a model in which Set2/H3K36me increases splicing efficiency by facilitating co-transcriptional spliceosome assembly. Moreover, our work reveals for the first time that different methylation states of H3K36 are important for transcript-specific splicing.
Results
Screen of histone H3 and H4 point mutants for effects on gene expression
We recently described pre-mRNA splicing-like phenotypes for a number of deletion mutants of histone-modifying factors with our gene expression reporter.44 Briefly, our reporter results in expression of the fluorescent proteins mCherry and GFP, serving as a proxy for in vivo levels of reporter pre-mRNA and spliced mRNA, respectively (Fig. 1E). Given the aforementioned findings, as well as the growing amount of work describing links between histone modifications and pre-mRNA splicing, we analyzed a collection of 486 histone H3 and H4 substitution and deletion mutants with our reporter via high-throughput flow cytometry.45 The resulting histone mutant data were incorporated into the deletion collection dataset consisting of nearly 5000 unique deletion mutants44 and re-clustered (raw data and processed data provided as Supporting Data sets S1 and S2-S4, respectively). Our clustering analysis pipeline vectorizes the data to compare the shape of the 2-dimensional reporter data in a non-directed manner.44 As with the deletion collection, the majority of histone H3 and H4 mutants did not differ significantly from wild-type (Fig. S1). Interestingly, a subset of histone point mutants clustered with deletion mutants exhibiting characteristic red-shifts as a result of an increase in the mCherry/GFP (unspliced/spliced) ratio (Fig. 1; global clustering behavior depicted in Fig. S1), suggesting a potential pre-mRNA splicing defect. It is important to note that mCherry/GFP expression is affected by processes other than pre-mRNA splicing. For instance we observe a red-shift in mutants of nonsense mediated decay and green-shifts in mutants of mRNA export and specific mRNA decay/pathways.44 Therefore, orthogonal assays are necessary to confirm the specific gene expression pathway(s) affected.
Figure 1.
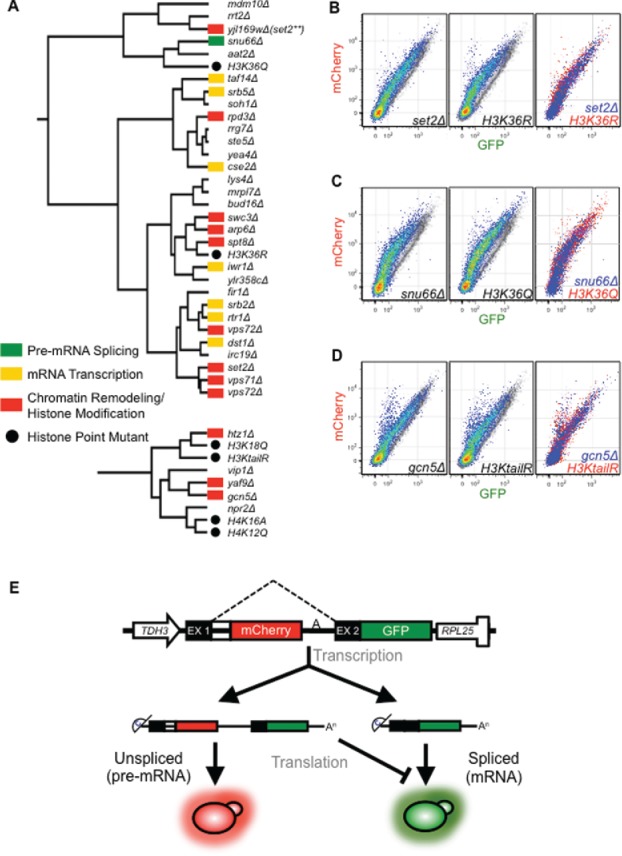
Specific histone mutants phenocopy loss of functionally relevant gene expression factors. Gene expression reporter phenograph data from the collection of histone point mutants were integrated with the deletion collection data set and hierarchically clustered with complete linkage and a centered absolute correlation similarity metric, resulting in a small number of histone point mutants clustering with deletion mutants within gene expression pathways (A). Nodes are annotated with the colors indicated in the legend. Global clustering behavior of the histone mutants is depicted in Fig. S1. Gene expression reporter phenograph overlays for deletion and histone mutants of interest (B-D) are depicted. Mutants are shown in pseudo-color on the left and middle panels and overlaid onto a grayscale wild-type phenograph for comparison. For the rightmost panel the phenograph for the deletion mutant (blue) and histone mutant (red) are overlaid to demonstrate similarity. The H3KtailR mutant is H3K9,14,18,23R. Note: there are 2 vps72Δ strains in the deletion mutant collection. (E). Gene Expression reporter schematic. Expression is driven by the constitutive TDH3 promoter. The sequence features of the pre-mRNA are modeled based on the inefficiently spliced CYH2 gene. The intron is marked by the dashed lines. The mCherry ORF is in frame with exon I, thus production of mCHerry results from mRNAs that have retained an intron. The GFP is in frame with exon 2 driving GFP expression only when the intron is removed.
Of the histone mutants examined, point mutants at H3K36 (H3K36Q and H3K36R) clustered within a clade of primarily chromatin modifier and transcription-related deletion mutants (Fig. 1A top). The H3K36R mutant and the strain lacking the H3K36 methyltransferase SET2 (set2Δ) cluster in the same large red-shifted clade and have highly similar phenographs (Fig. 1B). Interestingly, a strain with a deletion of a dubious ORF (YJL169W), with sequence that overlaps the C-terminal ∼120nts of the SET2 gene, also falls within this red-shifted clade and has a similar phenograph to snu66Δ. It is currently not known how this small C-terminal deletion impacts SET2 expression, but it inhibited splicing of the gene expression reporter more than the full SET2 deletion. Additionally, the H3K36Q mutant is even more severely red-shifted and clusters with the pre-mRNA splicing deletion mutant snu66Δ46,47 (Fig. 1C). The clustering analysis of histone H3K36 mutants and set2Δ with respect to pre-mRNA splicing defects suggests that SET2/H3K36me is important for pre-mRNA splicing.
In addition to H3K36, we also found that the H3K9R, K14R, K18R and K23R quadruple mutant (H3KtailR) mimicking a constitutively deacetylated H3 tail, clusters with functionally relevant chromatin-modifying and histone point mutants (Fig. 1A bottom). Most striking is the similarity of this mutant with the gcn5Δ deletion (Fig. 1D). This similar reporter phenograph and clustering behavior was expected, as Gcn5 is known to acetylate the tail of H348 and contribute to co-transcriptional spliceosome recruitment.27,28 The YAF9 deletion mutant, also clustering in this small clade, is a component of the SWR1 complex that replaces histone H2A (HTA1/HTA2) with H2A.Z (HTZ1).49 Additionally, Yaf9 is a component of the NuA4 histone acetyltransferase complex, known to acetylate the N-terminal lysines of H4,50 consistent with the clustering behavior of the 2 H4 mutants H4K16A and H4K12Q (Fig. 1A bottom). Using our splicing reporter system, we were able to cluster specific histone modifier mutants with their respective histone point mutants, thereby validating the sensitivity and specificity observed with our reporter across a wide-range of gene expression mutants.44
Mutations that alter H3K36 methylation exhibit genetic interactions with genes that encode splicing factors
Our gene expression reporter assay data point to a potential role for Set2 and H3K36 methylation in pre-mRNA splicing (Fig. 1). To further investigate this hypothesis, we tested for genetic interactions between genes that encode RNA splicing factors and SET2 or JHD1, which encodes a demethylase enzyme that removes the mono- and di-methyl groups from H3K36.51-54 We generated double mutants that harbor both a deletion of a splicing protein gene and a deletion of either SET2 or JHD1 using SGA methodology.55 Deletion of SET2 resulted in reduced fitness in a strain lacking the IST3 gene that encodes a component of the U2 snRNP56 (Fig. 2A top panel). This negative genetic interaction is exacerbated at higher temperatures (37°C); the growth of the set2Δ ist3Δ double mutant is slowed significantly when compared to the set2Δ or ist3Δ single mutant strains. In addition, at 37°C SET2 loss also reduced fitness in a strain lacking ISY1 (Fig. 2A, lower panel), which encodes a component of the Prp19 complex that functions during the catalytic steps in splicing.57-59 In contrast, jhd1Δ displays mild positive genetic interactions with both ist3Δ and isy1Δ (Fig. 2B). For example, the jhd1Δ isy1Δ strain grows better than the jhd1Δ or isy1Δ single mutant strains, and this suppression is more apparent at 37°C (Fig. 2B, lower panel). Interestingly, deletion of the SNU66 splicing factor gene, which encodes a component of the tri-snRNP,46 displays opposite genetic phenotypes. There is a positive genetic interaction observed between set2Δ and snu66Δ at both 37°C (Fig. 2A, lower panel) and 16°C (Fig. S2A). Neither deletion of SET2 or JHD1 had a considerable impact on the fitness of strains lacking either MUD1, a component of the U1 snRNP,60,61 or MUD2, a commitment complex protein62,63 (Fig. S2B, upper panels) – both of which act early in splicing to commit an RNA to the splicing pathway. Taken together with results from the reporter assay, these genetic data further implicate both Set2 and Jhd1 in pre-mRNA splicing.
Figure 2.
