

Abstract
Since highly active antiretroviral therapy improved long‐term survival of acquired immunodeficiency syndrome (AIDS) patients, AIDS cardiomyopathy has become an increasingly relevant clinical problem. We used human immunodeficiency virus (HIV)‐1 transgenic (Tg26) mouse to explore molecular mechanisms of AIDS cardiomyopathy. Tg26 mice had significantly lower left ventricular (LV) mass and smaller end‐diastolic and end‐systolic LV volumes. Under basal conditions, cardiac contractility and relaxation and single myocyte contraction dynamics were not different between wild‐type (WT) and Tg26 mice. Ten days after open heart surgery, contractility and relaxation remained significantly depressed in Tg26 hearts, suggesting that Tg26 mice did not tolerate surgical stress well. To simulate heart failure in which expression of Bcl2‐associated athanogene 3 (BAG3) is reduced, we down‐regulated BAG3 by small hairpin ribonucleic acid in WT and Tg26 hearts. BAG3 down‐regulation significantly reduced contractility in Tg26 hearts. BAG3 overexpression rescued contractile abnormalities in myocytes expressing the HIV‐1 protein Tat. We conclude: (i) Tg26 mice exhibit normal contractile function at baseline; (ii) Tg26 mice do not tolerate surgical stress well; (iii) BAG3 down‐regulation exacerbated cardiac dysfunction in Tg26 mice; (iv) BAG3 overexpression rescued contractile abnormalities in myocytes expressing HIV‐1 protein Tat; and (v) BAG3 may occupy a role in pathogenesis of AIDS cardiomyopathy.
Keywords: in vivo hemodynamics, excitation–contraction coupling, surgical stress, adenovirus
Introduction
Prior to the introduction of highly active antiretroviral therapy (HAART), a major cause of death in human immunodeficiency virus (HIV)‐infected patients was the development of HIV‐related primary heart muscle disease as evidenced by severe left ventricular (LV) dysfunction and signs and symptoms of heart failure (HF).1 Indeed, LV dysfunction was seen in 3–41% of HIV‐1‐infected individuals and individuals with HIV‐1 cardiomyopathy had worse survival when compared to patients with ischemic‐ or nonischemic‐dilated cardiomyopathy.2, 3 The improved virologic control achieved in HIV‐1 patients on HAART has only decreased the prevalence of HIV‐associated HF by about 30%, and the incidence still remains unacceptably high. HIV‐1 remains a significant risk factor for the development of HF and the risk increases as HIV‐1 positive patient's age. Little is known about the pathobiology of HIV‐1 in the heart.
The hemizygous NL4‐3Δgag/pol transgenic (Tg26) mouse contains an internal deletion of the gag/pol coding sequence that renders the viral construct replication‐deficient, noninfectious, and useful to study acquired immunodeficiency syndrome (AIDS) cardiomyopathy.4 Tg26 mice exhibit HIV‐1‐associated nephropathy, wasting, and skin diseases that phenotypically resemble their clinical counterparts in AIDS.5 Tissue extracts from Tg26 animals demonstrate gp41, gp120, and gp160 env proteins as well as Nef. In addition, Tg26 hearts exhibit fourfold increase in percent of mitochondria that is damaged, increased atrial natriuretic factor but decreased sarco(endo)plasmic reticulum Ca2+‐ATPase (SERCA2) expression, making it a clinically relevant model to study the pathogenesis of AIDS cardiomyopathy.
The Bcl2‐associated athanogene 3 (BAG3) protein is a 575 amino acid antiapoptotic protein that is constitutively expressed in the heart, skeletal muscle, and some cancers and serves as a cochaperone of both the constitutively and nonconstitutively expressed heat shock proteins.6, 7 BAG3 stabilizes the sarcomere through regulation of filamin clearance and production and by binding to CapZ.8 BAG3 plays an important role in the development or progression of HF.9 The potential interplay between BAG3 and HIV‐1 stems from our previous studies demonstrate that during the course of productive HIV‐1 replication in microglial cells of the brain, expression of BAG3 is induced.10 In cardiac myocytes, uptake of soluble Tat secreted by productively and/or latently infected cells, upon association with BAG3, may interfere with BAG3's function on several pathways involved in cardiac myocyte homeostasis and function. The current study is undertaken to utilize Tg26 mice to evaluate if depressed cardiac function occurs under clinically relevant conditions, and to explore the role of BAG3 in the development of AIDS cardiomyopathy.
Methods
Tg26 mice and animal care
Tg26 is a well‐described murine line of NL4‐3Δgag/pol that expresses HIV‐related proteins such as Tat and Nef, and develops nephropathy and skin lesions in the hemizygotes.11 Hemizygous Tg26 mice (FVB/n backcrossed to C57BL/6J for three generations) were used throughout this study. Nontransgenic littermates were used as wild‐type (WT) controls. Mice were housed and fed on a 12 hours:12 hours light‐dark cycle at Temple University Animal Facility and were supervised by veterinary staff members. Standard care was provided to all mice used for experiments. All protocols applied to the mice in this study were approved and supervised by the Institutional Animal Care and Use Committee at Temple University.
