

Abstract
For cells on the path to carcinogenesis, the key to unlimited growth potential lies in overcoming the steady loss of telomeric sequence commonly referred to as the ‘end-replication problem’ that occurs with each cell division. Most human tumors have reactivated telomerase, a specialized reverse transcriptase that directs RNA-templated addition of telomeric repeats on to chromosomal termini. However, ∼10% of tumors maintain their telomeres through a recombination-based mechanism, termed alternative lengthening of telomeres or ALT. Here we demonstrate that telomeric DNA undergoes a high rate of a particular type of recombination visualized cytogenetically as sister chromatid exchange (SCE), and that this rate is dependent on genotype. A novel model of ALT is presented in which it is argued that telomeric exchanges, if they are unequal and occur at a sufficiently high frequency, will allow cells to proliferate indefinitely without polymerase-mediated extension of telomeric sequence.
INTRODUCTION
Normal human cells enter a senescent state after a finite number of population doublings known as the Hayflick limit, after which they remain alive but unresponsive to growth stimuli (1). Growth cessation has been linked to progressive loss of telomeric DNA with each cell division (2). This shortening has been attributed to an inability of DNA polymerases to replicate to the very end of a template during lagging-strand synthesis (3), the processing of telomeres to create 3′ overhangs (4) and oxidative damage to telomeric DNA (5). A plausible model of senescence proposes that, once one or more telomeres have shortened beyond a critical limit, they fail to form protective end structures and, appearing to the cell as DNA double-strand breaks (DSBs), trigger a DNA damage cell-cycle checkpoint response (6).
In contrast to normal cells, cancer cells proliferate indefinitely, and this characteristic may be a necessary condition for tumorigenesis (7). Inactivation of the p53 and Rb pathways allows proliferation beyond the Hayflick limit, but at the expense of escalating chromosomal instability, itself the result of further increases in unprotected chromosome ends. A crisis stage is reached when massive instability prohibits the generation of sufficient viable cells to sustain proliferation. A small percentage of cells avoid crisis by reestablishing chromosome end protection. Often these cells have activated telomerase allowing them to maintain adequate telomere length to form protective end structures (8). However, some telomerase-negative cells escape crisis by an as yet poorly understood mechanism referred to as ‘alternative lengthening of telomeres’ (ALT) (9,10). Occasionally tumors are found to possess neither both mechanisms of telomere length maintenance (11) or (12).
ALT is characterized by a lack of detectable telomerase activity and heterogeneous telomere lengths (13). Increasing evidence suggests ALT is recombination based. For example, ALT cells contain ALT-associated PML bodies (APBs), in which are found the promyelocytic leukemia (PML) protein and telomeric DNA, as well as telomere binding proteins such as TRF1 and TRF2, and recombination proteins (e.g. RAD50, RAD51, RAD52, MRE11, NBS1, BLM and WRN) (14). Taking these observations into account, recombination-based models of ALT have been proposed in which polymerase-mediated extension of one telomere uses the DNA of a second telomere as a template (15–18).
Sister chromatid exchange (SCE) is a common and readily quantifiable form of recombination in mammalian cells. The frequency of SCE is particularly high within the sub-telomeric regions of chromosomes (19). Here we extended SCE analysis into the telomere proper, and found that recombination between telomeres of sister chromatids also occurs at unusually high rates. Furthermore, we propose that clonal senescence can be delayed by unequal exchanges, and if such exchanges occur with sufficient frequency, an ALT-like phenotype ensues. This non-template copy mechanism challenges the assumption that progressive telomere shortening without replacement is necessarily incompatible with unlimited growth potential.
MATERIALS AND METHODS
Cells and culture conditions
Spontaneously transformed lung fibroblast cultures were derived from the following male mice: a repair proficient mouse (C57BLy6TacfBR-[KO]p53N4 WT), two mice having severe combined immunodeficiency (Tac:Icr:Ha(ICR)-scid), a p53 knockout mouse (C57BLy6TacfBR-[KO]p53N4HO), and two mice with a double p53−/−/scid mutation that were obtained by crossing p53 knockout mice with scid homozygous mice (20). Ku70- and Ku86-deficient mouse fibroblast cell lines, established from C57BLy6 knockout mice and transformed with the Abelson murine leukemia retrovirus (21,22), and an isogenic control, were gifts from Drs Gloria C. Li and David J. Chen. Spontaneously transformed lung fibroblasts were derived from PARP−/− male mice derived by gene targeting and a wild-type repair-proficient mouse (23) and were a gift from Dr David J. Chen. All cells were incubated at 37°C in 5% CO2 and cultured in alpha MEM supplemented with 20% fetal bovine serum and antibiotics. In some experiments, cells were exposed to either 200 μM IC86621 or to 3-aminobenzimide for 24 h before collecting mitotic cells as described below.
Chromosome orientation FISH
Chromosome orientation fluorescence in situ hybridization (CO-FISH) has been described in detail previously (24,25). Briefly, confluent cultures were subcultured into a medium containing a 3:1 ratio of 5′-bromo-2′-deoxyuridine (BrdU): 5′-bromo-2′-deoxycytidine (BrdC) (Sigma) at a total final concentration of 1 × 10−5 M and incubated at 37°C for 24 h (one cell cycle). Colcemid (0.2 μg/ml) was added during the last 4 h to accumulate mitotic cells. Cultures were trypsinized and cells suspended in 75 mM KCl at 37°C for 15 min before fixing in 3:1 methanol:acetic acid. Fixed cells were dropped onto cold, wet glass microscope slides. The slides were treated with 0.5 mg/ml RNase A for 10 min at 37°C (26), stained with 0.5 μg/ml Hoechst 33258 (Sigma) in 2× SSC (0.3 M NaCl, 0.03 M sodium citrate) for 15 min at room temperature and then exposed to 365 nm UV light (Stratalinker 1800 UV irradiator) for 25–30 min. Enzymatic digestion of the BrdU/BrdC-substituted DNA strands with 3 U/μl of Exonuclease III (Promega) in buffer supplied by the manufacturer (50 mM Tris–HCl, 5 mM MgCl2, and 5 mM dithiothreitol, pH 8.0) was allowed to proceed for 10 min at room temperature. An additional denaturation in 70% formamide, 2× SSC at 70°C for 1 min was performed in order to ensure complete removal of the newly replicated bromo-substituted strands and followed by dehydration in a cold ethanol series (70, 85, 100%). Probes to telomeric DNA were made by synthesizing oligomers having either the sequence (TTAGGG)7 or (CCCTAA)7. Thirty-five picomoles of oligomer was labeled by terminal deoxynucleotidal transferase tailing (Boehringer Mannheim) with Cy3-dCTP according to the manufacturer's instructions. A hybridization mixture containing 0.4 μg/ml probe DNA in 30% formamide and 2× SSC was applied to slides. Following an overnight hybridization in a moist chamber at 37°C, the slides were washed 5 times for 15 min each in 2× SSC at 42°C and mounted in a glycerol solution containing 1 mg/ml of the antifade compound p-phenylenediamine HCl and 0.1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) to counterstain chromosomal DNA (27).
