

ABSTRACT
RBM10 is an RNA binding protein and alternative splicing regulator frequently mutated in lung adenocarcinomas. Recent results indicate that RBM10 inhibits proliferation of lung cancer cells by promoting skipping of exon 9 of the gene NUMB, a frequent alternative splicing change in lung cancer generating a negative regulator of Notch signaling. Complementing these observations, we show that knock down of RBM10 in human cancer cells enhances growth of mouse tumor xenografts, confirming that RBM10 acts as a tumor suppressor, while knock down of an oncogenic mutant version of RBM10 reduces xenograft tumor growth. A RBM10 mutation found in lung cancer cells, V354E, disrupts RBM10-mediated regulation of NUMB alternative splicing, inducing the cell proliferation-promoting isoform. We now show that 2 natural RBM10 isoforms that differ by the presence or absence of V354 in the second RNA Recognition Motif (RRM2), display similar regulatory effects on NUMB alternative splicing, suggesting that V354E actively disrupts RBM10 activity. Structural modeling localizes V354 in the outside surface of one α-helix opposite to the RNA binding surface of RBM10, and we show that the mutation does not compromise binding of the RRM2 domain to NUMB RNA regulatory sequences. We further show that other RBM10 mutations found in lung adenocarcinomas also compromise regulation of NUMB exon 9. Collectively, our previous and current results reveal that RBM10 is a tumor suppressor that represses Notch signaling and cell proliferation through the regulation of NUMB alternative splicing.
KEYWORDS: Lung cancer, NUMB, Notch, RBM10, splicing
Introduction
Evidence for a role of RNA binding proteins in tumor progression is accumulating.1 In particular, a significant fraction of RNA processing factors have been shown to be up- or down-regulated or mutated in a variety of cancers.2,3 Concurrently, prominent alterations in alternative pre-mRNA splicing, the process by which primary transcripts undergo alternative patterns of intron/exon removal to generate mRNAs with different protein coding capacity or distinct regulation, have been observed in tumors.3,4 These alterations can contribute to virtually every aspect of tumor progression, including the control of cell division, programmed cell death, cell metabolism, tumor angiogenesis or tumor metastasis.4
In a few cases it has been possible to establish links between changes in alternative splicing of specific genes and alterations in expression or mutation of particular splicing factors associated with altered cell phenotypes. For example, SRSF1 (previously known as SF2/ASF) is a splicing regulator of the SR family that is frequently up-regulated in various tumors and its overexpression is sufficient to transform immortalized mouse fibroblasts. Expression of an oncogenic isoform of the S6K1 kinase promoted by SRSF1 recapitulates SRSF1-mediated transformation, thus providing a compelling argument that splicing regulation is at the basis of SRSF1 transforming activity.5
Another example is RBM10, an RNA binding protein frequently mutated in lung adenocarcinomas.6 Recent results indicate that RBM10 is an alternative splicing regulator7,8 that modulates alternative splicing of the Notch regulator gene NUMB.9 In NUMB, RBM10 promotes skipping of exon 9 and the synthesis of an mRNA encoding a NUMB isoform that inhibits accumulation of the Notch receptor.9,10 The Notch pathway is critical for lung cancer progression, as evidenced by the therapeutic effects of inhibitors of β-secretase, the enzyme that cleaves the receptor as a necessary step for its activation.11,12 Of relevance, higher levels of NUMB exon 9 inclusion (the pro-proliferative isoform) are among the most frequent splicing alterations in lung cancer.13 Thus, an alternative splicing switch frequent in lung cancer, affecting the function of a key cell proliferation pathway, is regulated by RBM10, one of the most frequently mutated genes in lung adenocarcinomas.
Here we discuss these findings and present evidence that RBM10 can act as a suppressor of mouse tumor xenografts and that a RBM10 mutation found in lung cancer cells actively disrupts its function as a regulator of NUMB alternative splicing without affecting RNA binding of the RRM2 motif.
RBM10 represses mouse xenograft tumor formation
HeLa cell lines stably expressing either shRNAs against RBM10 or control shRNA 9 were injected subcutaneously into CB17SC-M nude mice. Expression of the shRNAs against RBM10 led to significant and specific decrease in RBM10 protein levels in the stable cell lines.9 Injections were performed on both lateral dorsal sides of the animal and tumor formation, and progression was monitored weekly. Tumors formed by cells expressing control shRNA were detectable 2 weeks after injection (Fig. 1A, black line). In contrast, tumors formed by cells expressing either of 2 different shRNAs against RBM10 were detected already one week after injection (Fig. 1A, gray and pale gray lines). Xenograft tumors from RBM10-depleted cells continued developing and remained of significantly larger size than control xenografts. These results indicate that cells depleted of RBM10 are more efficient in xenograft tumor formation and therefore that RBM10 has properties of a tumor suppressor.
Figure 1.
