

Abstract
DNA strand breaks containing 3′-phosphoglycolate (3′-PG) ends are the major lesions induced by ionizing radiation. The repair of this lesion is not completely understood and several activities are thought to be involved in processing of 3′-PG ends. In this study we examined activities in human whole cell extracts (WCE) responsible for removal of 3′-PG. Using a radiolabelled oligonucleotide containing a single nucleotide gap with internal 5′-phosphate and 3′-PG ends, we demonstrate that the major 3′-PG activity in human WCE is Mg2+ dependent and that this activity co-purifies with AP endonuclease 1 (APE1) over phosphocellulose and gel filtration chromatography. Furthermore, immunodepletion of APE1 from active gel filtration fractions using APE1 specific antibodies reveals that the major activity against 3′-PG in human WCE is APE1.
INTRODUCTION
Oxidative DNA damage can be induced by ionizing radiation, by reactive oxygen species and by some antitumour agents such as bleomycin (1,2). Oxidative damage to the sugar moiety of DNA results in DNA fragmentation and the subsequent generation of DNA breaks with damaged ends (3,4). These terminal 3′-blocking groups, mostly 3′-phosphate or 3′-PG, must be removed and the single nucleotide gap created is repaired by proteins of base excision repair (BER). Repair involves one nucleotide incorporation and ligation by a DNA polymerase and a DNA ligase, respectively.
Previous works suggest that the major human AP endonuclease (APE1) is also involved in removal of 3′-phosphoglycolate (3′-PG) in human cells (5–8). APE1 belongs to a large family of nucleases homologous to exonuclease III that mediate the removal of 3′-PG in Escherichia coli (9). These enzymes act by cleaving the phosphodiester bond immediately 5′ to the phosphoglycolate residue creating a 3′-hydroxyl end. APE1 has been cloned (10,11) and recombinant APE1 was shown to be able to remove 3′-PG from strand breaks generated by γ-radiation and radiomimetic chemicals (7,12,13). However, analysis of 3′-PG activities in human cell extracts revealed a more complex picture. Demple's group described two distinct human DNA diesterases that hydrolyse 3′ end blocking deoxyribose fragments from oxidized DNA, while Winters et al. purified three chromatographically distinct human enzyme activities from HeLa cells using bleomycin treated DNA as a substrate (8,14). Moreover, while Mitra's group, using bleomycin treated DNA as a substrate, demonstrated that APE1 is the limiting activity for repair of 3′-blocking damage at DNA single-strand breaks, Winters et al., as adjudged by inhibition of APE1 with neutralizing APE1 antibodies, concluded that removal of 3′-PG is not dependent upon APE1 activity (15,16). The picture becomes even more obscure with the finding that tyrosyl–DNA phosphodiesterase (Tdp1) is also able to remove 3′-PG. Yeast Tdp1 was originally isolated as an activity that hydrolyses the phosphodiester bond linking tyrosine to a 3′ DNA end, formed as intermediates in DNA relaxation by topoisomerase I (17). The human homologue (hTdp1) was subsequently cloned and was shown to have 3′-PG activity (18).
Consequently, further studies are required to clarify the major activity in human cell extracts involved in repair of 3′-PG. In this study we used an oligonucleotide substrate containing a single-strand break with a 1 nt gap flanked by 5′-phosphate and 3′-PG ends and fractionated cell extract to address this question.
MATERIALS AND METHODS
Materials
Synthetic oligodeoxyribonucleotides were purchased from MWG-Biotech and gel purified on a 20% polyacrylamide gel. The 3′-PG substrate (5′-CAATAGAGTAACACGGpg-3′) was synthesized by Eurogentec using the method of Urata and Akagi (19), in which the 3′-phosphoglyceryl residue is oxidized sequentially by sodium periodate (NaIO4) and sodium chlorite (NaClO2). The oligonucleotide was purified by high-performance liquid chromatography (HPLC) and the presence of 3′-phosphoglycolate was confirmed by mass spectroscopy. [γ-32P]ATP (3000 Ci/mmol) was purchased from PerkinElmer Life Sciences. Recombinant human APE1 and DNA polymerase β (Pol β) were purified as described previously (20).
Antibodies
Tdp1 antibodies were kindly provided by K. Caldecott, Genome Damage and Stability Centre, University of Sussex, UK. Antibodies against rat Pol β and human APE1 were raised in rabbit and affinity-purified as described (21).