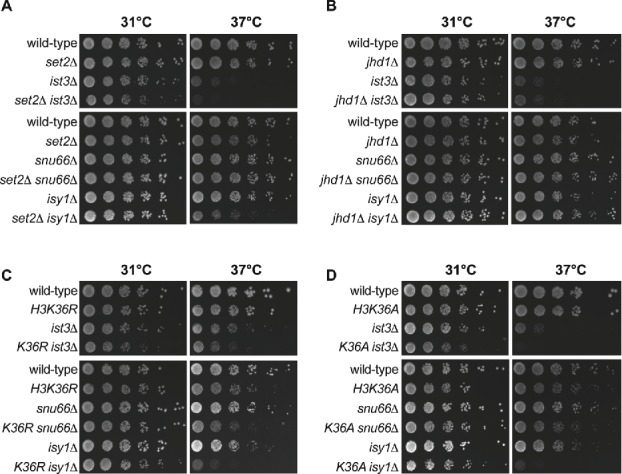
Mutations that alter H3K36 methylation state are genetically implicated in RNA splicing. RNA splicing factors display genetic interactions with SET2 and JHD1 (A and B). Deletion of SET2 combined with a deletion of a gene encoding a splicing factor leads to a range of synthetic growth defects (A). Deletion of JHD1 partially suppresses the growth defect of select strains harboring a deletion of a splicing factor gene (B). (C and D) RNA splicing factors display synthetic genetic interactions with H3K36R (C) and H3K36A (D). Serial dilutions of WT, single and double mutant strains grown on rich media at 31°C and 37°C. Plates photographed after 48 hours growth (note that the ist3Δ 37°C panel was photographed after 72 hours growth).
Our gene expression reporter data and genetic data support a role for Set2 and Jhd1 in pre-mRNA splicing, suggesting that H3K36 methylation state, not just the physical presence of the Set2 or Jhd1 proteins, is important for RNA splicing. To test whether H3K36 methylation underlies the genetic interactions we observed, we generated double mutants that contain both a deletion of a splicing gene and one of 3 point mutations in H3K36 that abolish methylation (H3K36A, H3K36R, or H3K36Q) and monitored their growth phenotypes (Figs. 2C and D, Fig. S4A). As expected, mutations that abolish H3K36 methylation directly (H3K36R and H3K36A; for H3K36Q see Fig. S4A) exhibit negative genetic interactions with ist3Δ and isy1Δ. For example, the H3K36A ist3Δ double mutant has reduced fitness relative to either the H3K36A or ist3Δ single mutation, particularly at 37°C (Fig. 2D, center panel). Like the SET2 deletion, we do not observe negative genetic interactions between the histone point mutations and deletion of MUD1 or MUD2 (Fig. S2B, lower panels). Interestingly, we observed one unexpected genetic interaction that does not phenocopy the SET2 deletion mutant phenotype; H3K36R and H3K36A both exhibit negative genetic interactions with snu66Δ (Figs. 2C and D, lower panels). Combined with the reporter assay, these genetic data provide further evidence for a specific role for H3K36 methylation state in RNA splicing.
H3K36 methylation loss results in gene-specific pre-mRNA splicing defects
The gene expression reporter results presented in Fig. 1 led us to ask whether the inability to methylate H3K36 results in a pre-mRNA splicing defect of endogenous yeast introns. We performed RT-qPCR for a number of introns within yeast strains lacking Set2 and Jhd1, as well as yeast harboring alanine, glutamine, or arginine substitutions at H3K36. The intron-containing genes we analyzed included DBP2, TEF5, and RPS21B, which have previously been analyzed in studies investigating co-transcriptional splicing.14,27-29,64 Yeast were grown at the permissive temperature prior to RNA extraction and the splicing mutant snu66Δ served as a positive control.
The first characteristic of the RT-qPCR results that stands out is the gene-specific nature of the splicing defects between strains (Fig. 3). For example, we observe very little change in pre-mRNA splicing efficiency in the TEF5 intron with H3K36 mutants, yet find nearly a 2-fold increase in the pre-mRNA/total ratio for DBP2 (Fig. 3 and Table S4; p < 0.01 for H3K36A and p < 0.05 for H3K36Q). Additionally for SRB2, we observed a 3-fold splicing defect across all 3 histone point mutants (p < 0.01), yet for RPS21B, we only observed a splicing defect for the glutamine substitution (p < 0.01). The splicing defects we observe are similar in magnitude to those observed when other histone modifications are perturbed.27-30,64 In addition, gene-specific splicing defects in budding yeast have been described previously12-14 and could possibly be a result of features of the gene or intron such as the length of the gene, and/or its transcription frequency. Nevertheless, we do observe a trend of decreased splicing efficiency upon removal of Set2, as compared to the wild-type strain (Fig. 3 and Table S4; p < 0.01 for DBP2, SRB2 and RPS21B). Conversely, when we delete the gene encoding the demethylase Jhd1, a defect is only observed for the DBP2 intron (Fig. 3 and Table S4; p < 0.01), supporting the idea that methylated H3K36 generally supports efficient pre-mRNA splicing, and that dynamic methylation-demethylation is important for at least DBP2 splicing. For the histone point mutants, we observed the strongest overall splicing defects with the glutamine substitution, followed by the arginine and alanine mutations respectively. While the splicing phenotypes of the histone point mutants generally phenocopied the SET2 deletion as anticipated, we observed some differences. For instance, the splicing of DBP2 is inhibited more by deletion of SET2 (>3 fold decrease in splicing; Fig. 3 and Table S4) than the histone point mutants, while the splicing of SRB2 is more severely impacted by mutation of the histone. These differences may be due to reduced dose of H3 and H4 in the histone point mutant strains45 or could indicate that Set2 or the H3K36 lysine residue has an additional role in splicing outside of histone methylation. Collectively, these data support a dynamic role for H3K36me in pre-mRNA splicing.
Figure 3.
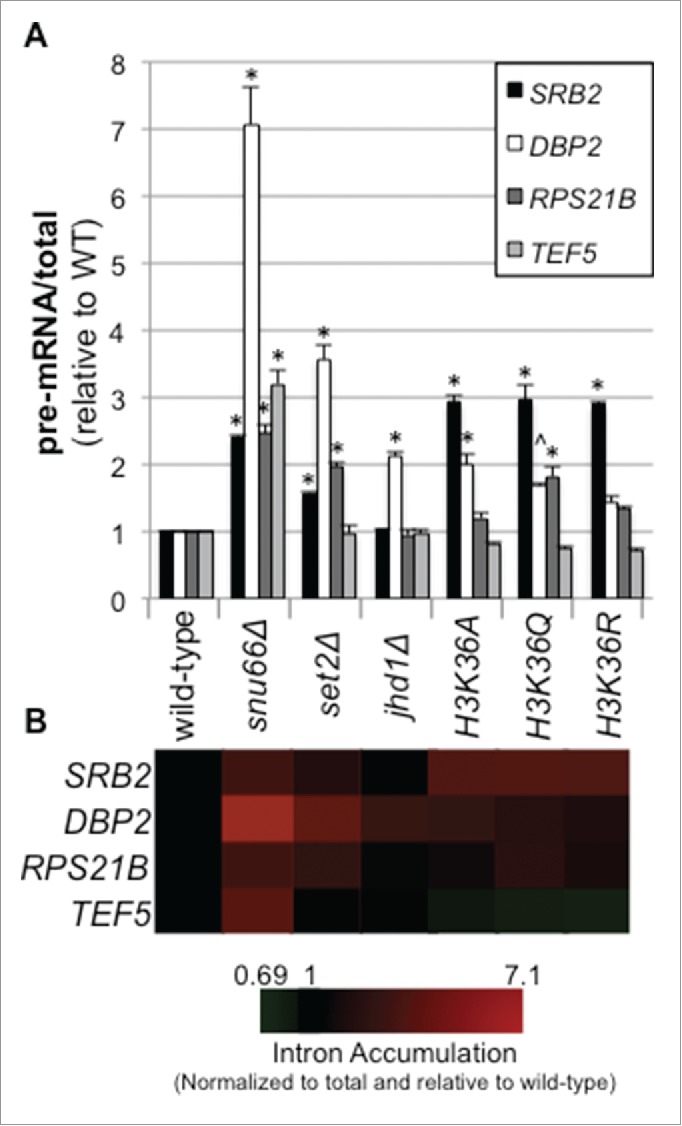
Genetic modulation of H3K36 methylation state results in gene-specific pre-mRNA splicing defects. RNA was harvested from the indicated yeast strains at early/mid log phase. RT-qPCR analysis was performed, determining the pre-mRNA and total levels of the indicated intron containing transcripts. Reported values were calculated using a standard curve calculations and are relative to wild-type ratio; ANOVA p-values and Tukey p-values for gene-specific pair-wise strain comparisons reported in Table S4 (A). The data represent biological triplicates and error bars represent one standard error of measurement. * indicates p <0.01 and ˆ indicates p<0.05 when comparing pre-mRNA/total in the given mutant strain to wild-type. For other pair-wise comparisons see Table S4 (B) The data are also shown in heat map form using Cluster and TreeView.
H3K36 methylation loss exacerbates the splicing defects found in pre-mRNA splicing mutants
Based on the results of the genetic analyses we predicted that deletion of SET2 or point mutation of H3K36 would exacerbate the splicing defect observed in strains lacking splicing factor genes. We tested this for a subset of our double mutant strains, focusing on those that had the most pronounced growth defects, first using the fluorescent gene expression reporter (Fig. S3). As expected from our genetic analyses, deletion of SET2 enhances the splicing defect observed in the isy1Δ strain (double mutant shifted toward the mCherry-axis compared to singles) and suppresses the splicing defect in the snu66Δ strain. Additionally, the H3K36A and H3K36R mutation exacerbates the splicing defect in isy1Δ, lea1Δ, snu66Δ, bud13Δ and ist3Δ (Fig. S3).
We expanded our analysis to endogenous pre-mRNAs using RT-qPCR to monitor splicing efficiency (Fig. 4). The splicing phenotypes, in general, mirror those observed using the reporter, although there are some gene-specific effects. Consistent with results using the reporter, set2Δ enhanced the splicing defect observed in the isy1Δ strain (e.g., SRB2 and RPS21B splicing; p < 0.01; Table S5), and strikingly, suppressed the splicing defect observed in the snu66Δ strain for all 4 of the pre-mRNAs monitored (Fig. 4A and Table S5; p < 0.01). Deletion of JHD1 also has gene-specific effects, but notably improved the splicing of DBP2 and TEF5 in the ist3Δ strain (Fig. 4B and Table S5; p < 0.01). Additionally, we observe exacerbated splicing defects for SRB2 and RPS21B in the jhd1Δ snu66Δ strain (Fig. 4B and Table S5; p < 0.01 and < 0.05, respectively), as predicted based on our genetic results.