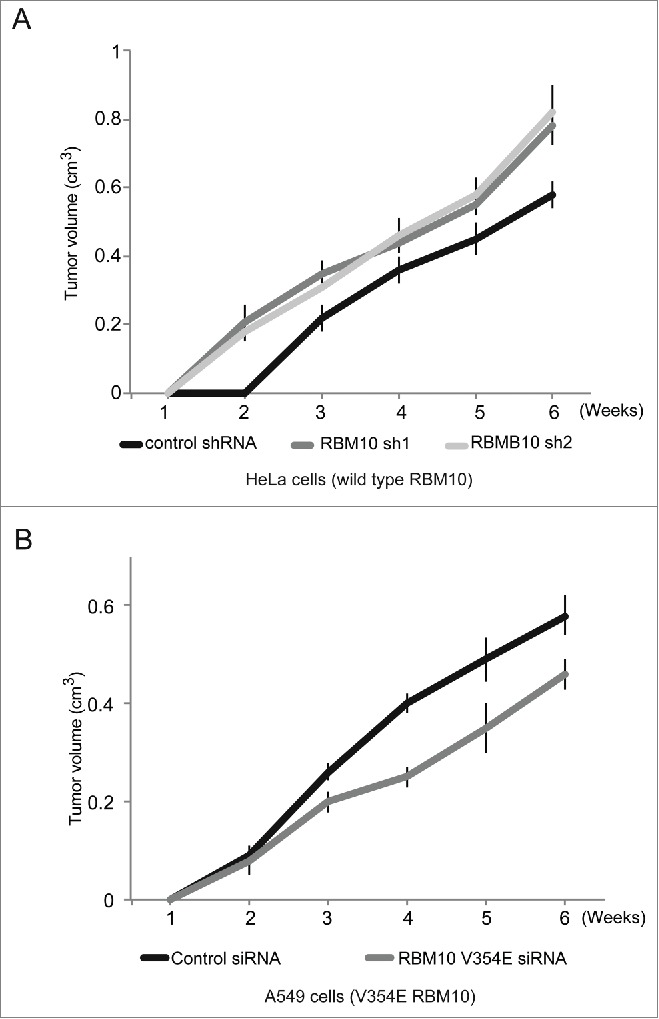
Effect of RBM10 depletion on in mouse xenograft tumor formation. Xenograft tumor formation assays were performed by injecting cells subcutaneously into CB17SC-M nude mice. (A) Evolution of xenograft tumors formed after injection of HeLa cells stably infected with 2 different shRNAs directed against RBM10 (gray and pale gray lines) or control shRNA (black line). (B) Evolution of tumors formed after injection of A549 cells transiently transfected with either siRNA against RBM10 V354E mutant (gray line) or control siRNA (black line). X and y axis represent weeks after injection and tumor volume, respectively. Error bars represent standard deviation from at least 3 different animals for each condition.
Similar experiments were carried out using lung adenocarcinoma A549 cells, which contain a V354E substitution in RBM10 that compromises its function in NUMB alternative splicing regulation.9 Because the RBM10 gene is located in the X chromosome and A549 cells are derived from a male patient (and therefore contain a single copy of the X chromosome), V354E is the only RBM10 variant expressed in these cells. In this case, cells were transiently transfected with a pool of siRNAs against RBM10 or a control scrambled siRNA. Tumors were detectable one week after injection (Fig. 1B, black and gray lines), but tumors formed upon depletion of RBM10 V354E (gray line) remained of smaller size than tumors induced by control cells (black line) throughout the experiment. This result is consistent with reduced cell growth and tumor formation upon depletion of the oncogenic (V354E) version of RBM10.
Collectively, the results of the xenograft experiments are consistent with a function of wild type RBM10 as a tumor suppressor and also with an oncogenic function of the V354E mutant version of RBM10 found in the A549 lung cancer cell line. The contrasting effects of depletion of wild type and mutant RBM10 further highlight the key role that RBM10 plays in the control of cell and tumor growth.
Role of RBM10 valine 354 variants in numb alternative splicing regulation
Analysis of transcriptome/proteome databases and our own sequencing of RBM10 cDNAs revealed the use of 2 consecutive alternative 5′ splice sites leading to mRNAs encoding versions of RBM10 with (Swiss prot identifier P98175-1) or without (Swiss prot identifier P98175-2) valine 354 (Fig. 2A). Given the relevance of mutation of valine 354 to glutamic acid in the regulation of NUMB splicing, cell growth and tumor formation9 (Fig. 1), we compared the activities of RBM10 containing or not valine 354 with that of the oncogenic mutant V354E. HeLa cells were co-transfected with a vector expressing the different RBM10 variants and the RG6-NUMB minigene construct,9 a modified version of a minigene14 containing NUMB exon 9 and 100 / 50 base pairs of the 5′ / 3′ flanking intronic sequences, respectively, inserted between 2 exons and their associated intronic sequences of a tropomyosin gene. RNA was isolated 24 hours post-transfection and the ratio between NUMB exon 9 inclusion and skipping analyzed by reverse transcription (RT)-PCR using minigene-specific sequences in the flanking exons and quantified by capillary electrophoresis.