Analysis
Scoring of telomeric SCE was restricted to the long (q) arms of mouse metaphase chromosomes, as the telocentric nature of the short (p) arms places these telomeres very close together, complicating interpretation of the hybridization pattern. Metaphase chromosomes were viewed and photographed on either a Zeiss Axiophot or an Olympus Provis AX-70 microscope, both equipped for epifluorescence. DAPI and Cy3 exicitor/dichroic/barrier filter sets (Omega Optical) were used to detect counterstained chromosomes and telomere signals, respectively. Digital images were acquired with the Olympus microscope outfitted with a Photometrics SenSys CCD camera and processed using PowerGene MacProbe analysis software (Applied Imaging). DAPI and Cy3 channels were pseudocolored (blue and red, respectively) before being merged to produce the images shown.
Pairwise comparisons for statistical significance were made by t-tests. Differences between genetic backgrounds or treatment conditions are reported in Results only when p-values were less than 0.05.
Differential staining technique for sister chromatid exchange (SCE)
Genomic SCEs were visualized using the standard Fluorescence-Plus-Giemsa technique (28). FPG staining requires that cells undergo two rounds of replication in the presence of BrdU, producing metaphase chromosomes with differentially substituted chromatids. Slides are prepared by standard cytogenetic techniques (above), stained in Hoechst 33258 (0.5 μg/ml) in 2× SSC (pH 7) for 15 min, rinsed with dH2O and allowed to air dry. Slides are then mounted with 22 × 50 mm coverslips and 2× SSC, and exposed to UV light for 25–30 min. Slides are exposed to 2× SSC at 60°C for 30 min, rinsed in dH2O and air-dried. Lastly, slides are stained for 20–25 min in 2% Giemsa dissolved in Sorensen's Buffer pH 6.8 or PBS, rinsed in dH2O and air-dried. For analysis, metaphase spreads were viewed with bright field microscopy for harlequin staining and each color switch scored as an SCE.
RESULTS
The method illustrated in Figure 1 was used to investigate recombination rates in telomeric DNA. With standard fluorescence in situ hybridization (FISH), a telomeric probe produces four signals, one on each end of the two chromatids of a mitotic chromosome. In contrast, the strand-specific nature of chromosome orientation FISH (CO-FISH) typically yields just two signals, one at each end of the chromosome. However, a sister chromatid exchange within telomeric DNA has the effect of splitting the probe hybridization causing the signal to appear on both chromatids on one chromosome end.
Figure 1.
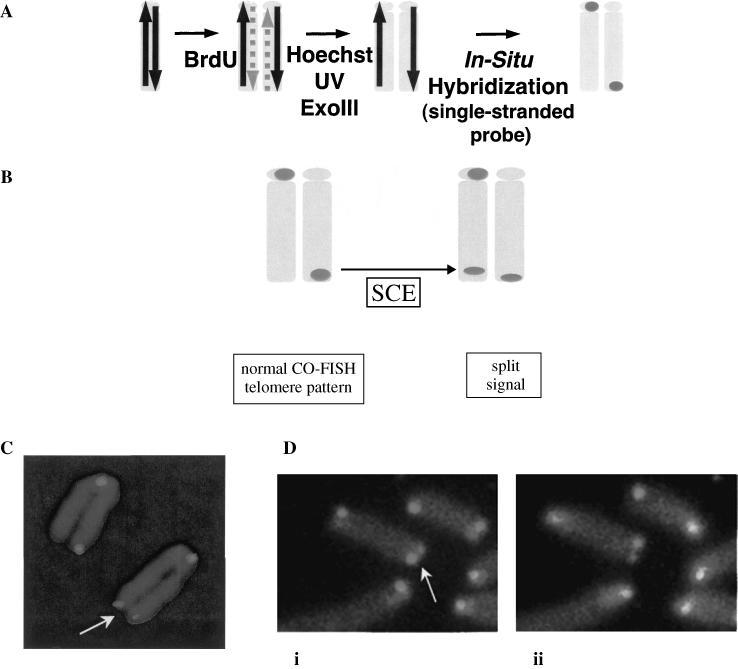
Method for detecting SCE within telomeres. (A) Mitotic cells are collected after culture in bromo-substituted nucleotides for a single cell cycle. Fixed cells on microscope slides are stained with the DNA-binding fluorescent dye Hoechst 33258. Exposure to UV light nicks the substituted strand, and exonuclease III digests it. The process effectively removes the newly synthesized DNA strands and leaves behind the two parental strands that are now located on sister chromatids. A single-stranded probe hybridizes to complementary telomeric DNA on one chromatid of each chromosome arm producing a two-signal pattern instead of the four signals seen with ordinary FISH. (B) The expected effect of an SCE within telomeric DNA is to split the hybridization signal. (C) An example of a three-signal hybridization pattern like that expected of a T-SCE. (D) Sequential CO-FISH detection of a T-SCE with the C-rich telomere probe (i), then the G-rich telomere probe (ii), demonstrating the reciprocal pattern with each. This pattern is only produced by true SCE.