Figure 4.
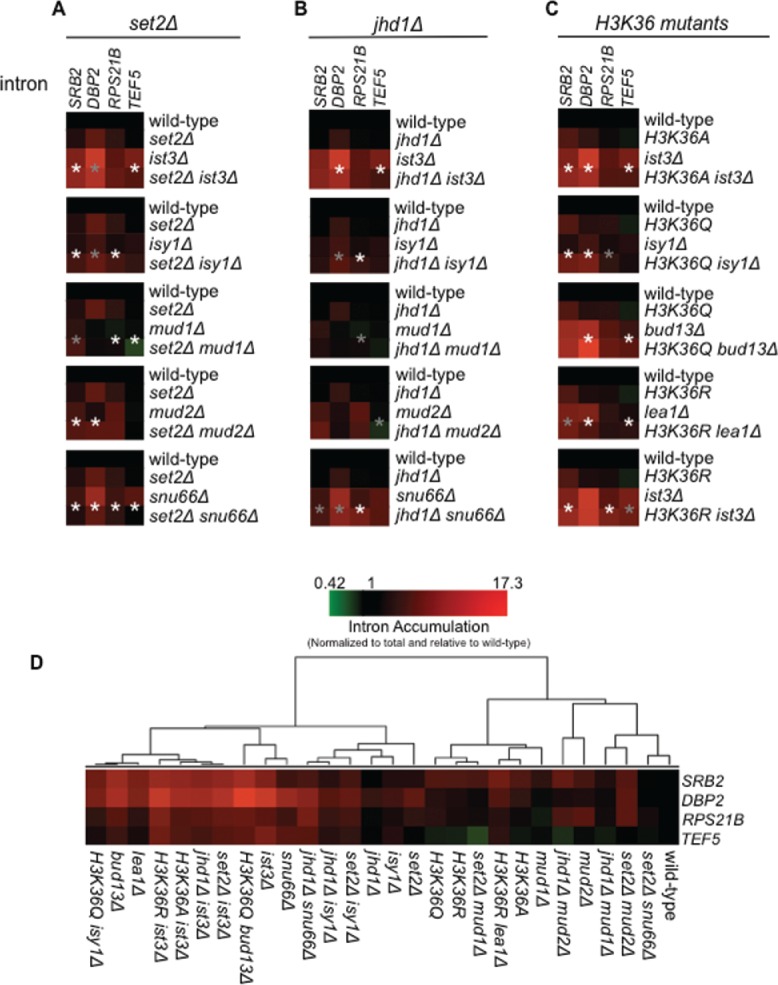
Synthetic-genetic effects on the splicing of endogenous introns. Heat map representation of RT-qPCR data for the strains designated to the right of each heat map for the introns labeled on the top (A). Splicing efficiency/intron accumulation was calculated as a ratio of pre-mRNA/total relative to wild-type and described on a green to red color scale, with black being set at a value of 1.0 (equal with wild-type). Values less than one indicate increased splicing efficiency while values greater than 1 indicate a splicing defect. Values represent the average from 3 biological replicates. Legend shown in the bottom right reflects the colors and values. ANOVA p-values and Tukey p-values for gene-specific pair-wise strain comparisons reported in Table S5 (B). Asterisks represent the p-values for the comparison of the splicing factor deletion mutant to the double mutant (i.e., ist3Δ versus set2Δ ist3Δ; gray asterisk indicates p < 0.05. white asterisk indicates p < 0.01). (B) Hierarchical clustering of the RT-qPCR data for all the strains is shown.
The H3K36 mutations also exacerbated the splicing defects observed in several splicing mutant strains, including ist3Δ, bud13Δ, lea1Δ and isy1Δ (Fig. 4C). The severity of the splicing defect is dependent on the pre-mRNA and the splicing mutants examined. For example, H3K36R worsened the splicing defect of RPS21B in the ist3Δ strain (p < 0.01), but does so to a much lesser degree in the lea1Δ strain (Fig. 4C and Table S5). TEF5 splicing is affected in a slightly different manner. H3K36 mutations exacerbate splicing of TEF5 in bud13Δ and lea1Δ strains (Fig. 4C and Table S5; p < 0.01). On the other hand the TEF5 splicing defect is partially suppressed when H3K36 is mutated in an ist3Δ background (Fig. 4C and Table S5; p < 0.05). We also clustered all the RT-qPCR data, similar to microarray data analysis, to observe the overall behavior and relationship of the double mutants (Fig. 4D). Strikingly, the deletion of set2Δ in the snu66Δ strain results in a major change of clustering behavior compared to snu66Δ, moving from a group with the strongest splicing defects all the way back to baseline (wild-type), demonstrating the strong suppression once again. Although there are exceptions, the data support a general theme of exacerbated splicing defects in splicing factor/H3K36me double mutants. These data strongly support the idea that H3K36 methylation has gene-specific effects on RNA splicing.
Association of Set2 with the transcribing Polymerase and H3K36 dimethylation is required for efficient mRNA splicing
We next wanted to determine what aspects of Set2 function were responsible for the observed splicing defects. Using a full-length expression construct of SET2, driven by its endogenous promoter65 we found that exogenous SET2 rescues splicing defects in a set2Δ strain (Fig. 5A and 5B and Table S6). In contrast, a catalytic mutant of Set2 incapable of H3K36me2/3 that also significantly reduces H3K36me166 (set2 H199L) was not able to rescue the splicing defect – thus demonstrating that Set2 activity is necessary for regulated splicing in yeast. Surprisingly, using a mutation in the SET domain of Set2 (set2 R195C) that diminishes H3K36me3 without altering H3K36me1/2, we found most of the splicing phenotypes observed in set2Δ could be suppressed, with the exception of the DBP2 transcript (Fig. 5B). This observation suggests that H3K36me3 is mostly dispensable for splicing in S. cerevisiae, a scenario that is different from human cells where H3K36me3, catalyzed by SETD2, is a critical regulator of splicing.31-34 However, we note it is formally possible that H3K36me1 may contribute to some splicing functions, as the H199L mutant still contain H3K36me1. Nevertheless, our data suggests that H3K36me2 largely drives most of the splicing phenotypes.
Figure 5.
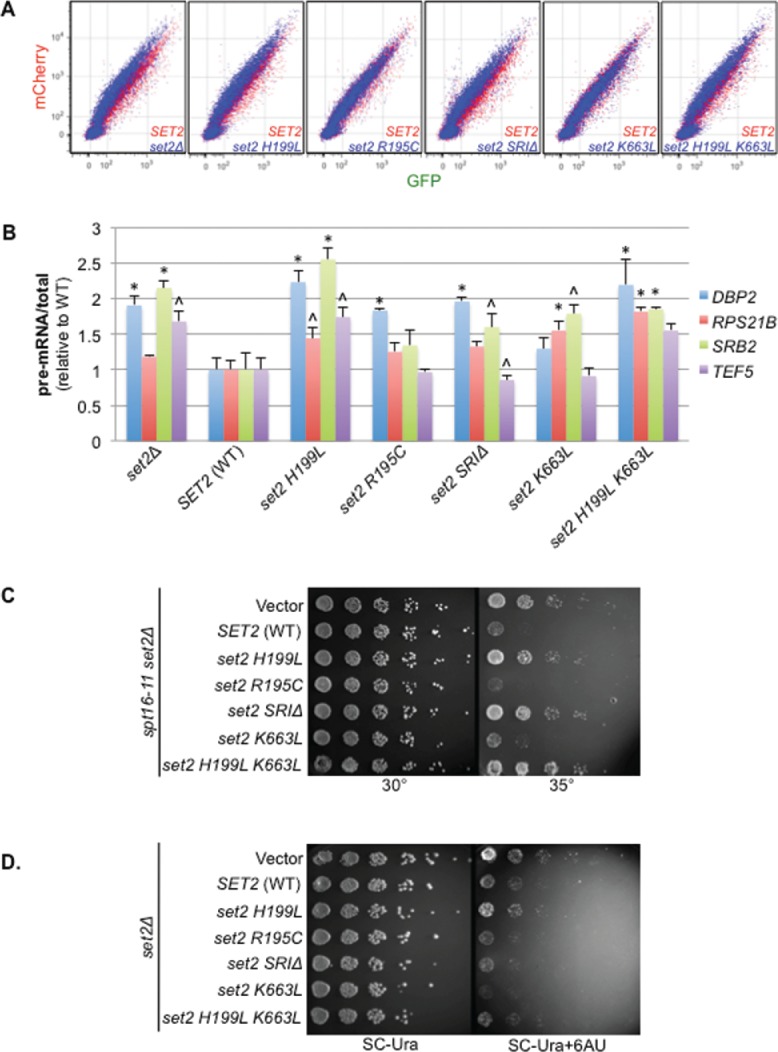
Identification of pre-mRNA splicing and transcription elongation defects in set2 mutants. (A). Expression of the gene expression reporter in a set2Δ strain carrying plasmids with set2 mutants (blue) compared to set2Δ covered by a wild-type SET2 plasmid (red). A shift towards the mCherry axis is indicative of a pre-mRNA splicing defect. (B) RT-qPCR analysis of the levels of four endogenous introns. Values shown are the ratio of pre-mRNA to total and are made relative to the wild-type (WT) SET2 strain. The average of 3 biological triplicates is used and error bars represent one SEM; ANOVA p-values and Tukey p-values for gene-specific pair-wise strain comparisons reported in Table S6. * indicates p < 0.01 and ˆ indicates p<0.05 when comparing pre-mRNA/total in the given mutant strain to SET2 (WT) strain. For other pair-wise comparisons see Table S6. (C and D) Improper transcription elongation in a set2Δ does not explain all the splicing defects observed in a set2Δ. Five-fold serial dilution of indicated strains were spotted and incubated at higher temperature (C) or spotted on 6-AU (transcription elongation inhibitor) (D) to monitor how various mutations of Set2 influence set2Δ phenotypes. Catalysis by Set2 is important for both resistance to 6-AU and suppressing the bypass of spt16-11, while interaction with RNAPII is dispensable for suppressing 6-AU resistance.