Figure 2.
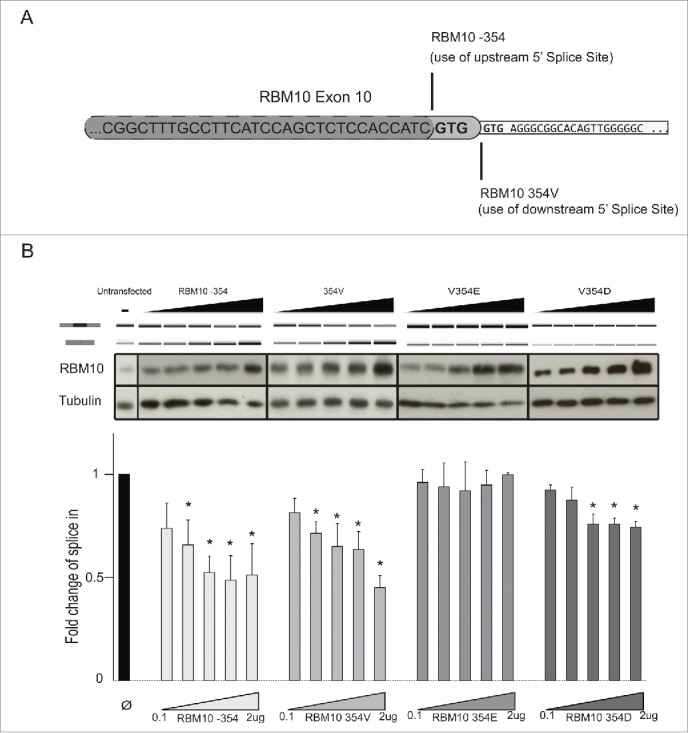
Activity of RBM10 RRM2 variants. (A) Sequences of the alternative 5′ splice sites in RBM10 exon 10 (UCSC Genome Browser coordinates ChrX:47038514-47039897) leading to protein isoforms containing or lacking valine 354. (B) Activity of RBM10 variants on NUMB exon 9 repression. Transient co-transfection assays were carried out using the pRG6-NUMB reporter minigene and vectors expressing RBM10 without (RBM10 −354) or with (RBM10 354V) valine 354, a V354E mutant found in A549 cells9 or a V354D mutant. Upper panel: RT-PCR analysis of exon 9 inclusion / skipping using primers located in the flanking exons. Middle panel: protein gel blot analysis of RBM10 levels and β-tubulin as a loading control. Lower panel: quantification of the ratio between exon 9 inclusion and skipping obtained from RT-PCR analyses. Bars represent standard deviation from 3 independent biological replicas of the experiment.
Overexpression of both alternatively spliced RBM10 proteins, with or without valine 354, promoted NUMB exon 9 skipping to a similar extent while, consistent with,9 overexpression of V354E failed to regulate NUMB exon 9 splicing (Fig. 2B, upper panel, results quantified in the lower graph). All three variants of RBM10 protein accumulated to similar levels in these assays (Fig. 2B, middle panel).
Taken together, these results indicate that, while the presence of valine 354 is not critical for the function of RBM10 as a regulator of NUMB exon 9 splicing, substitution of valine by glutamic acid abolishes this activity. To test whether the effect of the V354E is related to the presence of a negative charge, valine was replaced by aspartic acid. The V354D mutant also displayed reduced regulatory activity on NUMB exon 9 splicing, although some exon skipping was observed at the higher levels of protein expression (Fig. 2B). These results argue that a negatively charge at position 354 disrupts RBM10 function, although the longer side chain of glutamic acid is more disruptive.
To obtain further insights on the molecular basis for these observations, we took advantage of the solution structure of the RRM2 domain of the highly related factor RBM5 15 and used NMR models of maximal folding stability as templates to generate structural models of RBM10 RRM2, with valine 354 and with valine replaced by glutamic or aspartic acid. Mustang structural superimposition of RBM10 RRM2-Valine (2M2B) provided Root-Mean-Square Deviation (RMSD) of atomic positions of 1.913 A over 67 aligned residues with 70.15% sequence identity. Fig. 3 shows the predicted location of valine / glutamic / aspartic acid 354 in the structure of the RRM2, which adopts a typical RRM fold.16 The model predicts that valine 354 is located in the external edge of one of the 2 α-helices of the RRM fold, on the back side of (and oriented in opposite direction to) the β-sheet that typically holds the RNA binding surface of the domain. Replacement of valine by glutamic acid or aspartic acid does not significantly modify this structural model, given the solvent exposure of this residue (ΔΔG of −0.17 and −0.33 Kcal/mol, respectively, calculated using FoldX side chain modeling,17 well below the threshold for disruption of α-helices; similar conclusions were reached using the SwitchP algorithm24). Absence of valine 354 would shorten the helix, likely without a strong impact on the overall fold of the domain (see below).