Substrate labelling
Oligonucleotides were 5′ end labelled with [γ-32P]ATP using T4-polynucleotide kinase and unincorporated label removed on a Sephadex G-25 spin column. To prepare the 3′-PG substrate, an oligonucleotide 5′-CAATAGAGTAACACGGCpg-3′ was annealed to the hairpin oligonucleotide 5′-pCGACCAGTCCCTGCCTTTTGGCAGGGACTGGTCGGCCGTGTTACTCTATTG-BIOT-3′ at 90°C for 3–5 min followed by slow cooling to room temperature. To prepare the 5′-phosphate nick containing-substrate, the oligonucleotide 5′-CAATAGAGTAACACGGC-3′ was labelled and annealed to the hairpin substrate as above.
Fractionation of cell extracts
HeLa cell pellets were purchased from Paragon, USA. Whole cell extracts (WCE) were prepared by the method of Manley et al. (22), dialysed overnight against buffer containing 25 mM Hepes–KOH, pH 7.9, 100 mM KCl, 12 mM MgCl2, 0.1 mM EDTA, 17% glycerol and 2 mM DTT and aliquots frozen at −80°C for the repair assays. An aliquot of this extract (100 mg protein) was then purified by phosphocellulose chromatography using a step elution of 150 mM KCl (PC-FI) and 1 M KCl (PC-FII) as previously described (23). Proteins (10 mg) from the PC-FII fraction were further separated by gel filtration on a Superdex-75 column (Amersham) in a buffer containing 50 mM HEPES, pH 7.9, 150 mM KCl, 1 mM EDTA, 1 mM DTT and 1 mM phenylmethylsulfonyl fluride (PMSF). Fractions of 0.5 ml were collected and tested for the ability to remove 3′-PG as described in the repair assays section.
Repair assays
Repair assays contained 250 fmol oligonucleotide per reaction in 20 μl Reaction buffer containing 50 mM Hepes–KOH, pH 7.8, 50 mM KCl, 10 mM MgCl2, 0.5 mM EDTA, 1.5 mM DTT, 2.5% glycerol, 20 μM dCTP, 20 μM dATP, 20 μM dGTP, 20 μM dTTP, 2 mM ATP, 25 mM phosphocreatine (diTris salt, Sigma), 2.5 μg creatine phosphokinase (type I, Sigma), 0.25 mM NAD+ and 1 μg of carrier DNA (single-stranded 30mer oligonucleotide). Where indicated, dNTPs were removed from the reaction and MgCl2 was substituted with 10 mM EDTA. Reactions were incubated for the time indicated at 30°C and 20 μl formamide loading dye added (95% formamide, 0.02% xylene cyanol, 0.02% bromophenol blue) and the samples heated to 95°C for 5 min. Products were subsequently analysed by 20% denaturing PAGE and gels exposed to intensifying screens at 4°C prior to analysis by phosphorimaging.
Repair assays using whole cell extracts
For assays using WCE, streptavidin magnetic beads were blocked in 5% milk and subsequently washed with Binding buffer (20 mM Tris–HCl pH 7.5, 0.5 M NaCl, 1 mM EDTA) using a magnetic separator rack. Beads were then incubated with either the 3′-PG gap or the nick-containing radiolabelled hairpin substrates complete with a 3′-biotinylated moiety, at room temperature with agitation for 30 min in Binding buffer and the DNA beads were subsequently washed with Wash buffer (25 mM Hepes, pH 7.9, 100 mM KCl, 12 mM MgCl2, 1 mM EDTA, 5% glycerol and 2 mM DTT). The DNA beads (250 fmol DNA per reaction) were incubated with 100 μg WCE in 50 μl Reaction buffer at 30°C for the time indicated prior the addition of 12.5 μl of 500 mM EDTA to stop the reaction. Beads were washed twice with 50 μl 10 mM Tris–HCl, pH 8.0, 100 mM EDTA and resuspended in 20 μl formamide loading dye. Samples were heated to 95°C for 5 min prior to separation of the DNA on a 20% denaturing polyacrylamide gel and the gel exposed to intensifying screens at 4°C prior to analysis by phosphorimaging.