Intriguingly, the splicing defects observed in set2Δ were dependent on both the SRI domain in Set2, which couples it to the transcribing polymerase (set2 SRIΔ), and on H3K36me2 (set2 H199L and set2 R195C; Fig. 5B and Table S6). Another mutant in the SRI domain, K663L, predicted to disrupt Set2-RNA Pol II interaction (Hacker et. al., submitted for publication) did not result in significant alteration in the H3K36 methylation status, however this mutation does cause a decrease in the splicing of the several of the endogenous pre-mRNAs (Figs. S4B and 5B and Table S6). Furthermore, we noticed that the combined set2 H199L K663L mutant, which has lost all forms of methylation (Fig. S4B), is equally as incompetent in rescuing the splicing defect as the set2 H199L mutant (Fig. 5B), strongly suggesting that splicing in yeast is dependent, to a large extent, on H3K36me2 and a functional SRI domain (i.e., Set2-RNA Pol II interaction). Collectively, our findings show that catalysis and interaction of Set2 with RNA Pol II is critical for proper mRNA splicing.
Defective transcription elongation in set2Δ does not completely explain Set2-dependent splicing defects
Set2/H3K36me has been shown to impact chromatin structure through the regulation of histone exchange and activation of deacetylases.67 Given the intimate connection between transcription elongation and splicing,8,10,11,21,22 we next wondered if the splicing defects observed in the set2Δ strain were a consequence of alterations in transcriptional elongation. To answer this question, we took advantage of 2 well-established transcriptional elongation defects observed in the set2Δ: bypass of a FACT chromatin-reorganizing complex mutant, spt16-11,68 at semi-permissive temperature and resistance to the transcription elongation inhibitor 6-Azauracil (6-AU). Wild-type yeast display growth defects when treated with 6-AU or when the positive transcription regulator Spt16 is mutated (spt16-11; see Figs. 5C and 5D), indicative of a transcription elongation defect. We and others have shown that set2Δ bypasses the slow growth phenotype of spt16-11 at semi-permissive temperature (of 35°C) 68,69 and that set2Δ is resistant to 6-AU.36
Since FACT is a positive regulator of transcription elongation, and 6-AU results in elongation defects, bypass of spt16-11 mutant and resistance to 6-AU suggests Set2 as a negative regulator of transcription elongation. Consistent with this notion, loss of SET2 results in hyper-acetylation in open reading frames,39,40 thereby resulting in more open chromatin and more nascent transcription.3 Using the set2 mutants described above, we attempted to identify mutations that impact transcription elongation by testing for rescue of the transcriptional elongation phenotypes observed in set2Δ in the presence of 6-AU and in the spt16 mutant allele. Full-length SET2 but not the catalytic mutant (set2 H199L) or set2 SRIΔ was able to suppress the set2Δ phenotypes66 (Figs. 5C and 5D). In this scenario we also observed that the set2 H199L K663L double mutant behaved like the set2 H199L mutant, suggesting that the driver for the observed phenotypes are H3K36me2. These data suggest that both the SRI domain and H199L are critical for the negative regulation of transcription elongation by Set2. We also tested whether the set2 mutations implicated in transcription elongation altered RNA splicing. Both the set2 SRIΔ and set2 H199L reduce the splicing efficiency of endogenous pre-mRNAs (Fig. 5B and Table S6), consistent with a model in which transcription elongation and RNA splicing are coupled. Notably, the set2 R195C mutant, which selectively inhibits H3K36me3 (Fig. S4B), and the set2 K663L mutant are both 6-AU sensitive and cannot suppress the spt16-11 growth defect. These data suggest that H3K36me3 and certain set2 mutants (specifically set2 K663L) are dispensable for Set2-mediated negative regulation of transcriptional elongation. Surprisingly, we found the splicing of DBP2 is reduced in the set2 R195C mutant compared to the wild-type strain (Fig. 5B and Table S6; p < 0.01), and that the set2 K663L mutation results in splicing defects for the SRB2 and RPS21B when compared to a wild-type strain (p < 0.05 and p < 0.01, respectively). These data suggest that the splicing defects observed for these pre-mRNAs are not solely due to alteration of elongation alone. Taken together, these data indicate that in addition to the canonical function of regulating chromatin structure during transcription elongation, Set2-dependent H3K36 methylation regulates splicing, in a manner that may be uncoupled from its role in transcription elongation.
Deletion of SET2 reduces association of the U2 and U5 snRNPs with pre-mRNA during transcription
Our genetic analyses support the notion that the splicing effects of a SET2 deletion are not simply due to a defect in transcription elongation, which could have been possible since changes in the kinetics of elongation are known to impact splicing efficiency.8,10,11,21,22 Importantly, our observation is reinforced by studies from the Struhl lab that show Set2 loss has no effect on RNA Pol II elongation rate or processivity.70 Given that Set2/H3K36me2 is likely functioning directly in splicing, we reasoned that Set2 and/or H3K36 methylation might be important for recruiting splicing proteins to facilitate co-transcriptional spliceosome assembly10,11 (i.e., a recruitment model).
To directly test whether Set2 helps in recruiting RNA splicing proteins during transcription, we monitored the association of Prp42-HA (U1 snRNP), Lea1-HA (U2 snRNP) and the Snu114-HA (U5 snRNP) on chromatin in a set2Δ strain by chromatin immunoprecipitation (ChIP). Previous studies indicate that all 3 of these HA-tagged proteins can associate with pre-mRNA co-transcriptionally.5-8 We specifically monitored association of these snRNPs with the DBP2 pre-mRNA, which is the only pre-mRNA that we identified as being H3K36me3 dependent, RPS21B pre-mRNA, which is the only pre-mRNA that relies heavily on Set2 interaction with RNA Pol II for splicing, and SRB2, which requires H3 mono- and di- methylation for splicing (Fig. 6A). Wild-type and set2Δ cells were grown at the permissive temperature then shifted to 37°C for 30 minutes prior to ChIP analysis as we observed some of our strongest genetic interactions between SET2 and splicing proteins at 37°C. We used RT-qPCR to monitor splicing defects of the pre-mRNAs in the set2Δ strain at 37°C. (Fig. S5B). Importantly, deletion of SET2 does not appreciably alter the protein levels of the HA-tagged splicing factors used in this analysis (Fig. S5A) nor does it cause a significant change in the expression levels of other splicing proteins.71 Utilizing antibodies that recognize either the HA-tag found in the splicing proteins or the Rpb3 subunit of RNA Pol II, we found that deletion of SET2 resulted in a significant decrease in association of Lea1-HA in both the intron regions of the RPS21B and SRB2 genes (p < 0.05) as well as the some of the exon regions of the pre-mRNA (Fig. 6B). The RNA Pol II association with the RPS21B gene was lowered by less than 20% in the set2Δ strain compared to wild-type Lea1-HA strain (p < 0.05), but it is unlikely that this small reduction alone can account for the nearly 50% reduction in association of Lea1-HA with the pre-mRNA. We were unable to detect any significant reduction in RNA Pol II association with the SRB2 gene. RNA Pol II association with the DBP2 increased in exon region of the gene (p < 0.05) in the set2Δ strain, which is consistent with a previous report of RNA Pol II accumulation at the 3′ end of the SCC2 gene in the absence of SET272 (Fig. 6B). An increase in RNA pol II association might be due to a pause in RNA Pol II induced by SET2 deletion. Interestingly, according to a kinetic coupling model RNA Pol II pausing could allow time for enhanced snRNP association,10,11 however, we observe a modest albeit insignificant reduction in the association of Lea1-HA along the DBP2 gene (Fig. 6B).
Figure 6.
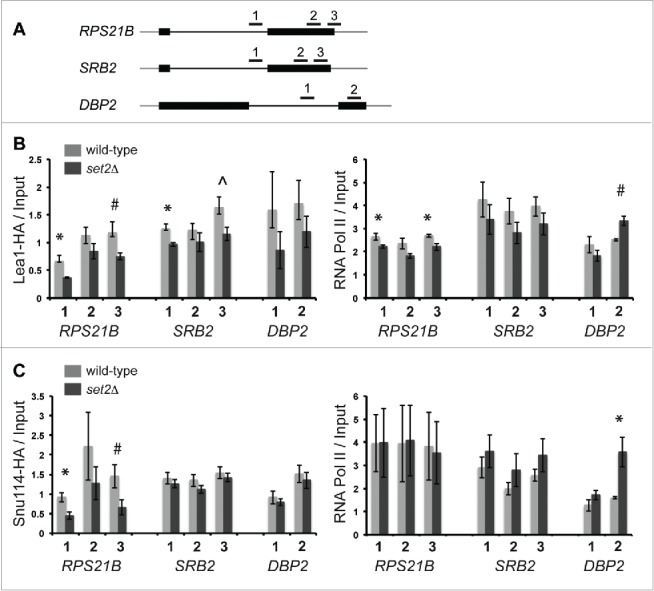
Deletion of SET2 impacts the recruitment of the U2 snRNP and U5 snRNP. Schematic diagram of the RPS21B, SRB2 and DBP2 genes and the primer sets used in the chromatin immunoprecipitation (ChIP) assays (A). Chromatin immunoprecipitations were carried out using α-HA or α-RNA pol II antibodies in a Lea1-HA strain strain (“wild-type”) or a Lea1-HA set2Δ strain (set2Δ) (B) or Snu114-HA (“wild-type”) or a Snu114-HA set2Δ strain (set2Δ) (C) strain grown at 30°C then shifted to 37°C for 30 min. Shown are the average of HA-tagged protein or RNA polymerase bound relative to input at the indicated regions (note that each IP sample was normalized to an averaged relative amount for an intergenic region). Error bars represent +/− SEM for each strain and primer set, n = 3–4 biological replicates (see Materials and Methods for details). * indicates p < 0.05 and ˆ and # indicate approaching significance (ˆ p < 0.07, # p < 0.09) comparing the set2Δ strain to wild-type.