Figure 3.
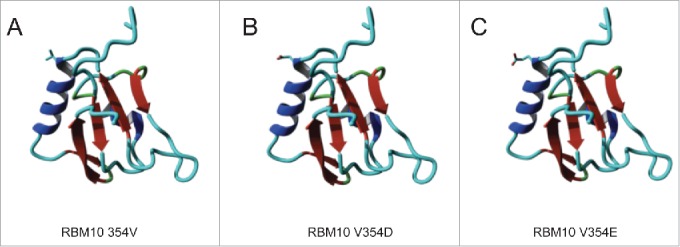
Structural modeling of RBM10 RRM2 and its variants. Molecular modeling is based upon the structure of the equivalent domain in RBM515. The location of the side chain for valine (A)/ aspartic acid (B) / glutamic acid (C) at position 354 are indicated.
To directly assess RNA binding by RRM2 and its variants, wild type (with or without valine 354) and V354E mutant RRM2 domains were expressed in and purified from E. coli and binding to a 16 nucleotide, radioactively-labeled RNA corresponding to a high affinity RBM10 binding site, the polypyrimidine tract of NUMB intron 8,9 was tested using electrophoretic gel retardation assays. The results indicate that the presence of valine at position 354 causes a relatively small reduction in the apparent RNA binding dissociation constant from approx. 500 nM to 1350 nM, but substitution of valine by glutamic acid does not affect RNA binding (Fig. 4), while the 2 proteins display opposite regulatory effects (Fig. 2).
Figure 4.
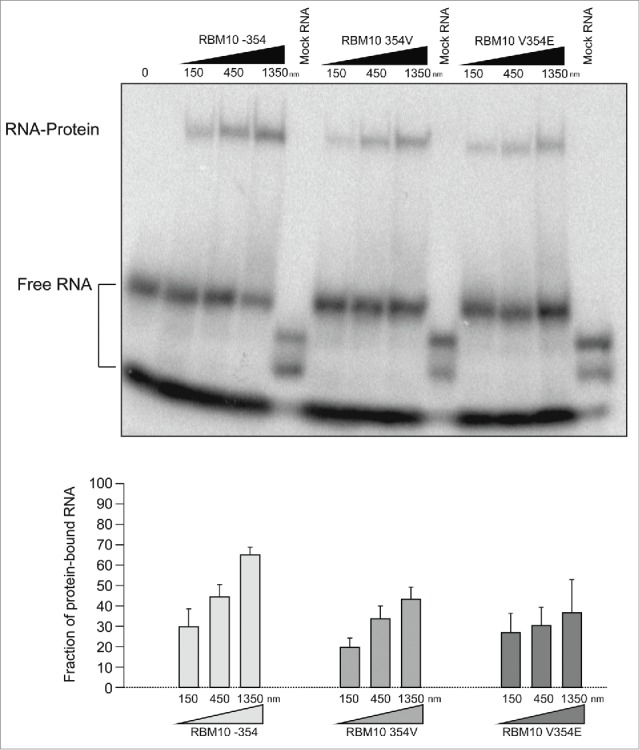
RNA binding by RBM10 RRM2 and its variants. (A) Mobility-shift assays corresponding to electrophoretic separation of free RNA and RNA-RRM2 domain complexes for the indicated RRM2 variants and RNAs, as indicated. (B) Quantification of the fraction of RNA in protein-RNA complexes in 3 independent mobility-shift assays carried out as in (A).
Collectively, the activity assays and structural modeling suggest that the loss of splicing regulation induced by the V354E mutation is not due to the loss of a specific RNA contact mediated by valine 354, but rather to the presence of a glutamic acid residue (at least partially due of its negatively charged side chain) in a surface domain of RRM2 which is not directly involved in RNA binding.
Other mutations detected in lung adenocarcinomas also reduce rbm10-mediated NUMB splicing regulation
A number of mutations identified in lung adenocarcinomas 6 Cosmic database 18 (cancer.sanger.ac.uk) introduce premature termination codons along the ORF and are predicted to generate truncated proteins. We have expanded the analysis of RBM10 variants with other truncating mutants that can help to define the functionality of the various domains of the protein (Table S1). Fig. 5 shows that C-terminal deletions of increasing length result in progressive loss of activity of the RBM10 variants. RBM10 truncated mutants lacking the C-terminal Zn finger and glycine patch are essentially non-functional. Interestingly, proteins spanning only the RRMs and first Zn finger (or shorter variants), and therefore lacking the OCRE domain,19 appear to promote exon 9 inclusion at the highest protein concentrations tested (Fig. 5), suggesting that these proteins exert a dominant-negative effect. We conclude that full function of RBM10 on exon 9 skipping requires the integrity of the protein.