In addition to SCE, other types of recombination could conceivably produce a three-telomere hybridization pattern like that shown in Figure 1. Some examples are; a telomeric fusion followed by breakage within telomeric DNA on one side of the fusion point, a large extrachromosomal telomeric DNA fragment joined to one telomere, or a break in one sister telomere producing a fragment that then joins to the other sister telomere. Each of these mechanisms requires ligation to a chromosome end, an event that should be prevented by functional telomeres. Nevertheless we sought to rule out these mechanisms experimentally. A true SCE produces a three-telomere hybridization pattern with either the G- or the C-rich telomere probe. In contrast, the alternative mechanisms produce a three-telomere hybridization pattern with only one of the probes. Presumptive telomeric SCEs (T-SCEs) were examined sequentially first with one telomere probe and then the other. All possessed a reciprocal hybridization pattern expected of true SCEs (Figure 1D).
In order to put telomeric recombination rates into perspective, T-SCE were compared to SCE in the genome as a whole. The later were quantified by the standard ‘fluorescence-plus-Giemsa’ technique, an assay that permits a genome-wide assessment of recombination rates occurring within the mitotic cell cycle (28). We will refer to these events as genomic SCEs (G-SCEs). In wild-type low passage primary mouse cells, T-SCEs occurred at a rate of 0.534 per cell or 0.0136 per chromosome (Table 1). G-SCE occurred at a rate of 40.1 per cell or 1.02 per chromosome (Table 2). To accurately compare the two types of exchange, the frequency of G-SCEs must be divided by two because they represent the cumulative number of exchanges occurring during the two cell cycles required to perform the assay. In addition, the T-SCE rate must be normalized to account for the fraction of the genome that is composed of telomeric DNA. Because mouse telomeres are ∼50 kb in length (29), and there are 40 chromosomes per cell, telomeric DNA composes 0.13% of the mouse genome. With these adjustments, the SCE rate in telomeres was found to exceed that in the genome as a whole by 20-fold. The true difference is likely to be even greater because T-SCEs were not scored in the short arms of telocentric chromosomes. These results demonstrate that telomeric DNA is especially susceptible to recombination.
Table 1. Frequency of sister chromatid exchange in the telomeres of mouse cells (T-SCE).
| Cell type | No. of cells scored | Average no. of Chromosomes per cell | Average no. of T-SCEs per chromosome | Ratio relative to control |
|---|---|---|---|---|
| WT | 82 | 39.3 | 0.0136 | 1.00 |
| scid | 154 | 39.3 | 0.0161 | 1.18 |
| p53−/− | 102 | 39.3 | 0.00698 | 0.513 |
| p53−/−/scid | 103 | 39.7 | 0.0221 | 1.63 |
| p53−/− (200 μM IC86621) | 30 | 71.8 | 0.00279 | 0.205 |
| Control (MLV)a | 100 | 64.3 | 0.0314 | 1.00 |
| Ku70−/− (MLV) | 100 | 52.3 | 0.0417 | 1.33 |
| Ku86−/− (MLV) | 99 | 71.9 | 0.0353 | 1.12 |
| PARP+/+ | 50 | 38.7 | 0.0227 | 1.00 |
| PARP+/+ (10 mM 3AB) | 50 | 37.4 | 0.0305 | 1.34 |
| PARP−/− | 48 | 43.5 | 0.0187 | 0.824 |
Table 2. Frequency of genomic SCE in mouse cells (G-SCE).
| Cell type | No. of cells scored | Average no. of chromosomes per cell | Average no. of SCE per cell | Average no. of G-SCEs per chromosome | Ratio relative to control |
|---|---|---|---|---|---|
| WT | 134 | 39.3 | 40.1 | 1.02 | 1.00 |
| scid | 38 | 38.6 | 32.6 | 0.845 | 0.828 |
| p53−/− | 74 | 39.3 | 32.3 | 0.822 | 0.806 |
| p53−/−/scid | 61 | 39.7 | 27.7 | 0.698 | 0.684 |
| Control (MLV) | 30 | 63.6 | 27.0 | 0.425 | 1.00 |
| Ku70−/− (MLV) | 30 | 56.6 | 20.2 | 0.357 | 0.840 |
| Ku86−/− (MLV) | 30 | 75.9 | 18.8 | 0.247 | 0.581 |
| PARP+/+ | 24 | 38.7 | 19.2 | 0.496 | 1.00 |
| PARP+/+ (10 mM 3AB) | 30 | 39.8 | 35.9 | 0.902 | 1.82 |
| PARP−/− | 5 | 43.0 | 55.0 | 1.28 | 2.58 |
To gain insight into the molecular mechanisms of telomeric recombination, the influence of several genetic mutations on SCE frequency was examined. The p53 pathway is disrupted in most cancers due to mutation in p53 itself, other genes in the p53 pathway, or interference by viral proteins (30). Experimental mouse models and the human Li Fraumeni syndrome confirm the importance of p53 as a tumor suppressor gene (31). In addition, p53 mutations modulate the tumorigenic potential of other genetic defects. Consistent with previous studies (32,33), we found no statistically significant impact of a p53 knockout mutation on G-SCEs. However, T-SCEs were reduced by about 2-fold compared to wild-type cells.
In addition to its better-known role in DSB repair by non-homologous end joining (NHEJ), DNA-dependent protein kinase (DNA-PK) also participates in the post-replicative process that creates functional telomeres (20,34,35). A mutation in the catalytic subunit of DNA-PK that is responsible for severe combined immunodeficiency (scid) also compromises telomere function, significantly increasing chromosomal end-to-end fusions over the experimentally undetectable rates of wild-type cells (20). The scid mutation in an otherwise wild-type background had little effect on T- or G-SCEs. However in a p53−/− background, the scid mutation elevated T-SCEs by 63% and lowered G-SCEs by 32%, suggesting an interdependence between these two mutations. In addition, the divergent influence of the scid mutation on T- and G-SCEs in this background suggests that there are differences in the processes that generate the two types of exchange. This conclusion is supported by several other observations below.
Together with DNA-PKcs, Ku70 and Ku86 comprise the DNA-PK holoenzyme (36). Mutations in these genes also impair chromosome end protection (20,37). A set of virally transformed mouse cells derived from wild-type and knockouts of Ku70 and Ku86 (21,22) were found to have higher levels of T-SCEs and fewer G-SCEs compared to primary cells, emphasizing the importance of matched controls. There was no clear dependence on either Ku70 or Ku86 other than that G-SCEs were particularly low in the Ku86 mutant.