U5 snRNP association with certain pre-mRNAs is also impacted by deletion of SET2. We observed a significant decrease in the association of Snu114-HA with the RPS21B gene (Fig. 6C; p < 0.05 for the intron amplicon), and importantly we see no change in RNA Pol II association in this strain compared to WT (Fig. 6C). U5 association with SRB2 or DBP2 is not significantly impacted by deletion of SET2. We do not observe a decrease in the association of U1 snRNP with chromatin in the set2Δ strain (Fig. S5C and S5D), consistent with a recent study.29 We therefore conclude that while Set2 may impact splicing of some pre-mRNAs by altering elongation rate, Set2 also plays a more direct role in U2 and U5 snRNP recruitment to pre-mRNAs during transcription to promote co-transcriptional spliceosome assembly.
Discussion
We utilized a fluorescent gene expression reporter to reveal particular histone residues and modifications that are important for pre-mRNA splicing. We focused on a specific role for H3K36 methylation, as both the loss of SET2 and mutation of H3K36 resulted in splicing defects, thus implying a direct function of Set2 in the RNA splicing process. We show that mutations altering H3K36 methylation genetically interact with splicing factors and are required for the splicing of a subset of introns in yeast. Moreover, we demonstrate that Set2 functions to promote RNA splicing by facilitating co-transcriptional recruitment of the U2 and U5 snRNPs to nascent RNA. Furthermore, our work reveals that splicing depends on the association of Set2 with the transcribing polymerase, and is particularly sensitive to the specific H3K36 methylation state. To our knowledge, this is the first report of transcript-linked H3K36me requirement in RNA splicing in yeast – thus implying a universal conservation of the linked role of H3K36me in this event.
Our gene expression reporter screen identified a variety of histone residues important for splicing efficiency. With the interpretation that the phenotypes observed are mediated through the loss of the histone modifications normally found at these residues, our work confirmed roles for H3 acetylation by Gcn5,27,28 H2BK123 ubiquitination by Bre1,29,30 and support a role for the previously identified modification enzymes Set1, Set229 and Bdf1 and Vps7264 in RNA splicing. Importantly, we uncovered novel players in splicing regulation. For example, we determined that the H4K16A and H4K12Q mutations in addition to H3K36 mutations also impact RNA splicing. H4K16 and H4K12 acetylation is enriched in transcriptionally active regions of eukaryotes and plays a role in regulating chromatin organization and dynamics.24,73
Previous work had identified a role for Set2 in splicing in yeast,29 however it was not determined whether this role was mediated through H3K36me (or specific methylation states of H3K36) nor was the underlining mechanism elucidated. Here we demonstrate that mutation of the H3K36 to A, R or Q causes a defect in RNA splicing and exacerbates the growth defect and splicing defect in yeast strains harboring deletions of splicing factors, thus demonstrating a role for methylation of H3K36 in RNA splicing. Moreover, our studies revealed that splicing is dependent on H3K36 di-methylation. H3K36 can also be acetylated by Gcn5.74 However our results indicate that the H3K36Q acetylation-mimic mutant has defects in RNA splicing demonstrating that acetylation of H3K36 is not required for the splicing of the pre-mRNAs used in this study. It is conceivable that the role of H3K36me2 in splicing is directly connected to the ability of this mark to maintain a suppressed chromatin environment through the recruitment of Rpd3S and Isw1b.37-41 Indeed, deletion of RPD3, which encodes a member of the Rpd3 complex,75-77 also leads to modest splicing defects, which could imply that histone deacetylation and chromatin remodeling (i.e., creation of a particular chromatin environment) plays a key role in snRNP recruitment. Another possibility is the potential role of H3K36 methylation in affecting some aspect of RNA Pol II elongation or termination. Indeed, recent studies in yeast show that slowing elongation improves pre-mRNA splicing efficiency and that fast elongation impairs splicing.8,21,22 Further, impaired termination has also been shown to impact proper splicing in metazoan.78 This possibility seems less likely as a recent report revealed that deletion of SET2 had no impact on RNA Pol II elongation or processivity.70 The ultimate consequence of Set2/H3K36me2 on some aspect of transcription termination that might impinge directly on the recruitment of splicing machinery to chromatin is currently unknown. As we showed that Set2 was required for the association of U2 and U5 on chromatin (Fig. 6), it may be that these splicing factors bind directly to H3K36-marked nucleosomes or the chromatin structure generated by H3K36me. The idea of direct methyl binding is consistent with recent studies in mammals showing that H3K36me3 by SETD2 is recognized by adapter proteins, which in turn recruits the U2 snRNP or splicing regulatory proteins to pre-mRNA to modulate alternative RNA splicing.31-33 Whether Set2 works directly to recruit the snRNPs via H3K36me or altered chromatin structure remains an interesting question for future studies.
Our biochemical data also showed that Set2 is not required for recruitment of the U1 snRNP (Fig. S5D), which is consistent with a recent report,29 and indicates that Set2 functions to affect splicing downstream of U1 binding. Our genetic data agree with this model. For example, deletion of SET2 is epistatic with deletion of MUD1, which encodes a U1 snRNP component (Fig. S2). Notably, we observe negative genetic interactions between mutations that abolish H3K36 me (SET2 and H3K36A/R) and IST3 (Fig. 2) and LEA1 (not shown), components of the U2 complex,56,79 as well as deletions of factors required for later steps of splicing (Figs. 2). One intriguing exception is that deletion of SNU66, a component of the U4/U6•U5 tri-snRNP,46,47 has positive genetic interactions with SET2 (Fig. 2), suggesting that deletion of SET2 introduces a stall in RNA splicing that can be bypassed when SNU66 is deleted. Thus, Snu66 function in splicing and transcription may extend beyond that of the other canonical splicing factors. Taken together our genetic data suggest that Set2 and H3K36me function to modulating splicing steps downstream of U1 snRNP binding, and support a model in which Set2 aids in the co-transcriptional association of the U2 and U5 snRNPs with RNA. Recruitment of U2 snRNP appears to be a common mechanism by which histone modification impacts RNA splicing in both yeast and metazoa. The Gcn5 histone acetylase and H3 acetylation is important for recruiting the U2 snRNP in yeast27,28 and in metazoa, H3K4 methylation by Set1 methyltransferase as well as H3K36me3 by SETD2, both function to recruit the U2 snRNP co-transcriptionally.31,33,35
Interestingly, in yeast, there appears to be a requirement for a cycle of histone modification and subsequent removal of the histone modification for efficient RNA splicing. For example, deletion of the Bre1 E3 ubiquitin ligase, as well as deletion of Ubp8 deubiquitinase, which both modulate H2B ubiquitination levels, cause a reduction in RNA splicing.30 Furthermore, H3 acetylation by Gcn5 and deacetylation by Hos2/3 are required for RNA splicing.27,28 Here we show that deletion of the H3K36me1/2 demethylase Jhd1 has modest positive genetic interactions with splicing factor genes (e.g.,, jhd1Δ ist3Δ; Fig. 2) and improves splicing of some introns in strains lacking splicing factor genes (e.g., DBP2 splicing in the jhd1Δ ist3Δ; Fig 4A), suggesting that Jhd1 might normally negatively regulate RNA splicing. However, we also observe that deletion of JHD1 alone causes an inhibition of DBP2 splicing when compared to a wild-type strain (Fig. 3), and it exacerbates the splicing of DBP2 in an isy1Δ strain (Fig. 4A), suggesting that Jhd1 can also stimulate splicing. Thus, while a clear role for Jhd1 in splicing remains to be resolved, it appears that demethylation of H3K36 is also required for the splicing of some endogenous transcripts, indicating that the dynamic cycle of H3K36 methylation and demethylation might also be required for RNA splicing.
A model in which histone modification and RNA splicing are tightly coordinated is rapidly emerging, and leaves open the exciting possibility that coordination may be bidirectional and that RNA splicing can influence histone modification, thus changing the epigenetic signature along a gene. Indeed, recent reports in metazoa have shown that perturbation of RNA splicing by either the deletion of the BPS or 3′SS or using the splicing inhibitor spliceostatin A, alters the pattern of distribution of H3K36me3, shifting it away from the intron region toward the 3′ end of a gene.80 In addition, a second study found that perturbing splicing using the splicing inhibitor meayamycin or by inhibiting splicing by knocking down the splicing factor SAP130 lead to a decreased association of both H3K36me3, as well as SETD2, with intron contain regions of chromatin.81 Whether or not splicing in yeast enhances the recruitment of Set2 or the pattern of H3K36me to reciprocally couple RNA splicing with histone modification remains to be determined.
Our study also uncovered novel, specific roles for different methylation states of H3K36 in yeast pre-mRNA splicing. Both mono and di-methylation of H3K36 are required for the efficient splicing of all of the transcripts that we analyzed, as well as the gene expression reporter (Fig. 5A and 5B; see set2 H199L). However, tri-methylation is only required for the efficient splicing of one of the 4 endogenous introns surveyed (DBP2 in set2 R195C; Fig 5B). In higher eukaryotes, H3K36me3 is enriched in exons relative to introns,25 and is implicated in alternative splicing.31-34 In yeast, the extent of H3K36me required for RNA splicing appears to be transcript specific and genome-wide studies will be necessary to determine the subsets of RNAs requiring specific modification states. In addition, it will be important to determine whether H3K36 mono- or di-methylation are required for splicing in metazoa. With the availability of new Drosophila histone point mutant platforms,43 it will be of interest to directly address the role of H3K36me in regulating canonical and alternative splicing in a metazoan.