Echocardiographic and hemodynamic analyses of cardiac function
Transthoracic two‐dimensional echocardiography was performed in anesthetized (2% inhaled isoflurane) Tg26 or WT mice (16– 18 weeks old) with a 12‐MHz probe as previously described.12, 13, 14, 15, 16 For in vivo hemodynamic measurements, a 1.4 French micromanometer‐tipped catheter (SPR‐671, Millar Instruments, Inc., Colorado Springs, CO, USA) was inserted into the right carotid artery and advanced into the LV of lightly anesthetized (tribromoethanol/amylene hydrate, Avertin; 2.5% wt/vol, 8 μL/g IP) mice with spontaneous respirations and placed on a heated (37°C) pad.12, 13, 14, 15, 16 Hemodynamic parameters including heart rate (beats/minute, bpm), LV end‐diastolic pressure (LVEDP), and maximal first time derivative of LV pressure rise (+dP/dt) and fall (−dP/dt) were recorded in closed‐chest mode, both at baseline and in response to increasing doses of isoproterenol (Iso; 0.1, 0.5, 1, 5, and 10 ng).12, 13, 14, 15, 16
Knockdown of BAG3 by Adv‐shRNA injection into LV
BAG3shRNA‐Ad construct was made using the BD Adeno‐X Expression Systems 2PT3674‐1 (Pr36024) and BD knockout RNAi Systems PT3739 (PR42756)(BD Biosciences‐Clontech, Palo Alto, CA, USA) as previously described.10 A double‐stranded DNA oligonucleotide against a specific BAG3 mRNA (5’‐AAG GUU CAG ACC AUC UUG GAA‐3’) was inserted in an RNAi‐ready pSIREN‐DNR vector designed to express a small hairpin RNA (shRNA) driven by the human Pol III–dependent U6 promoter. After ligation, this vector was used to transfer the shRNA expression cassette to the Adenoviral Acceptor Vector pLP‐Adeno‐X‐PRLS viral DNA (BD Adeno‐X Expression Systems 2) containing ΔE1/ΔE3 Ad5 genome by Cre‐loxP‐mediated recombination. An AdNull empty adenoviral vector was used as control. Adenoviruses were propagated in an HEK‐293 cell line, purified, and titered (plaque‐forming unit [pfu]) according to standard techniques.
After cleansing the skin with betadine solution, the left chest of anesthetized (2% inhaled isoflurane) mouse was opened, the heart exteriorized, and 25 μL (total volume) of Adv‐GFP (3.3 × 108 pfu) or Adv‐shRNA BAG3 (7.5 × 107 pfu) was directly injected to anterior and posterior LV wall and the apex. The heart was returned to the chest cavity and the wound sutured. The entire surgical procedure took <45 seconds. Typically >95% of animals survived the procedure. Survivors were allowed to recover for 7–10 days before hearts were excised and myocytes were isolated from areas of LV that fluoresced green (indicating successful virus‐mediated transfer).13
Isolation, adenoviral infection, and culture of adult murine cardiac myocytes
Cardiac myocytes were isolated from the green fluoresced areas of the septum and LV free wall of Tg26 or WT mice (16– 18 weeks old) according to the protocol of Zhou et al.17 and plated on laminin‐coated glass coverslips.18 Two hours after isolation, myocytes were infected with replication‐deficient Adv‐GFP (6.6 × 106 pfu/mL), Adv‐Tat (4.4 × 106 pfu/mL), Adv‐BAG3 (2.0 × 106 pfu/mL), Adv‐Tat + Adv‐BAG3 or Adv‐shRNA‐BAG3 (6.0 × 106 pfu/mL) in 1 mL of fetal bovine serum (FBS)‐free Eagle minimal essential medium (MEM) containing 0.2% bovine serum albumin, creatine (5 mM), carnitine (2 mM), taurine (5 mM), NaHCO3 (4.2 mM), penicillin (30 mg/L), gentamicin (4 mg/L), insulin–transferrin–selenium supplement, and 2,3‐butanedione monoxime (BDM, 10 mM) for 3 hours. An additional mL of MEM (with same supplements) was then added, and myocytes were cultured for 48 hours before measurements. Media was changed daily. We have previously demonstrated that under our culture conditions, adult mouse myocytes cultured for up to 48 hours maintained rod‐shape morphology, t‐tubule organization, and normal contractile function.19 Before measurements of myocyte contraction, cells were bathed with MEM without BDM and returned to the incubator (37°C) for 30 minutes. Coverslips containing cultured myocytes were then taken out of the incubator, mounted in Dvorak‐Stotler chamber, and bathed in fresh media before measurements. For the sake of brevity, myocytes infected with Adv‐GFP, Adv‐BAG3, Adv‐Tat, and Adv‐shRNA‐BAG3 are referred to as GFP, BAG3, Tat, and shBAG3 myocytes, respectively.