The dependence of telomeric recombination on DNA-PK activity was tested further using a highly specific inhibitor designated IC86621 (38). In the p53 null background, IC86621 decreased T-SCEs, an effect opposite to that of eliminating DNA-PK activity through the scid mutation. Opposing effects of chemical inhibition versus genetic deficiency on homologous recombination (HR) rates were seen in a previous study (39). These authors attributed elevated recombination in scid cells to release from a negative regulatory effect of DNA-PK on HR. Drug inhibition differs from genetic deficiency in that DNA-PK remains intact but is rendered non-functional. DNA-PK binds DSB ends (36) until liberated through auto-phosphorylation (40). In the presence of the inhibitor, DNA-PK may remain bound, impairing both NHEJ and HR.
PARP1 [poly(ADP-ribose) polymerase] functions as a negative regulator of recombination (23,41,42). As a result, G-SCE rates increase substantially in PARP1−/− cells (43). Consistent with these reports, we found G-SCEs to be more than twice as frequent in PARP1−/− cells. Inhibition of PARP activity by 10 mM 3-aminobenzimide also increased G-SCEs in PARP1+/+ cells by 82%. Interestingly, T-SCEs were not elevated in PARP1−/− cells compared to the PARP1+/+ control, indicating that PARP1 does not serve as a negative regulator of intrachromosomal telomeric recombination. Tankyrase, a telomere-bound protein possessing PARP activity (44) may serve this function.
When compared on a per unit DNA length basis, T-SCE frequencies were much higher than G-SCE frequencies under all conditions examined (Table 3). The greater susceptibility of telomeric DNA to oxidative damage (5) may contribute to high T-SCE frequencies. However, the ratio of the two SCE types ranged from a low of about 13 to a high of more than 200, emphasizing that T-SCEs are not merely a subset of G-SCEs. Despite a superficial resemblance between the two types of exchange, the underlying molecular mechanisms have different genetic dependencies. Interestingly, the frequencies of both genomic intrachromosomal homologous recombination and G-SCE are the same in ALT and telomerase-positive cells (45,46), suggesting that ALT may be mediated by increased telomere-specific recombination rather than a general increase in recombination rates. This underscores the need for telomere-specific recombination assays. Because none of the cell lines used here had the heterogeneous telomere lengths that characterize either ALT cells or cell lines having both ALT and active telomerase (47,48), the T-SCE rates we observed presumably represent values typical of non-ALT cells.
Table 3. Ratio of normalized T-SCE to G-SCE.
| Cell type | T-SCE/G-SCE |
|---|---|
| WT | 19.9 |
| scid | 28.4 |
| p53−/− | 12.7 |
| p53−/−/scid | 47.2 |
| Control (MLV) | 110 |
| Ku70−/− (MLV) | 174 |
| Ku86−/− (MLV) | 213 |
| PARP+/+ | 68.4 |
| PARP+/+ (10 mM 3AB) | 50.4 |
| PARP−/− | 24.6 |
DISCUSSION
G-SCE occurs spontaneously in every cell type examined. Several mutations are known to modulate G-SCE frequencies, but no known viable mutation abolishes them. Thus G-SCEs appear to be mediated by a process vital to cell survival. Homologous recombination associated with replication seems the most likely, if unproven, mechanism for SCE because exchanges take place between identical sequences (49). Recombinational repair or bypass is thought to be triggered when a replication fork encounters a break (or other non-coding lesion) in one of the parental template strands. DNA polymerase stalls, and the resulting 3′-ended strand is then paired with its complement in the other nascent sister chromatid. In the case of a break in the leading-strand template, this pairing may be brought about through simple reversal of the replication fork by branch migration. This would be followed by templated repair synthesis, and resolved by forward branch migration to restore and restart the replication fork. For the case of a break in the lagging-strand template, the discontinuous synthesis of Okazaki fragments probably results in a frank DSB, with two double-stranded ends, left in the wake of the replication fork. Repair of this lesion would be accomplished by a process more closely resembling the classic ‘double-strand break repair’ (DSBR) model for HR proposed by Szostak et al. (50) or more recently proposed ‘synthesis-dependent strand annealing’ (SDSA) models (51). The intermediates of DSBR or SDSA pathways (Holliday junctions or related structures) would be resolved in some instances such that a crossover between the two nascent sister chromatids results, thereby creating a cytogenetically visible SCE. Visible SCE's, however, likely under-represent the true frequency of HR interactions between sister chromatids, as most would be expected to resolve without crossover and so be cytogenetically undetectable.
If HR is indeed the causative mechanism, then SCEs occurring within unique DNA sequences are expected to exchange precisely equal amounts of DNA. However, within repetitive sequences, the invading strand can find homology at multiple points. Consequently, SCEs in the repetitive telomeric sequence need not exchange equal amounts of DNA, and may give rise to sister telomeres of unequal length. Although there is no net gain or loss of telomeric DNA, unequal T-SCEs can have a profound and counterintuitive effect on the proliferation of telomerase-negative cells. For illustration, Figure 2 presents the case of a cell approaching senescence. On the left, the shortest telomere in the cell allows it to undergo two additional divisions before the supply of telomeric DNA is exhausted. On the right, an unequal T-SCE occurs in an otherwise identical cell causing one sister telomere to capture nearly all of the telomeric DNA at the expense of the other. During mitosis the two sister chromatids, now possessing telomeres of unequal lengths, segregate into different daughter cells. One daughter cell senesces immediately because it lost most of its telomeric DNA in the exchange, but the other inherits an elongated telomere endowing it with an enhanced proliferative potential. Overall, the unequal T-SCE in this example has the effect of producing a colony that proliferates for two additional divisions yielding more than twice as many cells by the time it senesces.
Figure 2.