Immunodepletion of APE1
Protein A Sepharose CL-4B was allowed to swell for 1 h in phosphate-buffered saline (PBS) and following washes with PBS a 50% suspension was prepared. To 100 μl suspension was added 5 μl APE1 antibodies or 5 μl pre-immune serum and incubated for 2 h at 4°C with gentle shaking. The APE1–Sepharose was washed five times with 0.3 ml PBS and resuspended in 100 μl PBS. Fractions purified from phosphocellulose and gel filtration chromatography (20 μl) were mixed with 20 μl APE1–Sepharose and incubated for 2 h at 4°C with gentle shaking. The mixture was filtered through Spin-X columns at 4°C and aliquots taken and SDS–PAGE loading dye (25 mM Tris–HCl, pH 6.8, 2.5% mercaptoethanol, 1% SDS, 5% glycerol, 1 mM EDTA, 0.15 mg/ml bromophenol blue) added. The samples were heated to 90°C for 3 min before loading on a 10% SDS–polyacrylamide gel followed by transfer to a PVDF membrane and immunoblot analysis with the indicated antibodies. Aliquots were also taken for 3′-PG repair assays as described above.
RESULTS
Substrate characterization
A 16mer 3′-PG-containing oligonucleotide was generated by sequential oxidation of the 3′-phosphoglyceryl residue by sodium periodate (NaIO4) and sodium chlorite (NaClO2) and its structure confirmed by mass spectroscopy. The oligonucleotide was 5′ end labelled and compared, by 20% denaturing PAGE, to the corresponding parent 16mer oligonucleotide and a 17mer oligonucleotide (Figure 1A). The 3′-PG oligonucleotide has a distinct mobility shift, running slower than the parent oligonucleotide but faster than the corresponding 17mer. The 3′-PG oligonucleotide was used to construct a duplex oligonucleotide substrate, complete with a hairpin loop to protect the DNA ends from nuclease digestion, and containing a 5′-phosphate within the gap (Figure 1B). This substrate was further characterized by the ability of purified recombinant human APE1 to remove 3′-PG and to activate the 3′ end for DNA polymerase synthesis. In agreement with previous results (6,7,10) we have shown that purified recombinant APE1 is able to remove 3′-PG in a dose-dependent manner, (Figure 1C) and that this activity is magnesium dependent, as in the presence of EDTA no cleavage of 3′-PG can be observed (data not shown). The activity of APE1 on the 3′-PG-containing substrate also primes the DNA for synthesis by Pol β, as primer extension is observed only on the addition of both proteins, (Figure 1D, compare lanes 2 and 4) further confirming the structure of the substrate used.
Figure 1.
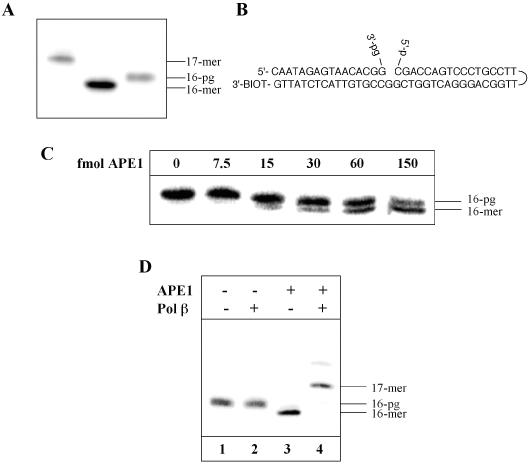
Characterization of the 3′-PG-containing oligonucleotide substrate. An oligonucleotide (16mer) containing a 3′-PG end was 5′ end labelled and compared by 20% denaturing PAGE to the parent 16mer and a 1 nt addition product 17mer (A). The 5′ end labelled 3′-PG oligonucleotide was annealed to an oligonucleotide (51mer) containing a hairpin loop and a 5′-phosphate residue to generate a substrate for use in repair assays (B). This substrate was incubated with APE1 (0–150 fmol) for 10 min at 30°C prior to the addition of formamide loading dye. An aliquot was analysed by 20% denaturing PAGE and phosphorimaging (C). Primer extension from 3′-PG ends by human Pol β (50 fmol) was also investigated in the presence and absence of APE1 (150 fmol) (D).