Figure 5.
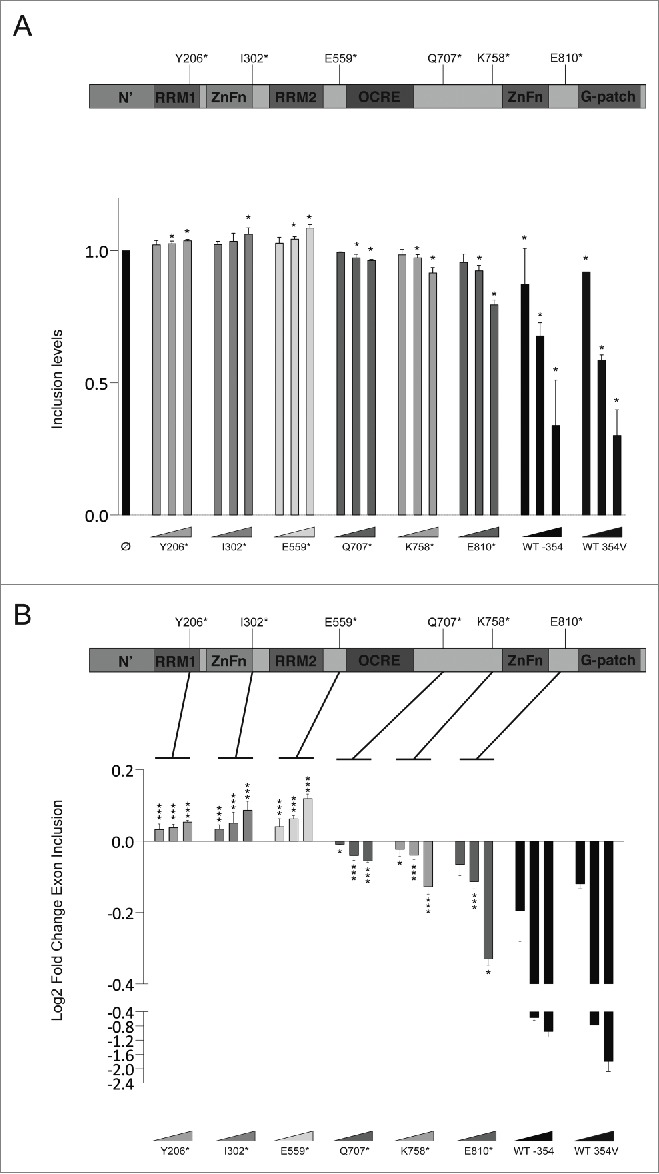
Activity of RBM10 truncated variants. (A) Representation of RBM10 protein domains (top) and normalized level of NUMB exon 9 inclusion upon expression of the indicated RBM10 truncated mutants for 3 independent experiments (top) (statistical significance of the effects of each construct compared to control was determined using a t-test). (B) Top panel represents RBM10 protein with their different domains Lower panel shows the log2 fold change of exon inclusion, t-test analysis performed for each of the constructs vs the WT protein.
Outlook
Several lines of evidence are consistent with the functional relevance of RMB10-mediated regulation of NUMB.9 First, RBM10 knock down leads to higher levels of exon 9 inclusion and hyperproliferation of a variety of cancer cell lines, as well to enhanced growth of xenograft tumors in mice. Second, the hyperproliferative effects of RBM10 knock down can be reversed by promoting exon 9 skipping using modified oligonucleotides that inhibit NUMB exon 9 splice sites. The latter result indicates that the effects of RBM10 on cancer cell growth ex vivo are mediated, in large part, by its effects on NUMB alternative splicing. Third, a RBM10 mutation identified in lung cancer cells prevents its function as a repressor of NUMB exon 9 and knock down of this mutant reduces growth of xenograft tumors in mice.
Also consistent with a key role of NUMB splicing regulation in lung cancer, recent results revealed that QKI, a KH domain-containing RNA binding protein frequently downregulated in lung cancer, promotes NUMB exon 9 skipping by preventing recognition of the branch point sequence by SF1/BBP.20 Therefore RBM10 and QKI may concertedly act to prevent cell growth by promoting skipping of NUMB exon 9, a regulatory circuit altered in lung cancer cells upon mutation or decreased expression of these splicing regulators.
Recent results argue that the presence or absence of valine 354 alters the structure of one of the α-helices in RRM2 21 and that preferential expression of the valine minus isoform in the small cell lung cancer GLC20 cell line correlates with higher levels of NUMB exon 9 inclusion. Our structural modeling, RNA binding and functional splicing regulation assays, however, indicate that the 2 isoforms are largely equivalent. Although substitution of valine 354 by glutamic acid did not compromise the intrinsic RNA binding affinity of RBM10 RRM2, further experiments will be needed to establish whether binding of RBM10 to the 3′ splice site region preceding NUMB exon 9 is compromised in the V354E mutant (e.g. by failure to assemble a repressive complex with other factors) or whether RNA binding still occurs in vivo but the mutation compromises other activities of RBM10 necessary for splicing repression.