Materials and methods
Yeast strains and plasmids
For a list of the strains used in this study please see Table S1 and for a description of the methods used to create new strains for this study please see the Supporting Methods section. Please refer to Table S2 for a list of plasmids used in this study.
Qualitative growth assays
Yeast cultures were grown to mid-log (OD585 0.4-0.8) and diluted to OD585 0.1, then further diluted by serial 5-fold dilutions, spotted on YEPD plates and incubated at 31°C or 37°C for 2-3 days.
Flow cytometry
Log-phase yeast harboring the reporter were fixed, washed and resuspended in 1X PBS prior to analysis with a Becton Dickinson LSRFortessa. For excitation of eGFP and mCherry, 488 nm and 561 nm lasers were used respectively. For detection a 505 LP and 530/30 BP were used for eGFP and a 600 LP and 610/20 BP for mCherry. For each sample 21,000 events were collected and 10,000 events are shown in the phenographs. Analysis and visualization was completed with FlowJo. Details on the binning and clustering analysis are previously described.44
RT-qPCR
A full description of these methods have been previously described.44 Briefly, log phase cells were harvested and total RNA was extracted using standard acid/phenol extraction using Phase Lock tubes (5 PRIME). RNA (5 µg) was DNase treated (5 U RQ1 DNase, Promega) and purified (RNA Clean and Concentrator kit; Zymo). For the RT reaction the following was added to RNA; 10X RT Buffer (0.5 M Tris-HCl pH 8.5) and random nonamer primers (25 µM; IDT). After annealing a RT master mix (1X RT Buffer, 3 mM MgCl2, 10 mM DTT, 0.5 mM dNTPs and 5 U/µl MultiScribe RT enzyme) was added. The RT reaction was incubated overnight at 42°C and the resulting cDNA was diluted. Each qPCR reaction contained 5 µl cDNA, 7.5 µl Power SYBR Green PCR Master Mix (ABI) and 2.5 µl primers (1.5 µM each; see above for sequences). Standard thermocycling, fluorescence detection and analysis (standard curve) were completed using the ViiA 7 Real-Time PCR System (Life Technologies). For the RT-qPCR shown in Fig. S5B, indicated yeast strain cultures were grown at 31°C to OD585 between 0.25-0.4 and shifted to 37°C for 30 min prior to cDNA synthesis. The percent unspliced RNA = relative copies intron/relative copies Exon2 × 100 (Fig. S5B). Primers are indicated in Table S2. Error bars represent one SEM. We tested for strain differences in splicing efficiency of each pre-mRNA within each yeast strain using ANOVA, with yeast strain as the independent factor, followed by Tukey tests for pair-wise differences between strains (ANOVA and Tukey p-values reported in Table S4, S5, and S6). Raw data were square root transformed in order to meet the assumption of homogeneous variances.
Transcriptional assays
Briefly, cells with indicated genotypes were grown overnight either in YPD or in selective medium. Saturated cultures were serially diluted (1:5) plated on relevant plates (YPD plates for spt16-11 strains; 6-AU containing plates for set2 mutants) and pictures were obtained after 2-3 days.
Site directed mutagenesis
Point mutants in FL Set2 were made using the Quikchange Site directed mutagenesis kit (from Agilent) as per manufacturer's directions. The primers sequences have been provided in Table S3.
Chromatin immunoprecipitation
ChIPs were performed as previously described,82 with the following modifications. Yeast cultures were grown at 31°C to OD585 between 0.25-0.4 and shifted to 37°C for 30 mins. The chromatin fraction was sonicated in a QSonia Q800 at intervals of 30sec. sonication, 20 sec. rest for 70 mins, followed by centrifugation at 4°C at 10,000 RCF for 10 mins. Protein G beads (10 μL; GE Healthcare) were pre-incubated with 3.5 μL of either α-Rbp3 (Neoclone W0012) or 7.5 μL α-HA (Roche 12CA5, Cat #11583816001) for 2 hrs at 4°C, then washed with1 ml Lysis buffer plus protease inhibitors (100 μg/ml aprotonin, leupeptin, antipain, and 200 μM PMSF). IPs were incubated for 2 hrs at 4°C. DNA pellets were resuspended in 50 μL TE. The total DNA was diluted 1/320 and the IP DNA was diluted 1/20. DNA was analyzed by qPCR using an Stratagene Mx3000P. Each 50 μL PCR reaction was assembled as above with 20 μL DNA (primers are listed in Table S3). A standard curve was generated using yeast genomic DNA. Each IP sample was run in duplicate, the calculated relative amounts were averaged and normalized to an averaged relative amount for an intergenic region (indicated in Table S3). Each IP was then normalized to the average total input sample; values are reported in Fig. 6. Note that the values reported in Fig. S5D are the averaged α-HA IP/α-RNA pol IP. In Fig. 6, for Lea1-HA ChIPs n = 3 biological replicates for DBP2 and SRB2 and n = 4 for RPS21B and for Snu114-HA ChIPs, n = 4 for DBP2 and n = 3 for RPS21B and SRB2. In Fig. S5, for the Prp42-HA ChIPs n = 3 for both Rps21b and Dbp2. Error bars represent SEM. The p-values reported were determined using 2-tailed student t-tests.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the members of the Kress, Stevens and Strahl laboratories and D. Cameron for helpful discussion and L. Butler for assistance with the statistical analyses. This work was supported by grants to S.W.S by the American Cancer Society (RSG-05-137-01-GMC), National Institutes of Health (GM084246) and a training fellowship for M.R.S from the Cancer Prevention & Research Institute of Texas (RP101501). T.L.K was supported by a Cottrell College Science Award from the Research Corporation for Science Advancement (#20186). This work was also supported by a grant from the National Institutes of Health (GM110058) to B.D.S. The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors do not claim any conflicts of interest.
References
- 1.Wahl MC, Will CL, Luhrmann R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009; 136:701-18; PMID:19239890; http://dx.doi.org/ 10.1016/j.cell.2009.02.009 [DOI] [PubMed] [Google Scholar]
- 2.Carrillo Oesterreich F, Preibisch S, Neugebauer KM. Global analysis of nascent RNA reveals transcriptional pausing in terminal exons. Mol Cell 2010; 40:571-81; PMID:21095587; http://dx.doi.org/ 10.1016/j.molcel.2010.11.004 [DOI] [PubMed] [Google Scholar]
- 3.Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 2011; 469:368-73; PMID:21248844; http://dx.doi.org/ 10.1038/nature09652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexander RD, Innocente SA, Barrass JD, Beggs JD. Splicing-dependent RNA polymerase pausing in yeast. Mol Cell 2010; 40:582-93; PMID:21095588; http://dx.doi.org/ 10.1016/j.molcel.2010.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kotovic KM, Lockshon D, Boric L, Neugebauer KM. Cotranscriptional recruitment of the U1 snRNP to intron-containing genes in yeast. Mol Cell Biol 2003; 23:5768-79; PMID:12897147; http://dx.doi.org/ 10.1128/MCB.23.16.5768-5779.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gornemann J, Kotovic KM, Hujer K, Neugebauer KM. Cotranscriptional spliceosome assembly occurs in a stepwise fashion and requires the cap binding complex. Mol Cell 2005; 19:53-63; PMID:15989964; http://dx.doi.org/ 10.1016/j.molcel.2005.05.007 [DOI] [PubMed] [Google Scholar]
- 7.Lacadie SA, Rosbash M. Cotranscriptional spliceosome assembly dynamics and the role of U1 snRNA:5′ss base pairing in yeast. Mol Cell 2005; 19:65-75; PMID:15989965; http://dx.doi.org/ 10.1016/j.molcel.2005.05.006 [DOI] [PubMed] [Google Scholar]
- 8.Lacadie SA, Tardiff DF, Kadener S, Rosbash M. In vivo commitment to yeast cotranscriptional splicing is sensitive to transcription elongation mutants. Genes Dev 2006; 20:2055-66; PMID:16882983; http://dx.doi.org/ 10.1101/gad.1434706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tardiff DF, Lacadie SA, Rosbash M. A genome-wide analysis indicates that yeast pre-mRNA splicing is predominantly posttranscriptional. Mol Cell 2006; 24:917-29; PMID:17189193; http://dx.doi.org/ 10.1016/j.molcel.2006.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bentley DL. Coupling mRNA processing with transcription in time and space. Nat Rev Genet 2014; 15:163-75; PMID:24514444; http://dx.doi.org/ 10.1038/nrg3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell 2011; 144:16-26; PMID:21215366; http://dx.doi.org/ 10.1016/j.cell.2010.11.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pleiss JA, Whitworth GB, Bergkessel M, Guthrie C. Rapid, transcript-specific changes in splicing in response to environmental stress. Mol Cell 2007; 27:928-37; PMID:17889666; http://dx.doi.org/ 10.1016/j.molcel.2007.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pleiss JA, Whitworth GB, Bergkessel M, Guthrie C. Transcript specificity in yeast pre-mRNA splicing revealed by mutations in core spliceosomal components. PLoS Biol 2007; 5:e90; PMID:17388687; http://dx.doi.org/ 10.1371/journal.pbio.0050090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kress TL, Krogan NJ, Guthrie C. A single SR-like protein, Npl3, promotes pre-mRNA splicing in budding yeast. Mol Cell 2008; 32:727-34; PMID:19061647; http://dx.doi.org/ 10.1016/j.molcel.2008.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, Kornblihtt AR. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell 2003; 12:525-32; PMID:14536091; http://dx.doi.org/ 10.1016/j.molcel.2003.08.001 [DOI] [PubMed] [Google Scholar]