Fractionation of 3′-PG activity from human WCE
We then tested human WCE for the ability to remove 3′-PG and subsequently undergo full repair of an oligonucleotide substrate. Using WCE we were able to demonstrate repair of an oligonucleotide containing an internal gap with 3′-PG and 5′-phosphate ends, with ∼30% of the substrate repaired within 8 min (Figure 2A). This is in comparison to the rapid repair of a nick substrate with a 3′-OH and a 5′-phosphate end, which is fully repaired within 4 min (Figure 2B). In WCE, removal of 3′-PG is followed by rapid addition of the first nucleotide by Pol β and ligation by DNA ligase. Correspondingly, intermediate repair products (16mer) arising as a result of 3′-PG removal activity do not accumulate during repair in WCE (Figure 2A). To visualize 3′-PG activity we blocked repair synthesis by removal of dNTPs from the reaction mixtures. Under these conditions we were able to detect removal of 3′-PG in WCE (Figure 2C). The 3′-PG activity in WCE is magnesium dependent as the cleavage of 3′-PG cannot be observed in the presence of EDTA (data not shown).
Figure 2.
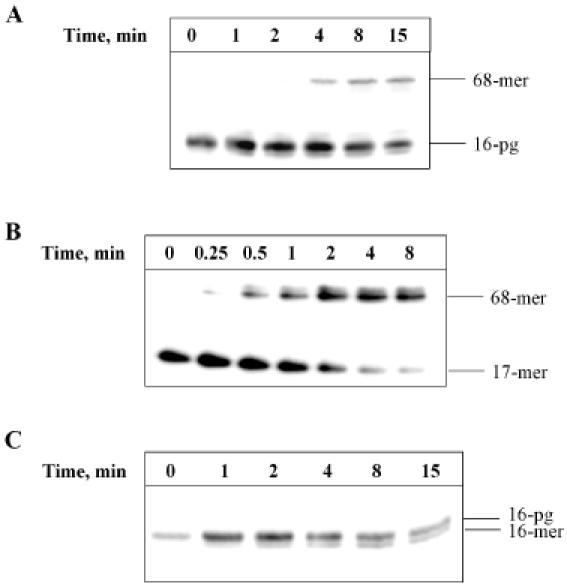
Repair of a 3′-PG-containing duplex oligonucleotide by human WCE. The 5′ end labelled 3′-PG oligonucleotide was annealed to an oligonucleotide (51mer) containing a hairpin loop and a 5′-phosphate residue and bound to streptavidin beads prior to incubation with 100 μg WCE for 0–15 min at 30°C. Reactions were stopped by the addition of 100 mM EDTA, the beads washed and the DNA resuspended in formamide loading dye prior to analysis by 20% denaturing PAGE and phosphorimaging (A). Repair of a 5′-phosphate nick-containing oligonucleotide was observed by incubation of the substrate with 100 μg WCE for 0–8 min at 30°C (B). 3′-PG specific activity in WCE was observed by conducting reactions of the 3′-PG duplex oligonucleotide substrate with WCE in the absence of dNTPs (C).
We proceeded to further characterize the 3′-PG activity from human WCE using phosphocellulose chromatography, eluting proteins off into two fractions using low (0.15 M KCl, PC-FI) and high salt (1 M KCl, PC-FII) elution (23). These fractions were tested for their ability to remove 3′-PG. The majority of the 3′-PG activity observed in human WCE is retained in the high-salt wash fraction PC-FII only (Figure 3A), which has also been found to contain the majority of BER proteins (23). No other residual activity for the removal of 3′-PG was observed in the low-salt wash fraction PC-FI. The proteins within fraction PC-FII, which contained the major 3′-PG activity, were further separated according to their size by gel filtration chromatography. The fractions were subsequently tested for their ability to remove 3′-PG and it was revealed that the activity was present predominantly within fractions 19–22, where proteins with a molecular weight of ∼40–60 kDa are eluted (Figure 3B). Western blot analysis of these fractions revealed that elution of APE1 and Tdp1 occurred within fractions 18–23 and 16–20, respectively (Figure 3C). We thus conclude that the 3′-PG activity co-purifies with APE1 protein.
Figure 3.