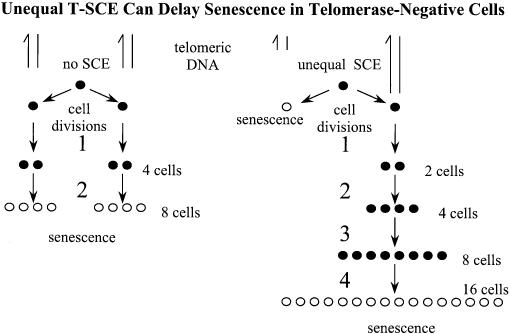
Unequal T-SCE delay senescence in telomerase-negative cells. This is an example of a single cell whose shortest telomere is just long enough to permit two cell divisions before senescence. As shown on the left side of the figure, if there is no T-SCE the cell produces an 8-cell colony at the time of clonal senescence. In contrast, the fate of the same cell would be quite different if an unequal T-SCE transferred most of the telomeric DNA to one daughter cell. The right side of the figure shows that one daughter cell senesces immediately, but the other cell proliferates for an additional two divisions and yields a 17-cell colony by the time proliferation ceases. Thus the effect of an unequal T-SCE is to delay clonal senescence.
In general, any inequality in sister telomere lengths will extend the proliferative life of a clone and yield a higher number of cells by the time proliferation ceases. Because telomeres shorten at the rate of ∼100 bp per cell division (2), each 100 bp gain of telomeric DNA prolongs growth by one cell division. For example, a 2 kb gain potentially extends proliferative growth by 20 divisions during which the colony would grow to more than 500 000 times the size of a clone in which there was no T-SCE. This example illustrates the profound impact of a single unequal T-SCE in the progenitor of a colony. Because telomeric exchange is a natural process that happens continually, in a real colony many more T-SCE would have occurred during clonal growth. Additional unequal T-SCEs in later cell divisions will further delay clonal senescence (or crisis). If repeated often enough, a clone might permanently escape limits on its growth potential. Unlike telomerase-immortalized cells, the cells in such a clone will remain mortal, i.e. individual cells remain susceptible to senescence or crisis.
Several observations are consistent with unequal T-SCEs contributing to ALT. We have shown here that T-SCEs occur spontaneously, and at high rates. Others have shown that, although the lengths of sister telomeres generally correlate well (52), they can be of unequal length (53). Unlike telomerase-induced immortality, unequal T-SCEs rescue only a portion of the cells in a culture from senescence, plausibly explaining the mixture of senescent and thriving cells seen in ALT cultures (54). Cells that acquire shorter telomeres in an exchange will senesce earlier or suffer growth retardation from instability. These cells are diluted by serial subculture. In contrast, cells inheriting longer telomeres will proliferate more successfully and eventually dominate the culture. This is consistent with observations of an increased average telomere length in ALT cells compared to pre-ALT cultures. The extra-chromosomal telomeric DNA fragments that have been observed in ALT cells (14) could be the result of occasional abortive exchanges. Either unequal T-SCE or abortive exchanges may cause the phenomenon of telomere rapid deletion, in which some telomeres undergo a large and rapid truncation (55). Unequal T-SCE and abortive exchanges also would generate the highly heterogeneous telomere lengths that are a defining characteristic of ALT cells (13).
Telomeres tend to cluster in interphase cell nuclei (56), and this close physical proximity may promote inter-chromosomal exchange (ICE), i.e. an exchange resembling SCE except that it occurs between the telomeres of different chromosomes. Telomeric-ICE (T-ICE) would have a capacity similar to unequal T-SCE to prolong clonal growth, as illustrated in Figure 3. In addition, T-ICE provides a potentially efficient mechanism for rescuing short telomeres and for equalizing telomere lengths. Following replication, chromosome ends appear as DSB ends until protective terminal caps are formed. In their unprotected state, chromosome ends may stimulate HR, much as DSBs do. Very short telomeres may cap poorly, making them the most recombinogenic, and the longest telomeres provide the largest target for strand invasion. Because an exchange can be initiated at any point within the long telomere, the most frequent outcome of a T-ICE is that the short telomere will acquire DNA at the expense of the long one.
Figure 3.
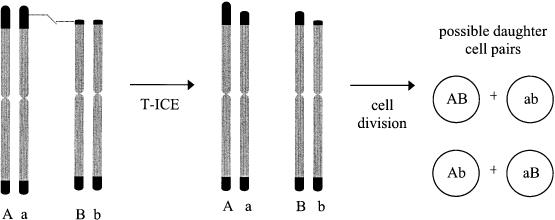
Unequal T-ICE may also delay senescence. The figure depicts an ICE between two telomeres of different chromosomes. In this example, the exchange lengthens the shortest telomere in the cell at the expense of truncating a longer telomere on a different chromosome. Four possible pairings of chromatids from the two chromosomes can be produced during cell division (AB, Ab, aB and ab). Colonies arising from each of the two possible daughter cell pairs have extended proliferative lives because one daughter cell in each pair inherits a lengthened version of the shortest telomere.
ICE also provides an explanation for another ALT-related observation—a marker placed into one telomere of an ALT cell was found to duplicate and re-localize to new chromosomes after cell division, but remained at chromosome ends (57). The authors attributed this phenomenon to a process starting with strand invasion initiated by an unmarked telomere into the marked telomere at a point centromeric to the marker, and copying of the marker through DNA synthesis primed by the invading strand. Resolution of the junction and fill-in synthesis complete transfer of the marker to a new chromosome. ICE occurring after replication also has the capacity to transfer a marker to the end of a new chromosome, and depending on how chromosomes segregate during mitosis, daughter cells may inherit 0, 1 or 2 copies of the marker (Figure 4). After an extended period of growth with further ICEs, the marker would have been lost from some cells, and would have multiplied in other cells and spread amongchromosomes.
Figure 4.