Myocyte‐shortening measurements
Myocytes adherent to coverslips were bathed in 0.6 mL of air‐ and temperature‐equilibrated (37°C), 4‐(2‐hydroxyethyl)‐1‐piperazine‐ethanesulfonic acid–buffered (20 mM, pH 7.4) medium 199 containing 1.8 mM [Ca2+]o. Measurements of myocyte contraction (2 Hz) were performed as previously described.18, 19
Immunoblotting
Mouse LV homogenates were prepared as previously described.18 Gradient (4–12%) gel was used to detect BAG3 in knockdown experiments, while 10% gel was used to assay for BAG3 in WT and Tg26 heart homogenates. Reducing conditions (5% β‐mercaptoethanol) were used. Rabbit antihuman BAG3 polyclonal antibody was obtained from Proteintech Group, Inc. (Chicago, IL, USA). Blots were washed and incubated with appropriate secondary antibody conjugated to horse radish peroxidase. Enhanced chemiluminescence (ECL, Amersham) was used for the detection of signals.
Statistics
All results are expressed as means ± SE. For analysis of a parameter (e.g., +dP/dt) as functions of group (WT, Tg26) and Iso, two‐way ANOVA was used to determine statistical significance. For analysis of BAG3 abundance, myocyte contraction amplitudes, myocyte shortening and relengthening velocities, and echocardiographic indices, one‐way ANOVA was used. A commercial software package (JMP version 10.0.2, SAS Institute; Cary, NC, USA) was used. In all analyses, p < 0.05 was taken to be statistically significant.
Results
Myocardial contractility and relaxation is normal in Tg26 mice under basal conditions
At 16–18 weeks of age, Tg26 mice had significantly lower LV mass, LV end‐diastolic, and end‐systolic volumes when compared to WT littermates (Table 1). However, contractility as determined by EF and FS was normal in Tg26 hearts (Table 1). This conclusion is supported by in vivo hemodynamic measurements since both basal and maximal (1 μM isoproterenol) +dP/dt were similar between WT and Tg26 mice (Figure 1; Table 1). Likewise, myocardial relaxation (−dP/dt) was not impaired in Tg26 mice (Table 1).
Table 1.
Echocardiographic and in vivo hemodynamic parameters of Tg26 mice
| WT | Tg26 | p | |
|---|---|---|---|
| Basal conditions | |||
| LV mass | 78.4 ± 2.9 (9) | 70.2 ± 1.4 (9) | 0.03 |
| LV volume diastolic, μL | 76.2 ± 2.5 | 63.4 ± 2.0 | 0.001 |
| LV volume systolic, μL | 26.0 ± 1.6 | 19.4 ± 1.4 | 0.006 |
| EF, % | 65.8 ± 1.8 | 69.6 ± 1.8 | 0.15 |
| FS, % | 36.0 ± 1.5 | 38.8 ± 1.4 | 0.20 |
| +dP/dt, mmHg/s | 5,689 ± 301 (4) | 6,073 ± 248 (4) | |
| Max +dP/dt, mmHg/s | 12,277 ± 144 | 12,432 ± 381 | 0.99 |
| −dP/dt, mmHg/s | 5,507 ± 299 | 5,986 ± 291 | |
| Max −dP/dt, mmHg/s | 9,222 ± 541 | 9,678 ± 332 | 0.87 |
| 10 days post Adv‐GFP injection into LV | |||
| +dP/dt, mmHg/s | 6,388 ± 524 (7) | 6,752 ± 399 (6) | |
| Max +dP/dt, mmHg/s | 11,946 ± 923 | 10,343 ± 409 | 0.015 |
| −dP/dt, mmHg/s | 6,236 ± 440 | 6,186 ± 301 | |
| Max −dP/dt, mmHg/s | 8,809 ± 668 | 7,847 ± 487 | 0.01 |
Values are means ± SE for number of mice in parentheses. WT, wild‐type; Tg26, hemizygous NL4‐3Δ gag/pol transgenic mice; LV, left ventricle; EF, ejection fraction; FS, fractional shortening; +dP/dt and –dP/dt, first time derivatives of LV pressure rise and fall, respectively. For +dP/dt and –dP/dt, two‐way ANOVA (group, Iso) was used to analyze data and P represents group x Iso interaction effects.
Figure 1.
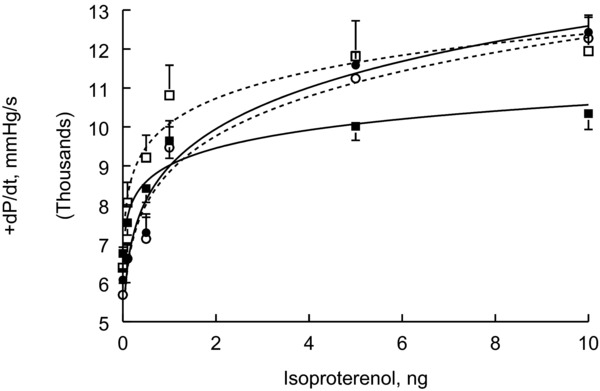
In vivo hemodynamics of WT and Tg26 mice, before and after open heart surgery. In vivo hemodynamics were measured in 18‐week‐old WT (•; n = 4) and Tg26 (o; n = 4) mice; and 10 days after open heart surgery in which Adv‐GFP was injected into three sites of LV during surgery. Shown are seven WT‐GFP (□) and six Tg26‐GFP (▪) hearts. Standard errors are not shown if they fall within the boundaries of the symbol. There are no differences in +dP/dt among WT, Tg26, and WT‐GFP hearts. Tg26‐GFP hearts performed significantly (p < 0.015) poorer when compared to WT‐GFP hearts.