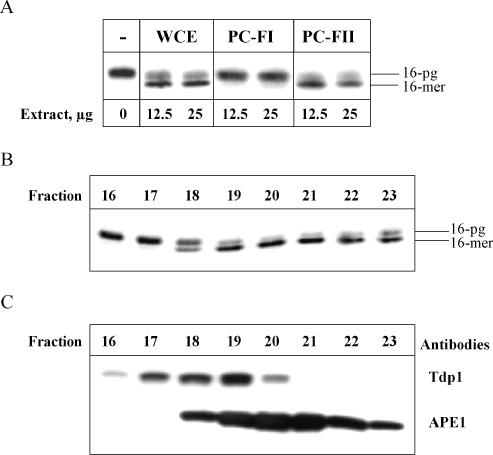
Purification of 3′-PG activity from human WCE using phosphocellulose and gel filtration chromatography. WCE was loaded on a phosphocellulose column and fractions were step eluted using 0.15 M (PC-FI) and 1 M (PC-FII) KCl. Fractions were tested for 3′-PG activity using a 5′ end labelled 3′-PG oligonucleotide annealed to an oligonucleotide (51mer) containing a hairpin loop and a 5′-phosphate residue. The oligonucleotide substrate was bound to streptavidin beads prior to incubation with WCE or phosphocellulose fractions (12.5 and 25 μg) in the absence of dNTPs for 10 min at 30°C. The beads were washed and the DNA subsequently resuspended in formamide loading dye and analysed by 20% denaturing PAGE and phosphorimaging (A). Proteins from PC-FII were further separated by gel filtration on a Superdex 75 column and the fractions obtained were analysed for 3′-PG activity using the 3′-PG duplex oligonucleotide substrate in the absence of dNTPs by 20% denaturing PAGE and phosphorimaging (B). Aliquots of the fractions were also analysed by SDS–PAGE and western blotting using APE1 and Tdp1 antibodies (C).
Co-precipitation of APE1 and 3′-PG activity
Gel filtration fractions containing the peak of 3′-PG activity (fractions 19–22) were chosen to immunodeplete APE1 using antibodies raised against APE1. Analysis by western blot reveals that although the levels of APE1 are slightly reduced by mock immunodepletion compared to the original fractions, using APE1 antibodies the fractions were completely depleted of the APE1 protein (Figure 4A). Furthermore, the levels of another similar sized protein, Pol β, were unaffected by the immunodepletion procedure. The original fractions, alongside the mock immunodepleted and immunodepleted fractions, were subsequently tested for their ability to remove 3′-PG. The mock immunodepletion protocol does not affect 3′-PG activity in fractions 20 and 21 (Figure 4B, lanes 4 and 5) compared to the original fractions (Figure 4B, lanes 2 and 3). However, using antibodies specific to APE1, the activity against 3′-PG is completely ablated (Figure 4B, lanes 6 and 7). Taken together, these results indicate that the major 3′-PG activity observed in human cells is APE1.
Figure 4.
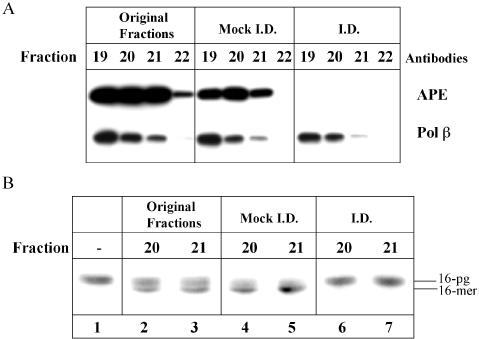
Immunoprecipitation of APE1 from gel filtration fractions containing 3′-PG activity. Fractions from gel filtration chromatography containing 3′-PG activity (fractions 19–22) were mock-immunodepleted and immunodepleted of APE1 using APE1-specific antibodies. Samples were analysed by SDS–PAGE and western blotting using antibodies against APE1 and Pol β (A). The original fractions, mock-immunodepleted and immunodepleted fractions 20 and 21 were tested for 3′-PG activity using a 5′ end labelled 3′-PG oligonucleotide annealed to an oligonucleotide (51mer) containing a hairpin loop and a 5′-phosphate residue. Samples were incubated for 10 min at 30°C in the absence of dNTPs prior to the addition of formamide loading dye and analysis by 20% denaturing PAGE and phosphorimaging (B).