Further analyses of the activity of additional RBM10 mutants identified in lung adenocarcinomas 6 on NUMB alternative splicing regulation may help to identify key residues and domains important for RBM10 function and provide insights into mechanisms of splicing regulation.
Materials and methods
Cell culture
HeLa, A549 and HEK293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) media, supplemented with 10% bovine fetal serum (FBS), 50units/ml penicillin and 50μl/ml streptomycin.
Xenografts and knock down assays
For xenograft experiments, HeLa cells were knocked down for RBM10 using lentivirus with pLKO.1 carrier vector containing the following shRNA sequences:
CCGGCTTCGCCTTCGTCGAGTTTAGCTCGAGCTAAA CTCGACGAAGGCGAAGTTTTTG and CCGGTCCAACGT GCGCGTCATAAAGCTCGAGCTTTATGACGCGCACGTT GGATTTT TG or a scrambled control (Sigma SHC002). Cells were cultured for 10–15 d after infection and selected with 2.5 µg/ml puromycin 24 h after infection. A549 cells were transiently transfected with a pool of siRNAs against RBM10 AAGAGCAACTTCTCCACATGT and AAGGACAGAGTGTTGGATGGC or control siRNA universal negative control from Sigma (SIC001). Substantial decreases in RBM10 expression were observed under these knock down conditions.9 Knockdown or control cells where injected subcutaneously into CB17SC-M nude mice. Injections were performed on both lateral dorsal sides of the animal and tumor formation and progression was monitored weekly as in.9
RBM10 overexpression
HeLa or HEK293 cells were transfected with Lipofectamine 2000 (Thermofisher, following manufacturer's instructions), transiently overexpressing RBM10 or its mutants variants, together with the RG6-NUMB minigene construct.9 RNA and protein samples were collected 24 h after transfection. RBM10 −354, RBM10 354V, RBM10 V354E, RBM10 V354D, RBM10 Y206*, RBM10 I302*, RBM10 E559*, RBM10 Q707*, RBM10 K758* and RBM10 E810* were cloned in p-DEST26 (Gateway p-DEST26, Thermo Fisher Scientific) for expression in mammalian cells as in Ref 9. RG6-NUMB contains the NUMB exon 9 plus 100 bp upstream and 50 bp downstream of the corresponding flanking intronic sequences, all these inserted between 2 exons and their associated intronic sequences of a tropomyosin gene.14
RNA extraction, RT-PCR and high-throughput capillary electrophoresis
RNA was extracted using Maxwell 16 LEV simple RNA tissue kit (Promega) following the manufacturer's instructions. 500 ng of RNA per sample where retrotranscribed using Superscript III Retro Transcriptase (Invitrogen, Life Technologies) following the manufacturer's instructions. PCR reactions were carried out using primers corresponding to minigene sequences (Fw: GGATTACAAGGATGACGATGACAAGGG, Rv: GTCACCTTCAGCTTCACGGTGTTGTG) and GOTaq DNA polymerase kit (GoTaq, Promega). High-Throughput Capillary Electrophoresis analysis and alternative splicing profiling were carried out as in.22
Western blot
Western blot analysis of RBM10 overexpression in HeLa and HEK293 cells was performed with α-RBM10 antibodies (Sigma Aldrich) and α-tubulin antibodies were used as loading control. Protein extraction and immunobloting were performed as previously described.23
The RBM10 mutant Y206* was not detectable by western blot. Given that the protein displays a dominant-negative effect, the N-terminal truncation of the protein likely prevents recognition by the anti-RBM10 antibody.
RRM purification
RBM10 RRM2 wild type and mutant variants were produced in Rosetta (DE3) pLysS competent cells (Novagen) as GST fusions. Bacteria were induced with 1 mM IPTG when the Optical Density reached 0.4–0.8 and grown overnight at 20°\C. after induction. GST-RBM10 RRM2 proteins were purified by (GST)-tag affinity chromatography using Glutathione Agarose beads (Sigma-Aldrich) following the manufacturer's protocol.
Electrophoretic mobility shift assays
10 fmol of terminally P32-labeled RNAs (the polypyrimidine tract of NUMB intron 8 GUUGUCUGCUCCCGUUGU identified in Ref. 9 and UCGUCGAUCGUCGA as a negative control) were incubated on ice for 15 min with GST-RBM10 RRM2 domains in the presence of 200 ng/µl of yeast tRNA. Samples were UV-crosslinked to stabilize protein-RNA interactions (400,000 μJoules/cm2), loaded onto native 5% polyacrylamide gels and electrophoresis was carried out at 200V and 4°C in 0.5% Tris-Borate-EDTA buffer for 80 min. The gel was subsequently dried before Phosphorimager analysis.