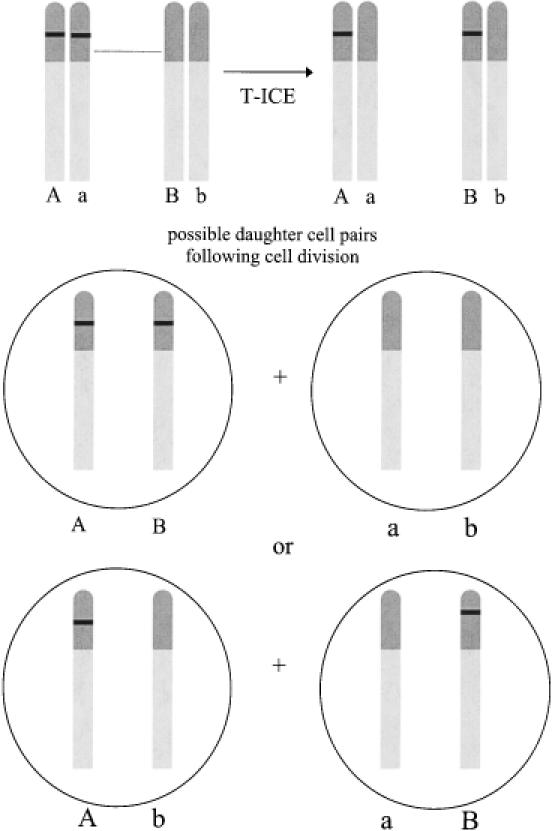
T-ICE can transfer markers to new chromosomes. A marker placed into telomeric DNA can be transferred to a new chromosome if an exchange occurs centromeric to the marker's location. Depending on segregation of chromatids during cell division, a cell may inherit a copy of the marker on both chromosomes (AB), the original chromosome only (Ab), the new chromosome only (aB) or neither chromosome (ab).
T-SCEs were observed in all genetic backgrounds examined, raising a question as to why ALT is a rare phenotype. The answer may lie in the frequency of events. Telomere length maintenance requires, at a minimum, that telomere erosion be balanced by extension. The ∼100 bp loss of telomeric DNA per cell cycle must be replaced by an equal or larger amount, although replacement need not occur in every cell cycle. If the exchange frequency is too low, then extension fails to keep up with loss. To permit unlimited proliferation, exchange frequencies must exceed a threshold. Consistent with this expectation, high rates of telomeric recombination were observed recently in two separate studies of human ALT cells (58,59), indicating that elevated T-SCE can serve as a useful marker of ALT. T-SCE frequencies can vary between genetic backgrounds, by at least 6-fold in our experiments, but raising the T-SCE frequency above the ALT threshold may require mutations in as yet unknown genes. Importantly, the long telomere tracts that occur in human ALT do not, in and of themselves, initiate increased levels of T-SCE, implying that additional factors are required to trigger the high rate of exchange seen in human ALT cells (59).
At present it is not known if ALT occurs through one or a combination of the proposed mechanisms. Nevertheless, these mechanisms have similar implications for both carcinogenesis theory and for therapy because all are recombination based. For example, if ALT is an inevitable result of hyper-recombination within telomeric DNA, then any change that increases telomeric exchange rates would favor extended proliferation. Thus ALT may not be associated with a single gene as in telomerase-mediated immortalization. Rather ALT may be a multigenic phenotype in which several genes participate to varying degrees in different tumors. In the treatment of ALT tumors, the equivalent of a telomerase inhibitor would be a drug that reduces telomeric exchange rates below the critical threshold frequency for telomere length maintenance. If ALT is multigenic, it may be difficult to find a single drug active against all ALT tumors. On the other hand, ALT cells contain some very short telomeres, so drugs directed against ALT may not have to be applied for long before the shortest telomeres become dysfunctional and the tumor begins to die. The methods used here for detection and quantification of telomere-specific recombination should aid in identifying ALT-associated genetic mutations, and for devising and testing novel therapies directed against ALT tumors.
Acknowledgments
ACKNOWLEDGEMENTS
The authors are grateful to Drs Robert L. Ullrich and Jac Nickoloff for their support, Drs Gloria C. Li and David J. Chen for cell lines used in this study, and Eli S. Williams for expert technical assistance. This research was supported by the Low Dose Radiation Research Program, Office of Biological and Environmental Research, U.S. Department of Energy awards W-7405-ENG-36 and ER-63239, and by National Cancer Institute awards CA77693 and CA43322. M.A.B. was the recipient of a postdoctoral fellowship award from the U.S. Army Breast Cancer Research Program (DAMD17-00-1-0367).
REFERENCES
- 1.Hayflick C.B. (1965) The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res., 37, 614–636. [DOI] [PubMed] [Google Scholar]
- 2.Harley C.B., Futcher,A.B. and Greider,C.W. (1990) Telomeres shorten during aging of human fibroblasts. Nature, 345, 458–460. [DOI] [PubMed] [Google Scholar]
- 3.Makarov V.L., Hirose,Y. and Langmore,J.P. (1997) Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell, 88, 657–666. [DOI] [PubMed] [Google Scholar]
- 4.Olovnikov A.M. (1971) Principle of marginotomy in template synthesis of polynucleotides. Dokl. Akad. Nauk SSSR, 201, 1496–1499. [PubMed] [Google Scholar]
- 5.von Zglinicki T., Saretzki,G., Docke,W. and Lotze,C. (1995) Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: a model for senescence? Exp. Cell Res., 220, 186–193. [DOI] [PubMed] [Google Scholar]
- 6.Itahana K., Dimri,G. and Campisi,J. (2001) Regulation of cellular senescence by p53. Eur. J. Biochem., 268, 2784–2791. [DOI] [PubMed] [Google Scholar]
- 7.Campisi J. (2001) Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol., 11, S27–S31. [DOI] [PubMed] [Google Scholar]
- 8.Shay J.W. and Bacchetti,S. (1997) A survey of telomerase activity in human cancer. Eur. J. Cancer, 33, 787–791. [DOI] [PubMed] [Google Scholar]
- 9.Bryan T.M., Englezou,A., Dalla-Pozza,L., Dunham,M.A. and Reddel,R.R. (1997) Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nature Med., 3, 1271–1274. [DOI] [PubMed] [Google Scholar]
- 10.Murnane J.P., Sabatier,L., Marder,B.A. and Morgan,W.F. (1994) Telomere dynamics in an immortal human cell-line. EMBO J., 13, 4953–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bryan T.M. and Reddel,R.R. (1997) Telomere dynamics and telomerase activity in in vitro immortalised human cells. Eur. J. Cancer, 33, 767–773. [DOI] [PubMed] [Google Scholar]