DISCUSSION
Ionizing radiation induces the generation of DNA strand breaks containing 3′-PG ends that must be removed prior to repair by a DNA polymerase and a DNA ligase (4). Conflicting reports suggest that APE1, Tdp1 or other, as yet unidentified, proteins are involved in removal of 3′-PG in human cells (5–8,12–16,18). One of the reasons for the controversial results on the enzymatic activity responsible for removal of 3′-PG may be the variations in substrates and experimental conditions used for measuring 3′-PG activity. A wide spectrum of substrates (from chemically synthesized oligonucleotide duplexes containing 3′-PG to bleomycin-treated plasmid DNA) and different reaction conditions have been employed by different groups. To resolve the existing controversy in the literature on the processing of 3′-PG, we used a double-stranded oligonucleotide substrate containing a strand break with 3′-PG and 5′-phosphate groups to identify the main protein involved in 3′-PG removal in human cell extracts. This substrate models 3′-PG-containing strand breaks arising after radiation-induced DNA damage. The oligonucleotide duplex substrate was carefully characterized by mass spectroscopy, ability to block DNA repair synthesis and by APE1 activation of the 3′ end for repair synthesis by purified human Pol β. All results unequivocally supported the presence of the phosphoglycolate group at the 3′ end of the substrate (Figure 1; and data not shown). However in our hands, electrophoretic mobility of the 16mer oligonucleotide containing phosphoglycolate (16-PG) was between 16mer and 17mer that disagreed with previously published data, suggesting that mobility of the 3′-PG-containing oligonucleotide was slightly slower compared to the same length oligonucleotides without 3′-PG (6,12,18). This may be due to differences in oligonucleotide composition and length, as well as different electrophoresis conditions used.
We next characterized 3′-PG activity in WCE and found that repair of the 3′-PG-containing substrate is Mg2+-dependent. We further purified this Mg2+-dependent 3′-PG activity and found that this activity is the major 3′-PG activity and that no other activity was isolated on separation ofproteins from WCE by phosphocellulose and gel filtra-tion chromatography. This is in contrast to previous datasuggesting that at least two other 3′-phosphodiesterase activities can be isolated, although these repair proteins are reported to be very unstable (8,14). As demonstrated by western blot analysis, the 3′-PG activity co-purifies with APE1. It was further confirmed to be the major 3′-PG activity by immunodepletion of APE1 from purified fractions.
Taken together, our data demonstrates that APE1 is the major 3′-PG activity in human cell extracts.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Keith Caldecott for providing Tdp1 antibodies and Dr B. Demple for critically reading the manuscript.
REFERENCES
- 1.Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature, 362, 709–715. [DOI] [PubMed] [Google Scholar]
- 2.Dedon P.C. and Goldberg,I.H. (1992) Free-radical mechanisms involved in the formation of sequence-dependent bistranded DNA lesions by the antitumor antibiotics bleomycin, neocarzinostatin, and calicheamicin. Chem. Res. Toxicol., 5, 311–332. [DOI] [PubMed] [Google Scholar]
- 3.Henle E.S. and Linn,S. (1997) Formation, prevention, and repair of DNA damage by iron/hydrogen peroxide. J. Biol. Chem., 272, 19095–19098. [DOI] [PubMed] [Google Scholar]
- 4.Demple B. and DeMott,M.S. (2002) Dynamics and diversions in base excision DNA repair of oxidized abasic lesions. Oncogene, 21, 8926–8934. [DOI] [PubMed] [Google Scholar]
- 5.Kane C.M. and Linn,S. (1981) Purification and characterization of an apurinic/apyrimidinic endonuclease from HeLa cells. J. Biol. Chem., 256, 3405–3414. [PubMed] [Google Scholar]