Single myocyte contraction dynamics is normal in Tg26 mice
Myocytes from WT and Tg26 mice were infected with Adv‐GFP, and contractility examined after 24 and 48 hours in culture. Under basal conditions, there were no differences in contraction amplitudes, shortening, and relengthening velocities between WT‐GFP and Tg26‐GFP myocytes (Table 2).
Table 2.
Contraction dynamics of single myocytes isolated from WT and Tg26 hearts
| WT‐GFP | Tg26‐GFP | p | |
|---|---|---|---|
| Day 1 | |||
| Contraction amplitude | 5.53 ± 0.49 (17) | 5.04 ± 0.27 (24) | 0.35 |
| Shortening velocity | 0.92 ± 0.09 | 0.99 ± 0.06 | 0.50 |
| Relengthening velocity | 0.78 ± 0.08 | 0.78 ± 0.05 | 0.95 |
| Day 2 | |||
| Contraction amplitude | 5.43 ± 0.51 (14) | 4.90 ± 0.37 (21) | 0.40 |
| Shortening velocity | 0.88 ± 0.10 | 0.80 ± 0.08 | 0.49 |
| Relengthening velocity | 0.81 ± 0.08 | 0.64 ± 0.06 | 0.08 |
Values are means ± SE for number of myocytes in parenthesis. Contraction amplitudes are in % resting cell length, while shortening and relengthening velocities are in % resting cell length/s.
Cardiac contractility in Tg26 mice does not fully recover after open heart surgery
To induce stress in Tg26 mice, we performed open heart surgery and injected Adv‐GFP into LV of both WT and Tg26 mice. Ten days after surgery, myocardial contractility (+dP/dt) in WT but not Tg26 mice recovered to that observed in nonoperated animals (Figure 1; Table 1). Similarly, myocardial relaxation was significantly compromised in Tg26 mice after open heart surgery (Table 1).
BAG3 down‐regulation by Adv‐shRNA significantly depresses cardiac contractility in Tg26 mice
We have recently reported that cardiac BAG3 levels were significantly reduced after myocardial infarction (MI) in pigs and after transverse aortic constriction (TAC) in mice.9 To simulate these pathological conditions, we attempted to knockdown endogenous BAG3 by shRNA. We initially infected isolated myocytes with Adv‐shRNA‐BAG3 and examined BAG3 expression and myocyte function after 2 days of culture. There was no significant knockdown of BAG3 protein after 2 days of Adv‐shRNA‐BAG3 infection (Figure 2 A). Lack of significant BAG3 knockdown after 2 days of culture was independently confirmed by the observation that contractility in WT‐shBAG3 myocytes was similar to control WT‐GFP myocytes (Table 3). These observations suggested that endogenous cardiac BAG3 turnover may be slow and it may require more than 2 days for shRNA‐BAG3 to knockdown BAG3 levels in mammalian hearts.
Figure 2.
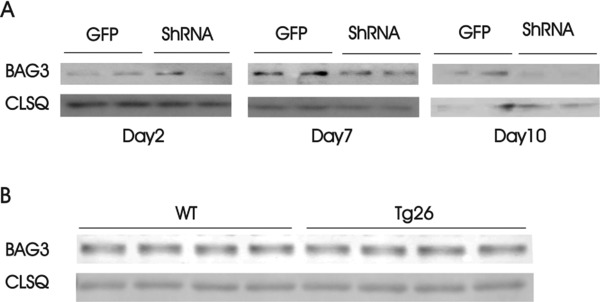
Knockdown of BAG3 in adult mouse hearts by Adv‐shRNA‐BAG3 and endogenous BAG3 levels in WT and Tg26 hearts. (A) Left panel: myocytes isolated from WT LV and septum were infected with Adv‐GFP or Adv‐shRNA‐BAG3, cultured for 2 days before BAG3 was measured (Methods section). Middle and right panel: Adv‐GFP or Adv‐shRNA‐BAG3 was injected directly into LV of WT mice, hearts were harvested after 7 (middle panel) and 10 days (right panel), and BAG3 measured in heart homogenates. (B) Homogenates were prepared from WT and Tg26 hearts and BAG3 measured. Calsequestrin was used as a loading control.
Table 3.
Contraction dynamics in WT‐GFP and WT‐shBAG3 myocytes after 48 hours
| WT‐GFP | WT‐shBAG3 | p | |
|---|---|---|---|
| Contraction amplitude | 5.66 ± 0.33 (33) | 6.15 ± 0.44 (17) | 0.98 |
| Shortening velocity | 0.88 ± 0.06 | 1.03 ± 0.09 | 0.30 |
| Relengthening velocity | 0.77 ± 0.05 | 0.91 ± 0.07 | 0.45 |
Values are means ± SE for number of myocytes in parenthesis. Single myocyte contraction is examined after 48 hours in culture. Contraction amplitudes are in % resting cell length, while shortening and relengthening velocities are in % resting cell length/s.