- 12.Ulaner G.A., Huang,H.Y., Otero,J., Zhao,Z., Ben-Porat,L., Satagopan,J.M., Gorlick,R., Meyers,P., Healey,J.H., Huvos,A.G. et al. (2003) Absence of a telomere maintenance mechanism as a favorable prognostic factor in patients with osteosarcoma. Cancer Res., 63, 1759–1763. [PubMed] [Google Scholar]
- 13.Bryan T.M., Englezou,A., Gupta,J., Bacchetti,S. and Reddel,R.R. (1995) Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J., 14, 4240–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henson J.D., Neumann,A.A., Yeager,T.R. and Reddel,R.R. (2002) Alternative lengthening of telomeres in mammalian cells. Oncogene, 21, 598–610. [DOI] [PubMed] [Google Scholar]
- 15.Teng S.C., Chang,J., McCowan,B. and Zakian,V.A. (2000) Telomerase-independent lengthening of yeast telomeres occurs by an abrupt Rad50p-dependent; Rif-inhibited recombinational process. Mol. Cell, 6, 947–952. [DOI] [PubMed] [Google Scholar]
- 16.Reddel R.R. (2003) Alternative lengthening of telomeres, telomerase, and cancer. Cancer Lett., 194, 155–162. [DOI] [PubMed] [Google Scholar]
- 17.Bosco G. and Haber,J.E. (1998) Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics, 150, 1037–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reddel R.R., Bryan,T.M. and Murnane,J.P. (1997) Immortalized cells with no detectable telomerase activity. A review. Biochemistry (Mosc.), 62, 1254–1262. [PubMed] [Google Scholar]
- 19.Cornforth M.N. and Eberle,R.L. (2001) Termini of human chromosomes display elevated rates of mitotic recombination. Mutagenesis, 16, 85–89. [DOI] [PubMed] [Google Scholar]
- 20.Bailey S.M., Meyne,J., Chen,D.J., Kurimasa,A., Li,G.C., Lehnert,B.E. and Goodwin,E.H. (1999) DNA double-strand break repair proteins are required to cap the ends of mammalian chromosomes. Proc. Natl Acad. Sci. USA, 96, 14899–14904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ouyang H., Nussenzweig,A., Kurimasa,A., Soares,V.D., Li,X.L., CordonCardo,C., Li,W.H., Cheong,N., Nussenzweig,M., Iliakis,G. et al. (1997) Ku70 is required for DNA repair but not for T cell antigen receptor gene recombination in vivo. J. Exp. Med., 186, 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nussenzweig A., Sokol,K., Burgman,P., Li,L.G. and Li,G.C. (1997) Hypersensitivity of Ku80-deficient cell lines and mice to DNA damage: the effects of ionizing radiation on growth; survival; and development. Proc. Natl Acad. Sci. USA, 94, 13588–13593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Murcia J.M., Niedergang,C., Trucco,C., Ricoul,M., Dutrillaux,B., Mark,M., Oliver,F.J., Masson,M., Dierich,A., LeMeur,M. et al. (1997) Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl Acad.Sci. USA, 94, 7303–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodwin E.H., Meyne,J. and Bailey,S.M. (1993) Strand-specific in situ hybridization reveals long-range molecular order in repetitive DNA. Cytogenet. Cell Genet., 63, 253–253. [Google Scholar]
- 25.Bailey S.M., Meyne,J. and Goodwin,E.H. (2001) Telomeres, DNA repair proteins and making ends meet. In Nickoloff,J.A. and Hoekstra,M.F. (eds), DNA Damage and Repair. Humana Press, Inc., Totowa, NJ, Vol. 3, pp. 359–375. [Google Scholar]
- 26.Hayata I. (1993) Removal of stainable cytoplasmic substances from cytogenetic slide preparations. Biotech. Histochem., 68, 150–152. [DOI] [PubMed] [Google Scholar]
- 27.Meyne J., Bailey,S.M. and Goodwin,E.H. (2002) Strand-specific fluorescence in situ hybridization: CO-FISH and COD-FISH, FISH technology. In Rautenstrauss,B.W. and Liehr,T. (eds), FISH Technology, Springer Laboratory Manual. Springer, pp. 262–271. [Google Scholar]
- 28.Perry P. and Wolff,S. (1974) New Giemsa method for the differential staining of sister chromatids. Nature, 251, 156–158. [DOI] [PubMed] [Google Scholar]
- 29.Zijlmans J., Martens,U.M., Poon,S.S.S., Raap,A.K., Tanke,H.J., Ward,R.K. and Lansdorp,P.M. (1997) Telomeres in the mouse have large inter-chromosomal variations in the number of T(2)AG(3) repeats. Proc. Natl Acad. Sci. USA, 94, 7423–7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sherr C.J. (2004) Principles of tumor suppression. Cell, 116, 235–246. [DOI] [PubMed] [Google Scholar]
- 31.Blackburn A.C. and Jerry,D.J. (2002) Knockout and transgenic mice of Trp53: what have we learned about p53 in breast cancer? Breast Cancer Res., 4, 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bouffler S.D., Kemp,C.J., Balmain,A. and Cox,R. (1995) Spontaneous and ionizing radiation-induced chromosomal abnormalities in p53-deficient mice. Cancer Res., 55, 3883–3889. [PubMed] [Google Scholar]
- 33.Bunz F., Fauth,C., Speicher,M.R., Dutriaux,A., Sedivy,J.M., Kinzler,K.W., Vogelstein,B. and Lengauer,C. (2002) Targeted inactivation of p53 in human cells does not result in aneuploidy. Cancer Res., 62, 1129–1133. [PubMed] [Google Scholar]
- 34.Bailey S.M., Cornforth,M.N., Kurimasa,A., Chen,D.J. and Goodwin,E.H. (2001) Strand-specific postreplicative processing of mammalian telomeres. Science, 293, 2462–2465. [DOI] [PubMed] [Google Scholar]
- 35.Bailey S.M., Brenneman,M.A., Halbrook,J., Nickoloff,J.A., Ullrich,R.L. and Goodwin,E.H. (2004) The kinase activity of DNA-PK is required to protect mammalian telomeres. DNA Repair (Amst.), 3, 225–233. [DOI] [PubMed] [Google Scholar]