- 16.de la Mata M, Lafaille C, Kornblihtt AR. First come, first served revisited: Factors affecting the same alternative splicing event have different effects on the relative rates of intron removal. RNA 2010; 16:904-12; PMID:20357345; http://dx.doi.org/ 10.1261/rna.1993510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fong N, Kim H, Zhou Y, Ji X, Qiu J, Saldi T, Diener K, Jones K, Fu XD, Bentley DL. Pre-mRNA splicing is facilitated by an optimal RNA polymerase II elongation rate. Genes Dev 2014; 28:2663-76; PMID:25452276; http://dx.doi.org/ 10.1101/gad.252106.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dujardin G, Lafaille C, de la Mata M, Marasco LE, Munoz MJ, Le Jossic-Corcos C, Corcos L, Kornblihtt AR. How slow RNA polymerase II elongation favors alternative exon skipping. Mol Cell 2014; 54:683-90; PMID:24793692; http://dx.doi.org/ 10.1016/j.molcel.2014.03.044 [DOI] [PubMed] [Google Scholar]
- 19.Ip JY, Schmidt D, Pan Q, Ramani AK, Fraser AG, Odom DT, Blencowe BJ. Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Genome Res 2011; 21:390-401; PMID:21163941; http://dx.doi.org/ 10.1101/gr.111070.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khodor YL, Rodriguez J, Abruzzi KC, Tang CH, Marr MT 2nd, Rosbash M. Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in drosophila. Genes Dev 2011; 25:2502-12; PMID:22156210; http://dx.doi.org/ 10.1101/gad.178962.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howe KJ, Kane CM, Ares M Jr. Perturbation of transcription elongation influences the fidelity of internal exon inclusion in saccharomyces cerevisiae. RNA 2003; 9:993-1006; PMID:12869710; http://dx.doi.org/ 10.1261/rna.5390803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braberg H, Jin H, Moehle EA, Chan YA, Wang S, Shales M, Benschop JJ, Morris JH, Qiu C, Hu F, et al.. From structure to systems: High-resolution, quantitative genetic analysis of RNA polymerase II. Cell 2013; 154:775-88; PMID:23932120; http://dx.doi.org/ 10.1016/j.cell.2013.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wozniak GG, Strahl BD. Hitting the 'mark': Interpreting lysine methylation in the context of active transcription. Biochim Biophys Acta 2014; 1839:1353-61; PMID:24631869; http://dx.doi.org/ 10.1016/j.bbagrm.2014.03.002 [DOI] [PubMed] [Google Scholar]
- 24.Smolle M, Workman JL. Transcription-associated histone modifications and cryptic transcription. Biochim Biophys Acta 2013; 1829:84-97; PMID:22982198; http://dx.doi.org/ 10.1016/j.bbagrm.2012.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hnilicova J, Stanek D. Where splicing joins chromatin. Nucleus 2011; 2:182-8; PMID:21818411; http://dx.doi.org/ 10.4161/nucl.2.3.15876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shieh GS, Pan CH, Wu JH, Sun YJ, Wang CC, Hsiao WC, Lin CY, Tung L, Chang TH, Fleming AB, et al.. H2B ubiquitylation is part of chromatin architecture that marks exon-intron structure in budding yeast. BMC Genomics 2011; 12:627-2164-12-627; PMID:22188810; http://dx.doi.org/ 10.1186/1471-2164-12-627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gunderson FQ, Johnson TL. Acetylation by the transcriptional coactivator Gcn5 plays a novel role in co-transcriptional spliceosome assembly. PLoS Genet 2009; 5:e1000682; PMID:19834536; http://dx.doi.org/ 10.1371/journal.pgen.1000682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gunderson FQ, Merkhofer EC, Johnson TL. Dynamic histone acetylation is critical for cotranscriptional spliceosome assembly and spliceosomal rearrangements. Proc Natl Acad Sci U S A 2011; 108:2004-9; PMID:21245291; http://dx.doi.org/ 10.1073/pnas.1011982108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herissant L, Moehle EA, Bertaccini D, Van Dorsselaer A, Schaeffer-Reiss C, Guthrie C, Dargemont C. H2B ubiquitylation modulates spliceosome assembly and function in budding yeast. Biol Cell 2014; 106:126-138; PMID:24476359; http://dx.doi.org/ 10.1111/boc.201400003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moehle EA, Ryan CJ, Krogan NJ, Kress TL, Guthrie C. The yeast SR-like protein Npl3 links chromatin modification to mRNA processing. PLoS Genet 2012; 8:e1003101; PMID:23209445; http://dx.doi.org/ 10.1371/journal.pgen.1003101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science 2010; 327:996-1000; PMID:20133523; http://dx.doi.org/ 10.1126/science.1184208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pradeepa MM, Sutherland HG, Ule J, Grimes GR, Bickmore WA. Psip1/ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet 2012; 8:e1002717; PMID:22615581; http://dx.doi.org/ 10.1371/journal.pgen.1002717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo R, Zheng L, Park JW, Lv R, Chen H, Jiao F, Xu W, Mu S, Wen H, Qiu J, et al.. BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol Cell 2014; 56:298-310; PMID:25263594; http://dx.doi.org/ 10.1016/j.molcel.2014.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simon JM, Hacker KE, Singh D, Brannon AR, Parker JS, Weiser M, Ho TH, Kuan PF, Jonasch E, Furey TS, et al.. Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome Res 2014; 24:241-50; PMID:24158655; http://dx.doi.org/ 10.1101/gr.158253.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sims RJ 3rd, Millhouse S, Chen CF, Lewis BA, Erdjument-Bromage H, Tempst P, Manley JL, Reinberg D. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol Cell 2007; 28:665-76; PMID:18042460; http://dx.doi.org/ 10.1016/j.molcel.2007.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strahl BD, Grant PA, Briggs SD, Sun ZW, Bone JR, Caldwell JA, Mollah S, Cook RG, Shabanowitz J, Hunt DF, et al.. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol Cell Biol 2002; 22:1298-1306; PMID:11839797; http://dx.doi.org/ 10.1128/MCB.22.5.1298-1306.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiao T, Hall H, Kizer KO, Shibata Y, Hall MC, Borchers CH, Strahl BD. Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes Dev 2003; 17:654-63; PMID:12629047; http://dx.doi.org/ 10.1101/gad.1055503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joshi AA, Struhl K. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to pol II elongation. Mol Cell 2005; 20:971-8; PMID:16364921; http://dx.doi.org/ 10.1016/j.molcel.2005.11.021 [DOI] [PubMed] [Google Scholar]
- 39.Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, et al.. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 2005; 123:581-92; PMID:16286007; http://dx.doi.org/ 10.1016/j.cell.2005.10.023 [DOI] [PubMed] [Google Scholar]
- 40.Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, Schuldiner M, Chin K, Punna T, Thompson NJ, et al.. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell 2005; 123:593-605; PMID:16286008; http://dx.doi.org/ 10.1016/j.cell.2005.10.025 [DOI] [PubMed] [Google Scholar]
- 41.Smolle M, Venkatesh S, Gogol MM, Li H, Zhang Y, Florens L, Washburn MP, Workman JL. Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat Struct Mol Biol 2012; 19:884-92; PMID:22922743; http://dx.doi.org/ 10.1038/nsmb.2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Venkatesh S, Smolle M, Li H, Gogol MM, Saint M, Kumar S, Natarajan K, Workman JL. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature 2012; 489:452-5; PMID:22914091; http://dx.doi.org/ 10.1038/nature11326 [DOI] [PubMed] [Google Scholar]
- 43.McKay DJ, Klusza S, Penke TJ, Meers MP, Curry KP, McDaniel SL, Malek PY, Cooper SW, Tatomer DC, Lieb JD, et al.. Interrogating the function of metazoan histones using engineered gene clusters. Dev Cell 2015; 32:373-86; PMID:25669886; http://dx.doi.org/ 10.1016/j.devcel.2014.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sorenson MR, Stevens SW. Rapid identification of mRNA processing defects with a novel single-cell yeast reporter. RNA 2014; 20:732-45; PMID:24671766; http://dx.doi.org/ 10.1261/rna.042663.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dai J, Hyland EM, Yuan DS, Huang H, Bader JS, Boeke JD. Probing nucleosome function: A highly versatile library of synthetic histone H3 and H4 mutants. Cell 2008; 134:1066-78; PMID:18805098; http://dx.doi.org/ 10.1016/j.cell.2008.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stevens SW, Barta I, Ge HY, Moore RE, Young MK, Lee TD, Abelson J. Biochemical and genetic analyses of the U5, U6, and U4/U6 × U5 small nuclear ribonucleoproteins from saccharomyces cerevisiae. RNA 2001; 7:1543-53; PMID:11720284 [PMC free article] [PubMed] [Google Scholar]
- 47.Gottschalk A, Neubauer G, Banroques J, Mann M, Luhrmann R, Fabrizio P. Identification by mass spectrometry and functional analysis of novel proteins of the yeast [U4/U6.U5] tri-snRNP. EMBO J 1999; 18:4535-48; PMID:10449419; http://dx.doi.org/ 10.1093/emboj/18.16.4535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grant PA, Duggan L, Cote J, Roberts SM, Brownell JE, Candau R, Ohba R, Owen-Hughes T, Allis CD, Winston F, et al.. Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: Characterization of an ada complex and the SAGA (spt/ada) complex. Genes Dev 1997; 11:1640-50; PMID:9224714; http://dx.doi.org/ 10.1101/gad.11.13.1640 [DOI] [PubMed] [Google Scholar]