Since isolated adult mouse LV myocytes maintain their rod‐shaped morphology and contractile phenotype only for a short time (48–72 hours) in culture,19 we injected Adv‐shRNA‐BAG3 into LV of WT and Tg26 hearts and examine BAG3 expression and cardiac contractility 10 days later. Using this approach, BAG3 levels in WT hearts decreased 7–10 days after Adv‐shRNA‐BAG3 injection (Figure 2 A). Specifically, BAG3 levels significantly decreased from 2.65 ± 0.56 in WT‐GFP (n = 5) to 1.21 ± 0.22 arbitrary units (a.u.) in WT‐shBAG3 (n = 5) hearts (p < 0.045). In addition, there were no significant (p = 0.16) differences in endogenous BAG3 levels between WT (3.96 ± 0.02; n = 4) and Tg26 (4.14 ± 0.11 a.u.) hearts (Figure 2 B).
In vivo hemodynamic measurements showed that Tg26‐shBAG3 hearts suffered significantly (p < 0.0001) larger decrements in +dP/dt than WT‐shBAG3 hearts (Figure 3). Down‐regulation of BAG3 in WT hearts by shBAG3 also resulted in a trend for a decrease in +dP/dt (p < 0.31; Figure 3).
Figure 3.
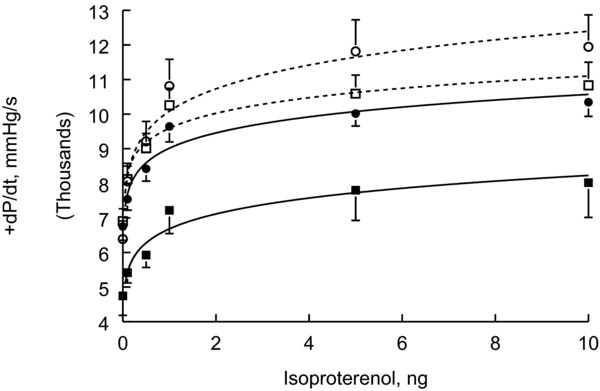
BAG3 down‐regulation depresses contractility in Tg26 hearts. Adv‐GFP or Adv‐shRNA‐BAG3 was injected into LV of WT and Tg26 hearts, and animals were allowed to recover for 10 days before in vivo hemodynamics were measured. Shown are 7 WT‐GFP (o), 9 WT‐shBAG3 (□), 6 Tg26‐GFP (•), and 6 Tg26‐shBAG3 (▪) hearts. Standard errors are not shown if they fall within the boundaries of the symbol. Contractility of Tg26‐shBAG3 hearts is significantly (p < 0.0001) depressed when compared to Tg26‐GFP hearts. Tg26‐GFP hearts also demonstrated significantly (p < 0.015) lower +dP/dt when compared to WT‐GFP hearts. WT‐shBAG3 hearts tended to contract lower than WT‐GFP hearts although the difference does not reach statistical significance.
BAG3 rescues contraction abnormalities in myocytes overexpressing Tat
The HIV‐1 protein Tat plays a critical role in the pathogenesis of the virus. It is well established that Tat is toxic to many cells and induces apoptosis by triggering several cell death signaling pathways and/or preventing prosurvival events.3, 5, 20 Overexpression of Tat in adult WT myocytes significantly reduced single myocyte contraction amplitude compared to control WT myocytes expressing GFP (Figure 4; Table 4). While overexpression of BAG3 in WT myocytes did not affect contractility at baseline, BAG3 overexpression rescued the deleterious effects of Tat in single myocyte contractility in that there were no differences in maximal contraction amplitudes, shortening, and relengthening velocities between WT‐GFP and WT‐Tat‐BAG3 myocytes (Figure 4; Table 4).
Figure 4.
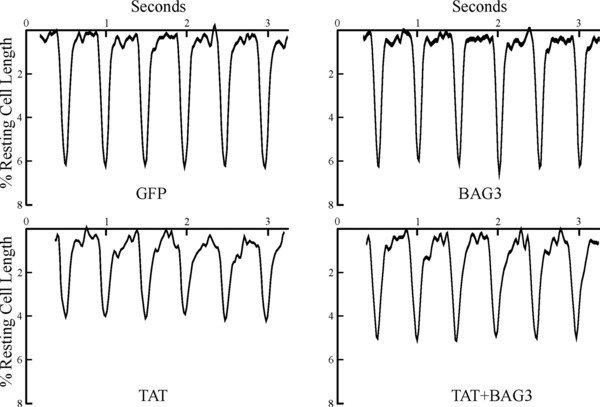
BAG3 overexpression rescues contractile abnormalities in adult myocytes overexpressing Tat. Isolated WT myocytes were infected with Adv‐GFP, Adv‐Tat, Adv‐BAG3, or Adv‐Tat + Adv‐BAG3, and culture for 48 hours before contractility measurements (Methods section). Myocytes were paced (2 Hz) to contract at 37°C and [Ca2+]o of 1.8 mM. Shown are steady‐state paced twitches from myocytes expressing GFP, BAG3, Tat, and Tat + BAG3. Composite results are summarized in Table 4.