- 36.Smith G.C.M. and Jackson,S.P. (1999) The DNA-dependent protein kinase. Genes Dev., 13, 916–934. [DOI] [PubMed] [Google Scholar]
- 37.Samper E., Goytisolo,F.A., Slijepcevic,P., van Vuul,P.P.W. and Blasco,M.A. (2000) Mammalian Ku86 protein prevents telomeric fusions independently of the length of TTAGGG repeats and the G-strand overhang. EMBO Rep., 1, 244–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kashishian A., Douangpanya,H., Clark,D., Schlachter,S.T., Eary,C.T., Schiro,J.G., Huang,H., Burgess,L.E., Kesicki,E.A. and Halbrook,J. (2003) DNA-dependent protein kinase inhibitors as drug candidates for the treatment of cancer. Mol. Cancer Ther., 2, 1257–1264. [PubMed] [Google Scholar]
- 39.Allen C., Halbrook,J. and Nickoloff,J.A. (2003) Interactive competition between homologous recombination and non-homologous end joining. Mol. Cancer Res., 1, 913–920. [PubMed] [Google Scholar]
- 40.Merkle D., Douglas,P., Moorhead,G.B., Leonenko,Z., Yu,Y., Cramb,D., Bazett-Jones,D.P. and Lees-Miller,S.P. (2002) The DNA-dependent protein kinase interacts with DNA to form a protein–DNA complex that is disrupted by phosphorylation. Biochemistry, 41, 12706–12714. [DOI] [PubMed] [Google Scholar]
- 41.Schultz N., Lopez,E., Saleh-Gohari,N. and Helleday,T. (2003) Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res., 31, 4959–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jeggo P.A. (1997) Dna repair : Parp : another guardian angel. Curr. Biol., 8, R49–R51. [DOI] [PubMed] [Google Scholar]
- 43.SimbulanRosenthal C.M., Haddad,B.R., Rosenthal,D.S., Weaver,Z., Coleman,A., Luo,R.B., Young,H.M., Wang,Z.Q., Ried,T. and Smulson,M.E. (1999) Chromosomal aberrations in PARP(−/−) mice: genome stabilization in immortalized cells by reintroduction of poly(ADP-ribose) polymerase cDNA. Proc. Natl Acad. Sci. USA, 96, 13191–13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith S., Giriat,I., Schmitt,A. and deLange,T. (1998) Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science, 282, 1484–1487. [DOI] [PubMed] [Google Scholar]
- 45.Bechter O.E., Zou,Y., Shay,J.W. and Wright,W.E. (2003) Homologous recombination in human telomerase-positive and ALT cells occurs with the same frequency. EMBO Rep., 4, 1138–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ford L.P., Zou,Y., Pongracz,K., Gryaznov,S.M., Shay,J.W. and Wright,W.E. (2001) Telomerase can inhibit the recombination-based pathway of telomere maintenance in human cells. J. Biol. Chem., 276, 32198–32203. [DOI] [PubMed] [Google Scholar]
- 47.Cerone M.A., Londono-Vallejo,J.A. and Bacchetti,S. (2001) Telomere maintenance by telomerase and by recombination can coexist in human cells. Hum. Mol. Genet., 10, 1945–1952. [DOI] [PubMed] [Google Scholar]
- 48.Perrem K., Colgin,L.M., Neumann,A.A., Yeager,T.R. and Reddel,R.R. (2001) Coexistence of alternative lengthening of telomeres and telomerase in hTERT-transfected GM847 cells. Mol. Cell Biol., 21, 3862–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thompson L.H. and Schild,D. (2001) Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat. Res., 477, 131–153. [DOI] [PubMed] [Google Scholar]
- 50.Szostak J.W., Orr-Weaver,T.L., Rothstein,R.J. and Stahl,F.W. (1983) The double-strand-break repair model for recombination. Cell, 33, 25–35. [DOI] [PubMed] [Google Scholar]
- 51.Paques F. and Haber,J.E. (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev., 63, 349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lansdorp P.M., Verwoerd,N.P., van de Rijke,F.M., Dragowska,V., Little,M.T., Dirks,R.W., Raap,A.K. and Tanke,H.J. (1996) Heterogeneity in telomere length of human chromosomes. Hum. Mol. Genet., 5, 685–691. [DOI] [PubMed] [Google Scholar]
- 53.Baird D.M., Rowson,J., Wynford-Thomas,D. and Kipling,D. (2003) Extensive allelic variation and ultrashort telomeres in senescent human cells. Nature Genet., 33, 203–207. [DOI] [PubMed] [Google Scholar]
- 54.Rogan E.M., Bryan,T.M., Hukku,B., Maclean,K., Chang,A.C., Moy,E.L., Englezou,A., Warneford,S.G., Dalla-Pozza,L. and Reddel,R.R. (1995) Alterations in p53 and p16INK4 expression and telomere length during spontaneous immortalization of Li-Fraumeni syndrome fibroblasts. Mol. Cell Biol., 15, 4745–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li B. and Lustig,A.J. (1996) A novel mechanism for telomere size control in Saccharomyces cerevisiae. Genes Dev., 10, 1310–1326. [DOI] [PubMed] [Google Scholar]
- 56.Weierich C., Brero,A., Stein,S., von Hase,J., Cremer,C., Cremer,T. and Solovei,I. (2003) Three-dimensional arrangements of centromeres and telomeres in nuclei of human and murine lymphocytes. Chromosome Res., 11, 485–502. [DOI] [PubMed] [Google Scholar]
- 57.Dunham M.A., Neumann,A.A., Fasching,C.L. and Reddel,R.R. (2000) Telomere maintenance by recombination in human cells. Nature Genet., 26, 447–450. [DOI] [PubMed] [Google Scholar]
- 58.Bechter O.E., Zou,Y., Walker,W., Wright,W.E. and Shay,J.W. (2004) Telomeric recombination in mismatch repair deficient human colon cancer cells after telomerase inhibition. Cancer Res., 64, 3444–3451. [DOI] [PubMed] [Google Scholar]
- 59.Londono-Vallejo J.A., Der-Sarkissian,H., Cazes,L., Bacchetti,S. and Reddel,R.R. (2004) Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res., 64, 2324–2327. [DOI] [PubMed] [Google Scholar]