Modeling
All energy calculations, repairing and modeling steps were carried out using FoldX.17 Side chains were modeled using the 2LKZ PDB structure as a template after repairing (only the NMR model with lower energy in the PDB was taken). SwitchP algorithm 24 was run on V, D and E mutant models and the switchability score was 0 for all of them.
Table 1.
RBM 10 wild type (WT) variants and truncated (*) mutants tested in functional alternative splicing assays. The identity, source, identifiers, tissue expression and protein domains composition of each variant is indicated. For truncation mutants, the amino acid replaced by a stop codon is indicated before an asterisk. Imielinsky, 2012 corresponds to Ref. 6; COSMIC corresponds to Ref. 18. NA: not applicable (tailored mutants). RRM = RNA Recognition Motif; ZnFn = Zinc Finger motif; OCRE= Octamer Repeats of Aromatic Residues domain; G-patch = Glycine patch.
| Mutation Code | Origin | ID | Tissue Found | Protein Domains Contained |
|---|---|---|---|---|
| Y206* | Imielinksy, 2012 | COSM369455 | Lung | RRM1 |
| I302* | Tailored mutation | NA | NA | RRM1, ZnFn1 |
| E559* | COSMIC | COSM612899 | Lung | RRM1, ZnFn1, RRM2 |
| Q707* | COSMIC | COSM612897 | Lung and Large Intestine | RRM1, ZnFn1, RRM2, OCRE |
| K758* | Tailored mutation | NA | NA | RRM1, ZnFn1, RRM2, OCRE |
| E810* | Imielinksy, 2012 | COSM366920 | Lung | RRM1, ZnFn1, RRM2, OCRE, ZnFn2 |
| WT 354V | Swiss-Prot | P98175-1 | RRM1, ZnFn1, RRM2, OCRE, ZnFn2, G-patch | |
| WT −354 | Swiss-Prot | P98175-2 | RRM1, ZnFn1, RRM2, OCRE, ZnFn2, G-patch |
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank members of our laboratories for technical advice, discussions and comments on the manuscript. EB was supported by a Marie Curie postdoctoral fellowship and JH by a La Caixa predoctoral fellowship. Work in JV and LS labs was supported by grants from Fundación Botín, Banco de Santander through its Santander Universities Global Division and by Consolider RNAREG, MICINN and AGAUR and we acknowledge support of the Spanish Ministry of Economy and Competitiveness, ‘Centro de Excelencia Severo Ochoa 2013–2017′, SEV-2012-0208.
References
- 1.Blackinton JG, Keene JD. Post-transcriptional RNA regulons affecting cell cycle and proliferation. Semin Cell Dev Biol 2014; 34:44-54; PMID:24882724; http://dx.doi.org/ 10.1016/j.semcdb.2014.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonnal S, Vigevani L, Valcarcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov 2012; 11:847-59; PMID:23123942; http://dx.doi.org/ 10.1038/nrd3823 [DOI] [PubMed] [Google Scholar]
- 3.Kaida D, Schneider-Poetsch T, Yoshida M. Splicing in oncogenesis and tumor suppression. Cancer Sci 2012; 103:1611-6; PMID:22691055; http://dx.doi.org/ 10.1111/j.1349-7006.2012.02356.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 2010; 24:2343-64; PMID:21041405; http://dx.doi.org/ 10.1101/gad.1973010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol 2007; 14:185-93; PMID:17310252; http://dx.doi.org/ 10.1038/nsmb1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al.. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012; 150:1107-20; PMID:22980975; http://dx.doi.org/ 10.1016/j.cell.2012.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Gogol-Doring A, Hu H, Frohler S, Ma Y, Jens M, Maaskola J, Murakawa Y, Quedenau C, Landthaler M, et al.. Integrative analysis revealed the molecular mechanism underlying RBM10-mediated splicing regulation. EMBO Mol Med 2013; 5:1431-42; PMID:24000153; http://dx.doi.org/ 10.1002/emmm.201302663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inoue A, Yamamoto N, Kimura M, Nishio K, Yamane H, Nakajima K. RBM10 regulates alternative splicing. FEBS Lett 2014; 588:942-7; PMID:24530524; http://dx.doi.org/ 10.1016/j.febslet.2014.01.052 [DOI] [PubMed] [Google Scholar]
- 9.Bechara EG, Sebestyen E, Bernardis I, Eyras E, Valcarcel J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol Cell 2013; 52:720-33; PMID:24332178; http://dx.doi.org/ 10.1016/j.molcel.2013.11.010 [DOI] [PubMed] [Google Scholar]
- 10.Pece S, Confalonieri S, RR P, Di Fiore PP. NUMB-ing down cancer by more than just a NOTCH. Biochimica et Biophysica Acta 2011; 1815:26-43; PMID:20940030; http://dx.doi.org/10.1016/j.bbcan.2010.10.001 [DOI] [PubMed] [Google Scholar]