Table 4.
BAG3 rescues contractile abnormalities in WT myocytes overexpressing Tat
| GFP | BAG3 | Tat | BAG3 + Tat | |
|---|---|---|---|---|
| Contraction amplitude | 5.66 ± 0.33 (33) | 6.15 ± 0.44 (17) | 4.27 ± 0.29* (22) | 5.54 ± 0.31# (21) |
| Shortening velocity | 0.88 ± 0.06 | 1.03 ± 0.09 | 0.69 ± 0.06* | 0.85 ± 0.07 |
| Relengthening velocity | 0.77 ± 0.05 | 0.91 ± 0.07 | 0.59 ± 0.05* | 0.71 ± 0.06 |
Values are means ± SE for number of myocytes in parenthesis. Single myocyte contraction is examined after 48 hours in culture. Contraction amplitudes are in % resting cell length, while shortening and relengthening velocities are in % resting cell length/s. *p < 0.04, GFP or BAG3 versus Tat; #p < 0.004, Tat versus BAG3 + Tat.
Discussion
Previous studies on AIDS cardiomyopathy utilizing different transgenic models have produced conflicting results with respect to cardiac dysfunction, or lack thereof. For example, high levels of cardiac‐specific Tat overexpression in hemizygous mice resulted in 46% increase in LV mass and reduction in FS from 40% to 28% at 90 days.21 By contrast, in vivo hemodynamic measurements in the same transgenic mice did not reveal any evidence of ventricular dysfunction until the mice were at least 6 months of age.22 In another HIV transgenic mouse in which gag/pol/env genes were deleted, accumulation of viral mRNA occurred in all tissues.23 At 16 weeks, there was no cardiac dysfunction as measured in an isolated working heart preparation.5 With respect to Tg26 mice, at 10 weeks, there were no differences in FS between WT and Tg26 hearts.4 At 12–16 weeks, when examined as in vitro working heart preparations under basal loading conditions (5 mL/minute cardiac output and 50 mmHg mean aortic pressure), both +dP/dt and –dP/dt in Tg26 hearts showed modest but statistically significant decreases compared to WT hearts. However, under conditions when preload and afterload were varied, there were no longer differences in +dP/dt and –dP/dt between WT and Tg26 hearts under maximum pressure, minimum pressure, or maximum volume loading conditions.4 Only under minimum volume, loading condition was –dP/dt (but not +dP/dt) significantly reduced in Tg26 hearts. The reasons for the discrepancies may relate to different genetic models, global versus cardiac‐specific expression of HIV‐1 proteins, age at which cardiac function was measured, and techniques used to evaluate cardiac contractility (in vivo echocardiography vs. in vitro isolated working heart preparations).
The first major observation is that under basal conditions, contractility of Tg26 hearts (16–18 weeks) was similar to that of WT hearts, whether measured noninvasively by echocardiography or invasively by conductance catheter in a closed‐chest preparation (Figure 1; Table 1). Similarly, Lewis et al.4 reported that at 10 weeks, FS was similar between WT and Tg26 hearts. At 12– 16 weeks, +dP/dt and –dP/dt were largely similar between WT and Tg26 measured in isolated working heart preparations. The only differences between our results and that of Lewis et al.4 are: (i) LV mass and LV volumes were smaller in Tg26 hearts at 16–18 weeks (Table 1), while heart weights and heart/body weights were not different between WT and Tg26 mice at 10 weeks4; and (ii) differences in +dP/dt and –dP/dt between WT and Tg26 isolated working heart preparations were observed under one specific loading condition and not across the entire range of preload and afterload conditions,4 whereas we observed no differences in hemodynamic performance in our in vivo measurements (Figure 1; Table 1). In addition, single myocyte contraction and relaxation properties were similar between WT and Tg26 myocytes (Table 2). The weight of current evidence indicates that under basal conditions, Tg26 hearts/myocytes exhibited normal contractility and relaxation compared to WT hearts/myocytes.
The second major observation is that 10 days after open heart surgery, WT but not Tg26 hearts recovered inotropic and lusitropic characteristics to preoperative levels (Figure 1; Table 1). This observation suggests that Tg26 mice do not tolerate surgical stress well and has clinical implications that chronic HIV‐1 infection may delay cardiac recovery following a major operation.