- 6.Suh D., Wilson,D.M.,III and Povirk,L.F. (1997) 3′-phosphodiesterase activity of human apurinic/apyrimidinic endonuclease at DNA double-strand break ends. Nucleic Acids Res., 25, 2495–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winters T.A., Henner,W.D., Russell,P.S., McCullough,A. and Jorgensen,T.J. (1994) Removal of 3′-phosphoglycolate from DNA strand-break damage in an oligonucleotide substrate by recombinant human apurinic/apyrimidinic endonuclease 1. Nucleic Acids Res., 22, 1866–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen D.S., Herman,V.G. and Demple,B. (1991) Two distinct human diesterases that hydrolyze 3′-blocking deoxyribose fragments from oxidised DNA. Nucleic Acids Res., 19, 5907–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Demple B. and Harrison,L. (1994) Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem., 63, 915–948. [DOI] [PubMed] [Google Scholar]
- 10.Demple B., Herman,T. and Chen,D.S. (1991) Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes. Proc. Natl Acad. Sci. USA, 88, 11450–11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robson C.N. and Hickson,I.D. (1991) Isolation of cDNA clones encoding a human apurinic/apyrimidinic endonuclease that corrects DNA repair and mutagenesis defects in E. coli xth (exonuclease III) mutants. Nucleic Acids Res., 19, 5519–5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaudhry M.A., Dedon,P.C., Wilson,D.M., Demple,B. and Weinfeld,M. (1999) Removal by human apurinic/apyrimidinic endonuclease 1 (Ape 1) and Escherichia coli exonuclease III of 3′-phosphoglycolates from DNA treated with neocarzinostatin, calicheamicin, and gamma-radiation. Biochem. Pharmacol., 57, 531–538. [DOI] [PubMed] [Google Scholar]
- 13.Xu Y.J., Kim,E.Y. and Demple,B. (1998) Excision of C-4′-oxidized deoxyribose lesions from double-stranded DNA by human apurinic/apyrimidinic endonuclease (Ape1 protein) and DNA polymerase beta. J. Biol. Chem., 273, 28837–28844. [DOI] [PubMed] [Google Scholar]
- 14.Winters T.A., Weinfeld,M. and Jorgensen,T.J. (1992) Human HeLa cell enzymes that remove phosphoglycolate 3′ end groups from DNA. Nucleic Acids Res., 20, 2573–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Izumi T., Hazra,T.K., Boldogh,I., Tomkinson,A.E., Park,M.S., Ikeda,S. and Mitra,S. (2000) Requirement for human AP endonuclease 1 for repair of 3′- blocking damage at DNA single-strand breaks induced by reactive oxygen species. Carcinogenesis, 21, 1329–1334. [PubMed] [Google Scholar]
- 16.Winters T.A., Russell,P.S., Kohli,M., Dar,M.E., Neumann,R.D. and Jorgensen,T.J. (1999) Determination of human DNA polymerase utilization for the repair of a model ionizing radiation-induced DNA strand break lesion in a defined vector substrate. Nucleic Acids Res., 27, 2423–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang S.W., Burgin,A.B.,Jr, Huizenga,B.N., Robertson,C.A., Yao,K.C. and Nash,H.A. (1996) A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc. Natl Acad. Sci. USA, 93, 11534–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inamdar K.V., Pouliot,J.J., Zhou,T., Lees-Miller,S.P., Rasouli-Nia,A. and Povirk,L.F. (2002) Conversion of phosphoglycolate to phosphate termini on 3′ overhangs of DNA double strand breaks by the human tyrosyl-DNA phosphodiesterase hTdp1. J. Biol. Chem., 277, 27162–27168. [DOI] [PubMed] [Google Scholar]
- 19.Urata H. and Akagi,M. (1993) A convenient synthesis of oligonucleotides with a 3′-phosphoglycolate and 3′-phosphoglycaldehyde terminus. Tetrahedron Lett. 34, 4015–4018. [Google Scholar]
- 20.Strauss P.R., Beard,W.A., Patterson,T.A. and Wilson,S.H. (1997) Substrate binding by human apurinic/apyrimidinic endonuclease indicates a Briggs-Haldane mechanism. J. Biol. Chem., 272, 1302–1307. [DOI] [PubMed] [Google Scholar]
- 21.Dianova I.I., Bohr,V.A. and Dianov,G.L. (2001) Interaction of human AP endonuclease 1 with flap endonuclease 1 and proliferating cell nuclear antigen involved in long-patch base excision repair. Biochemistry, 40, 12639–12644. [DOI] [PubMed] [Google Scholar]
- 22.Manley J.L., Fire,A., Samuels,M. and Sharp,P.A. (1983) In vitro transcription: whole-cell extract. Methods Enzymol., 101, 568–582. [DOI] [PubMed] [Google Scholar]
- 23.Dianov G.L., Jensen,B.R., Kenny,M.K. and Bohr,V.A. (1999) Replication protein A stimulates proliferating cell nuclear antigen-dependent repair of abasic sites in DNA by human cell extracts. Biochemistry, 38, 11021–11025. [DOI] [PubMed] [Google Scholar]