- 49.Zhang H, Richardson DO, Roberts DN, Utley R, Erdjument-Bromage H, Tempst P, Cote J, Cairns BR. The Yaf9 component of the SWR1 and NuA4 complexes is required for proper gene expression, histone H4 acetylation, and Htz1 replacement near telomeres. Mol Cell Biol 2004; 24:9424-36; PMID:15485911; http://dx.doi.org/ 10.1128/MCB.24.21.9424-9436.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Le Masson I, Yu DY, Jensen K, Chevalier A, Courbeyrette R, Boulard Y, Smith MM, Mann C. Yaf9, a novel NuA4 histone acetyltransferase subunit, is required for the cellular response to spindle stress in yeast. Mol Cell Biol 2003; 23:6086-102; PMID:12917332; http://dx.doi.org/ 10.1128/MCB.23.17.6086-6102.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim T, Buratowski S. Two saccharomyces cerevisiae JmjC domain proteins demethylate histone H3 Lys36 in transcribed regions to promote elongation. J Biol Chem 2007; 282:20827-35; PMID:17525156; http://dx.doi.org/ 10.1074/jbc.M703034200 [DOI] [PubMed] [Google Scholar]
- 52.Tu S, Bulloch EM, Yang L, Ren C, Huang WC, Hsu PH, Chen CH, Liao CL, Yu HM, Lo WS, et al.. Identification of histone demethylases in saccharomyces cerevisiae. J Biol Chem 2007; 282:14262-71; PMID:17369256; http://dx.doi.org/ 10.1074/jbc.M609900200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006; 439:811-6; PMID:16362057; http://dx.doi.org/ 10.1038/nature04433 [DOI] [PubMed] [Google Scholar]
- 54.Fang J, Hogan GJ, Liang G, Lieb JD, Zhang Y. The saccharomyces cerevisiae histone demethylase Jhd1 fine-tunes the distribution of H3K36me2. Mol Cell Biol 2007; 27:5055-65; PMID:17470555; http://dx.doi.org/ 10.1128/MCB.00127-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, et al.. Global mapping of the yeast genetic interaction network. Science 2004; 303:808-13; PMID:14764870; http://dx.doi.org/ 10.1126/science.1091317 [DOI] [PubMed] [Google Scholar]
- 56.Gottschalk A, Bartels C, Neubauer G, Luhrmann R, Fabrizio P. A novel yeast U2 snRNP protein, Snu17p, is required for the first catalytic step of splicing and for progression of spliceosome assembly. Mol Cell Biol 2001; 21:3037-46; PMID:11287609; http://dx.doi.org/ 10.1128/MCB.21.9.3037-3046.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dix I, Russell C, Yehuda SB, Kupiec M, Beggs JD. The identification and characterization of a novel splicing protein, Isy1p, of saccharomyces cerevisiae. RNA 1999; 5:360-8; PMID:10094305; http://dx.doi.org/ 10.1017/S1355838299981396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen CH, Tsai WY, Chen HR, Wang CH, Cheng SC. Identification and characterization of two novel components of the Prp19p-associated complex, Ntc30p and Ntc20p. J Biol Chem 2001; 276:488-94; PMID:11018040; http://dx.doi.org/ 10.1074/jbc.M006958200 [DOI] [PubMed] [Google Scholar]
- 59.Chan SP, Kao DI, Tsai WY, Cheng SC. The Prp19p-associated complex in spliceosome activation. Science 2003; 302:279-82; PMID:12970570; http://dx.doi.org/ 10.1126/science.1086602 [DOI] [PubMed] [Google Scholar]
- 60.Liao XC, Tang J, Rosbash M. An enhancer screen identifies a gene that encodes the yeast U1 snRNP A protein: Implications for snRNP protein function in pre-mRNA splicing. Genes Dev 1993; 7:419-28; PMID:8449403; http://dx.doi.org/ 10.1101/gad.7.3.419 [DOI] [PubMed] [Google Scholar]
- 61.Neubauer G, Gottschalk A, Fabrizio P, Seraphin B, Luhrmann R, Mann M. Identification of the proteins of the yeast U1 small nuclear ribonucleoprotein complex by mass spectrometry. Proc Natl Acad Sci U S A 1997; 94:385-90; PMID:9012791; http://dx.doi.org/ 10.1073/pnas.94.2.385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abovich N, Liao XC, Rosbash M. The yeast MUD2 protein: An interaction with PRP11 defines a bridge between commitment complexes and U2 snRNP addition. Genes Dev 1994; 8:843-54; PMID:7926772; http://dx.doi.org/ 10.1101/gad.8.7.843 [DOI] [PubMed] [Google Scholar]
- 63.Rain JC, Legrain P. In vivo commitment to splicing in yeast involves the nucleotide upstream from the branch site conserved sequence and the Mud2 protein. EMBO J 1997; 16:1759-71; PMID:9130720; http://dx.doi.org/ 10.1093/emboj/16.7.1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Albulescu LO, Sabet N, Gudipati M, Stepankiw N, Bergman ZJ, Huffaker TC, Pleiss JA. A quantitative, high-throughput reverse genetic screen reveals novel connections between pre-mRNA splicing and 5′ and 3′ end transcript determinants. PLoS Genet 2012; 8:e1002530; PMID:22479188; http://dx.doi.org/ 10.1371/journal.pgen.1002530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Du HN, Fingerman IM, Briggs SD. Histone H3 K36 methylation is mediated by a trans-histone methylation pathway involving an interaction between Set2 and histone H4. Genes Dev 2008; 22:2786-98; PMID:18923077; http://dx.doi.org/ 10.1101/gad.1700008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jha DK, Strahl BD. An RNA polymerase II-coupled function for histone H3K36 methylation in checkpoint activation and DSB repair. Nat Commun 2014; 5:3965; PMID:24910128; http://dx.doi.org/ 10.1038/ncomms4965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Venkatesh S, Workman JL. Set2 mediated H3 lysine 36 methylation: Regulation of transcription elongation and implications in organismal development. Wiley Interdiscip Rev Dev Biol 2013; 2:685-700; PMID:24014454; http://dx.doi.org/ 10.1002/wdev.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Biswas D, Dutta-Biswas R, Mitra D, Shibata Y, Strahl BD, Formosa T, Stillman DJ. Opposing roles for Set2 and yFACT in regulating TBP binding at promoters. EMBO J 2006; 25:4479-89; PMID:16977311; http://dx.doi.org/ 10.1038/sj.emboj.7601333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Biswas D, Takahata S, Xin H, Dutta-Biswas R, Yu Y, Formosa T, Stillman DJ. A role for Chd1 and Set2 in negatively regulating DNA replication in saccharomyces cerevisiae. Genetics 2008; 178:649-59; PMID:18245327; http://dx.doi.org/ 10.1534/genetics.107.084202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mason PB, Struhl K. Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol Cell 2005; 17:831-40; PMID:15780939; http://dx.doi.org/ 10.1016/j.molcel.2005.02.017 [DOI] [PubMed] [Google Scholar]
- 71.Lenstra TL, Benschop JJ, Kim T, Schulze JM, Brabers NA, Margaritis T, van de Pasch LA, van Heesch SA, Brok MO, Groot Koerkamp MJ, et al.. The specificity and topology of chromatin interaction pathways in yeast. Mol Cell 2011; 42:536-49; PMID:21596317; http://dx.doi.org/ 10.1016/j.molcel.2011.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, Strahl BD. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol 2005; 25:3305-16; PMID:15798214; http://dx.doi.org/ 10.1128/MCB.25.8.3305-3316.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011; 21:381-95; PMID:21321607; http://dx.doi.org/ 10.1038/cr.2011.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morris SA, Rao B, Garcia BA, Hake SB, Diaz RL, Shabanowitz J, Hunt DF, Allis CD, Lieb JD, Strahl BD. Identification of histone H3 lysine 36 acetylation as a highly conserved histone modification. J Biol Chem 2007; 282:7632-40; PMID:17189264; http://dx.doi.org/ 10.1074/jbc.M607909200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kasten MM, Dorland S, Stillman DJ. A large protein complex containing the yeast Sin3p and Rpd3p transcriptional regulators. Mol Cell Biol 1997; 17:4852-58; PMID:9234741; http://dx.doi.org/ 10.1128/MCB.17.8.4852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lechner T, Carrozza MJ, Yu Y, Grant PA, Eberharter A, Vannier D, Brosch G, Stillman DJ, Shore D, Workman JL. Sds3 (suppressor of defective silencing 3) is an integral component of the yeast Sin3[middle dot]Rpd3 histone deacetylase complex and is required for histone deacetylase activity. J Biol Chem 2000; 275:40961-6; PMID:11024051; http://dx.doi.org/ 10.1074/jbc.M005730200 [DOI] [PubMed] [Google Scholar]
- 77.Rundlett SE, Carmen AA, Kobayashi R, Bavykin S, Turner BM, Grunstein M. HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc Natl Acad Sci U S A 1996; 93:14503-508; PMID:8962081; http://dx.doi.org/ 10.1073/pnas.93.25.14503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.West S, Proudfoot NJ. Transcriptional termination enhances protein expression in human cells. Mol Cell 2009; 33:354-64; PMID:19217409; http://dx.doi.org/ 10.1016/j.molcel.2009.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Caspary F, Seraphin B. The yeast U2A'/U2B complex is required for pre-spliceosome formation. EMBO J 1998; 17:6348-58; PMID:9799242; http://dx.doi.org/ 10.1093/emboj/17.21.6348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim S, Kim H, Fong N, Erickson B, Bentley DL. Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc Natl Acad Sci U S A 2011; 108:13564-9; PMID:21807997; http://dx.doi.org/ 10.1073/pnas.1109475108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.de Almeida SF, Grosso AR, Koch F, Fenouil R, Carvalho S, Andrade J, Levezinho H, Gut M, Eick D, Gut I, Andrau JC, Ferrier P, Carmo-Fonseca M. Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nat Struct Mol Biol 2011; 18:977-83; PMID:21792193; http://dx.doi.org/ 10.1038/nsmb.2123 [DOI] [PubMed] [Google Scholar]
- 82.Strahl-Bolsinger S, Hecht A, Luo K, Grunstein M. SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev 1997; 11:83-93; PMID:9000052; http://dx.doi.org/ 10.1101/gad.11.1.83 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.