Mutations in BAG3 have been linked to hereditary cardiomyopathy24 and reductions in endogenous BAG3 levels are observed in porcine hearts post‐MI and murine hearts post‐TAC.9 In addition, compared to nonfailing human hearts, BAG3 protein (but not mRNA) levels in failing human hearts are significantly decreased.24 Thus, the third major finding is that BAG3 can be down‐regulated by shRNA in adult hearts (Figure 2 A). The observation that BAG3 levels were unchanged after 2 days of Adv‐shRNA‐BAG3 infection (Figure 2 A) suggests that the turnover of BAG3 in adult mouse myocytes is relatively slow. By comparison, antisense oligonucleotides targeted to the start codon (nucleotides −11 to +9) of guinea pig cardiac Na+/Ca2+ exchanger mRNA resulted in 40% decrease in Na+/Ca2+ exchanger expression after only 2 days of exposure.25
Down‐regulation of BAG3 tended to decrease contractility in WT hearts but significantly reduced +dP/dt in Tg26 hearts (Figure 3). This observation suggests that BAG3 may interact with HIV‐1 proteins and thereby afford protection in AIDS cardiomyopathy. To test this hypothesis, we expressed Tat in adult myocytes and observed decreases in contraction amplitudes, shortening and relengthening velocities when compared to control myocytes expressing GFP (Figure 4; Table 4). Our results are in agreement with previous reports in which myocytes isolated from cardiac‐specific hemizygous Tat overexpression mice exhibited significantly decreased peak shortening amplitude, and shortening and relengthening velocities when compared to myocytes isolated from WT littermates.26 More importantly, BAG3 overexpression normalized contraction amplitudes in myocytes expressing Tat (Figure 4; Table 4). Our observations that BAG3 knockdown impacted cardiac function much more negatively in Tg26 compared to WT hearts, and that BAG3 overexpression rescued contraction abnormalities in myocytes expressing Tat, suggest that BAG3 is likely an important factor in the pathogenesis of AIDS cardiomyopathy.
In summary, under basal conditions, Tg26 heart/myocyte function was similar to that observed in WT animals. Cardiac function did not fully recover in Tg26 mice 10 days after cardiac surgery. BAG3 down‐regulation significantly impaired cardiac contractility and relaxation in Tg26 mice, whereas BAG3 overexpression rescued contraction abnormalities in myocytes overexpressing Tat. We conclude that Tg26 mice do not tolerate surgical stress well and that BAG3 likely plays a key role in AIDS cardiomyopathy.
Sources of Funding
This work was supported in part by the National Institutes of Health GrantsRO1‐HL58672, RO1‐HL74854, and RO1‐HL123093 (JYC); PO1‐HL91799 (Project 2) and RO1‐HL123093 (AMF); RO1‐HL56025, RO1‐HL61690, RO1‐HL85503, PO1‐HL‐75443, and PO1‐HL‐91799 (WJK); R01‐HL105414 (DGT); and RO1‐HL123093 (JG & KK). Core facility services were provided by CNACNIMH P30MH092177 at Temple University School of Medicine.
Acting EIC – Scott A. Waldman, M.D., Ph.D.
References
- 1. Herskowitz A, Vlahov D, Willoughby S, Chaisson RE, Schulman SP, Neumann DA, Baughman KL. Prevalence and incidence of left ventricular dysfunction in patients with human immunodeficiency virus infection. Am J Cardiol. 1993; 71: 955–958. [DOI] [PubMed] [Google Scholar]
- 2. Herskowitz A. Cardiomyopathy and other symptomatic heart diseases associated with HIV infection. Curr Opin Cardiol. 1996; 11: 325–331. [DOI] [PubMed] [Google Scholar]
- 3. Felker GM, Thompson RE, Hare JM, Hruban RH, Clemetson DE, Howard DL, Baughman KL, Kasper EK. Underlying causes and long‐term survival in patients with initially unexplained cardiomyopathy. New Eng J Med. 2000; 342: 1077–1084. [DOI] [PubMed] [Google Scholar]
- 4. Lewis W, Grupp IL, Grupp G, Hoit B, Morris R, Samarel AM, Bruggeman L, Klotman P. Cardiac dysfunction occurs in the HIV‐1 transgenic mouse treated with zidovudine. Lab Invest. 2000; 80: 187–197. [DOI] [PubMed] [Google Scholar]
- 5. Lewis W. Use of the transgenic mouse in models of AIDS cardiomyopathy. AIDS. 2003; 17(Suppl 1): S36–S45. [DOI] [PubMed] [Google Scholar]