- 11.Maraver A, Fernandez-Marcos PJ, Herranz D, Canamero M, Munoz-Martin M, Gomez-Lopez G, Mulero F, Megias D, Sanchez-Carbayo M, Shen J, et al.. Therapeutic effect of gamma-secretase inhibition in KrasG12V-driven non-small cell lung carcinoma by derepression of DUSP1 and inhibition of ERK. Cancer Cell 2012; 22:222-34; PMID:22897852; http://dx.doi.org/ 10.1016/j.ccr.2012.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Westhoff B, Colaluca IN, D'Ario G, Donzelli M, Tosoni D, Volorio S, Pelosi G, Spaggiari L, Mazzarol G, Viale G, et al.. Alterations of the Notch pathway in lung cancer. Proc Natl Acad Sci U S A 2009; 106:22293-8; PMID:20007775; http://dx.doi.org/ 10.1073/pnas.0907781106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Misquitta-Ali CM, Cheng E, O'Hanlon D, Liu N, McGlade CJ, Tsao MS, Blencowe BJ. Global profiling and molecular characterization of alternative splicing events misregulated in lung cancer. Mol Cell Biol 2011; 31:138-50; PMID:21041478; http://dx.doi.org/ 10.1128/MCB.00709-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orengo JP, Bundman D, Cooper TA. A bichromatic fluorescent reporter for cell-based screens of alternative splicing. Nucleic Acids Res 2006; 34:e148; PMID:17142220; http://dx.doi.org/ 10.1093/nar/gkl967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song Z, Wu P, Ji P, Zhang J, Gong Q, Wu J, Shi Y. Solution structure of the second RRM domain of RBM5 and its unusual binding characters for different RNA targets. Biochemistry 2012; 51:6667-78; PMID:22839758; http://dx.doi.org/ 10.1021/bi300539t [DOI] [PubMed] [Google Scholar]
- 16.Daubner GM, Clery A, Allain FH. RRM-RNA recognition: NMR or crystallography…and new findings. Curr Opin Struct Biol 2013; 23:100-8; PMID:23253355; http://dx.doi.org/ 10.1016/j.sbi.2012.11.006 [DOI] [PubMed] [Google Scholar]
- 17.Schymkowitz JW, Rousseau F, Martins IC, Ferkinghoff-Borg J, Stricher F, Serrano L. Prediction of water and metal binding sites and their affinities by using the Fold-X force field. Proc Natl Acad Sci U S A 2005; 102:10147-52; PMID:16006526; http://dx.doi.org/ 10.1073/pnas.0501980102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, et al.. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015; 43:D805-11; PMID:25355519; http://dx.doi.org/ 10.1093/nar/gku1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Callebaut I, Mornon JP. OCRE: a novel domain made of imperfect, aromatic-rich octamer repeats. Bioinformatics 2005; 21:699-702; PMID:15486042; http://dx.doi.org/ 10.1093/bioinformatics/bti065 [DOI] [PubMed] [Google Scholar]
- 20.Zong FY, Fu X, Wei WJ, Luo YG, Heiner M, Cao LJ, Fang Z, Fang R, Lu D, Ji H, et al.. The RNA-binding protein QKI suppresses cancer-associated aberrant splicing. PLoS Genet 2014; 10:e1004289; PMID:24722255; http://dx.doi.org/ 10.1371/journal.pgen.1004289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tessier SJ, Loiselle JJ, McBain A, Pullen C, Koenderink BW, Roy JG, Sutherland LC. Insight into the role of alternative splicing within the RBM10v1 exon 10 tandem donor site. BMC Research Notes 2015; 8:46; PMID:25889998; http://dx.doi.org/10.1186/s13104-015-0983-525482508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tejedor JR, Papasaikas P, Valcarcel J. Genome-wide identification of Fas/CD95 alternative splicing regulators reveals links with iron homeostasis. Mol Cell 2015; 57:23-38; PMID:25482508; http://dx.doi.org/ 10.1016/j.molcel.2014.10.029 [DOI] [PubMed] [Google Scholar]
- 23.Paronetto MP, Minana B, Valcarcel J. The Ewing sarcoma protein regulates DNA damage-induced alternative splicing. Mol Cell 2011; 43:353-68; PMID:21816343; http://dx.doi.org/ 10.1016/j.molcel.2011.05.035 [DOI] [PubMed] [Google Scholar]
- 24.Diaz C, Corentin H, Thierry V, Chantal A, Tanguy B, David S, Jean-Marc H, Pascual F, Francoise B, Edgardo F. Virtual screening on an alpha-helix to beta-strand switchable region of the FGFR2 extracellular domain revealed positive and negative modulators. Proteins 2014; 82:2982-97; PMID:25082719; http://dx.doi.org/ 10.1002/prot.24657 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.