- 6. Rosati A, Graziano V, De Laurenzi V, Pascale M, Turco MC. BAG3: a multifaceted protein that regulates major cell pathways. Cell Death Dis. 2011; 2: e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McCollum AK, Casagrande G, Kohn EC. Caught in the middle: the role of Bag3 in disease. Biochem J. 2010; 425: e1–e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ulbricht A, Hohfeld J. Tension‐induced autophagy: may the chaperone be with you. Autophagy. 2013; 9: 920–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Knezevic T, Myers VD, Gordon J, Tilley DG, Sharp TE, 3rd , Wang J, Khalili K, Cheung JY, Feldman AM. BAG3: a new player in the heart failure paradigm. Heart Fail Rev. 2015; 20: 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rosati A, Khalili K, Deshmane SL, Radhakrishnan S, Pascale M, Turco MC, Marzullo L. BAG3 protein regulates caspase‐3 activation in HIV‐1‐infected human primary microglial cells. J Cell Physiol. 2009; 218(2): 264–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kopp JB, Klotman ME, Adler SH, Bruggeman LA, Dickie P, Marinos NJ, Eckhaus M, Bryant JL, Notkins AL, Klotman PE. Progressive glomerulosclerosis and enhanced renal accumulation of basement membrane components in mice transgenic for human immunodeficiency virus type 1 genes. Proc Natl Acad Sci U S A. 1992; 89: 1577–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang J, Chan TO, Zhang XQ, Gao E, Song J, Koch WJ, Feldman AM, Cheung JY. Induced overexpression of Na+/Ca2+ exchanger transgene: altered myocyte contractility, [Ca2+]i transients, SR Ca2+ contents and action potential duration. Am J Physiol Heart Circ Physiol. 2009; 297: H590–H601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang J, Gao E, Rabinowitz J, Song J, Zhang XQ, Koch WJ, Tucker AL, Chan TO, Feldman AM, Cheung JY. Regulation of in vivo cardiac contractility by phospholemman: role of Na+/Ca2+ exchange. Am J Physiol Heart Circ Physiol. 2011; 300: H859–H868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Song J, Gao E, Wang J, Zhang XQ, Chan TO, Koch WJ, Shang X, Joseph JI, Peterson BZ, Feldman AM, et al. Constitutive overexpression of phospholemman S68E mutant results in arrhythmias, early mortality and heart failure: potential involvement of Na+/Ca2+ exchanger. Am J Physiol Heart Circ Physiol. 2012; 302: H770–H781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang J, Gao E, Chan TO, Zhang XQ, Song J, Shang X, Koch WJ, Feldman AM, Cheung JY. Induced overexpression of Na+/Ca2+ exchanger does not aggravate myocardial dysfunction induced by transverse aortic constriction. J Card Fail. 2013; 19: 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang J, Gao E, Song J, Zhang XQ, Li J, Koch WJ, Tucker AL, Philipson KD, Chan TO, Feldman AM, et al. Phospholemman and β‐adrenergic stimulation in the heart. Am J Physiol Heart Circ Physiol. 2010; 298: H807–H815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou YY, Wang SQ, Zhu WZ, Chruscinski A, Kobilka BK, Ziman B, Wang S, Lakatta EG, Cheng H, Xiao RP. Culture and adenoviral infection of adult mouse cardiac myocytes: methods for cellular genetic physiology. Am J Physiol Heart Circ Physiol. 2000; 279: H429–H436. [DOI] [PubMed] [Google Scholar]
- 18. Tucker AL, Song J, Zhang XQ, Wang J, Ahlers BA, Carl LL, Mounsey JP, Moorman JR, Rothblum LI, Cheung JY. Altered contractility and [Ca2+]i homeostasis in phospholemman‐deficient murine myocytes: role of Na+/Ca2+ exchange. Am J Physiol Heart Circ Physiol. 2006; 291: H2199–H2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Song J, Zhang XQ, Wang J, Cheskis E, Chan TO, Feldman AM, Tucker AL, Cheung JY. Regulation of cardiac myocyte contractility by phospholemman: Na+/Ca2+ exchange vs. Na+‐K+‐ATPase. Am J Physiol Heart Circ Physiol. 2008; 295: H1615–H1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Homma S, Iwasaki M, Shelton GD, Engvall E, Reed JC, Takayama S. BAG3 deficiency results in fulminant myopathy and early lethality. Am J Pathol. 2006; 169: 761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Raidel SM, Haase C, Jansen NR, Russ RB, Sutliff RL, Velsor LW, Day BJ, Hoit BD, Samarel AM, Lewis W. Targeted myocardial transgenic expression of HIV Tat causes cardiomyopathy and mitochondrial damage. Am J Physiol Heart Circ Physiol. 2002; 282: H1672–H1678. [DOI] [PubMed] [Google Scholar]
- 22. Fang Q, Kan H, Lewis W, Chen F, Sharma P, Finkel MS. Dilated cardiomyopathy in transgenic mice expressing HIV Tat. Cardiovasc Toxicol. 2009; 9: 39–45. [DOI] [PubMed] [Google Scholar]
- 23. Tinkle BT, Ngo L, Luciw PA, Maciag T, Jay G. Human immunodeficiency virus‐associated vasculopathy in transgenic mice. J Virol. 1997; 71: 4809–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Feldman AM, Begay RL, Knezevic T, Myers VD, Slavov DB, Zhu W, Gowan K, Graw SL, Jones KL, Tilley DG, et al. Decreased levels of BAG3 in a family with a rare variant and in idiopathic dilated cardiomyopathy. J Cell Physiol. 2014; 229: 1697–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eigel BN, Hadley RW. Antisense inhibition of Na+/Ca2+ exchange during anoxia/reoxygenation in ventricular myocytes. Am J Physiol Heart Circ Physiol. 2001; 281: H2184–H2190. [DOI] [PubMed] [Google Scholar]
- 26. Chen F, Lewis W, Hollander JM, Baseler W, Finkel MS. N‐acetylcysteine reverses cardiac myocyte dysfunction in HIV‐Tat proteinopathy. J Appl Physiol. 2012; 113: 105–113. [DOI] [PubMed] [Google Scholar]