

Abstract
Cladribine, 2-chloro-2′-deoxyadenosine, is a highly efficacious, clinically used nucleoside for the treatment of hairy cell leukemia. It is also being evaluated against other lymphoid malignancies and has been a molecule of interest for well over half a century. In continuation of our interest in the amide bond-activation in purine nucleosides via the use of (benzotriazol-1yl-oxy)tris(dimethylamino)phosphonium hexafluorophosphate, we have evaluated the use of O6-(benzotriazol-1-yl)-2′-deoxyguanosine as a potential precursor to cladribine and its analogues. These compounds, after appropriate deprotection, were assessed for their biological activities, and the data are presented herein. Against hairy cell leukemia (HCL), T-cell lymphoma (TCL) and chronic lymphocytic leukemia (CLL), cladribine was the most active against all. The bromo analogue of cladribine showed comparable activity to the ribose analogue of cladribine against HCL, but was more active against TCL and CLL. The bromo ribose analogue of cladribine showed activity, but was the least active among the C6-NH2-containing compounds. Substitution with alkyl groups at the exocyclic amino group appears detrimental to activity, and only the C6 piperidinyl cladribine analogue demonstrated any activity. Against adenocarcinoma MDA-MB-231 cells, cladribine and its ribose analogue were most active.
Keywords: cladribine, nucleoside, guanosine, benzotriazole, (benzotriazol-1yl-oxy)-tris(dimethylamino)phosphonium hexafluorophosphate, BOP
1. Introduction
Cladribine, 2-chloro-2′-deoxyadenosine, has been a molecule of interest for well over five decades. The history of this compound dates back to 1960, when it was used in the synthesis of 2′-deoxyguanosine and 2′-deoxyinosine [1]. A decade later, cladribine was shown to be a poor substrate for adenosine deaminase that underwent phosphorylation by deoxycytidine kinase, finally resulting in the triphosphate, and inhibiting DNA synthesis rather than RNA synthesis [2,3]. Later still, it was shown to be a substrate for deoxyguanosine kinase, which is responsible for phosphorylation of purine nucleosides in mitochondria [3]. Several mechanisms have been proposed by which cladribine can cause mitochondrial damage and apoptotic cell death [4,5,6,7,8].
In contemporary medicine, cladribine is used in the treatment of lymphoid malignancies, most notably for its efficacy against hairy cell leukemia [9]. Cladribine is also being evaluated against several other indolent lymphoid malignancies, also in combination with other drug candidates [10,11].
The synthesis of cladribine has primarily relied on three major methods: (a) glycosylation reactions of a nucleobase with a sugar [12,13,14,15,16,17,18], and its variations; (b) deoxygenation of the C2′ hydroxyl group of a suitable nucleoside derivative [12,15,19,20]; (c) enzymatic glycosyl transfer reactions [21,22,23,24]; and (d) conversion of readily available nucleoside precursors (some utilizing nucleosides for glycosyl transfer reactions) [21,22,24,25,26]. Each of these methods has been used with varying levels of convenience and success. Among the many approaches, one convenient method is the selective displacement of a leaving group (chloride or aryl sulfonate) from the C6 position of a suitable purine nucleoside precursor. Despite the availability of this selective SNAr displacement, we find that no other N6-substituted cladribine analogues have been synthesized by such a method. Because of our interest in broadening the utilities of O6-(benzotriazol-1-yl)purine nucleoside derivatives, we elected to evaluate the synthesis of N6-substituted cladribine analogues via amide-bond activation of guanine nucleosides with (benzotriazol-1yl-oxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP).
2. Results and Discussion
For the modification of the nucleobases of inosine, and 2′-deoxyinosine (3 examples), BOP had been used for in situ activation of the amide moieties in these substrates, followed by SNAr displacement with amines [27,28]. It was proposed that reaction of the amide group with BOP proceeded via a phosphonium intermediate, which could be directly captured by a reactive amine [27]. On the other hand, with less reactive amines, the O-(benzotriazol-1-yl) intermediate can be formed by competitive capture of the phosphonium intermediate by the benzotriazol-1-yloxy anion [27].
In 2007, we first reported the isolation of O6-(benzotriazol-1-yl)inosine and -2′-deoxyinosine by amide-bond activation with BOP, in the absence of a nucleophile [29], an observation that was later reconfirmed by others [30,31]. These electrophilic nucleosides, which are stable to storage, are exceptionally good partners in SNAr reactions with oxygen, nitrogen, and sulfur nucleophiles [29]. Subsequently, we demonstrated that O6-(benzotriazol-1-yl)inosine and -2′-deoxyinosine can also be prepared via the use of PPh3/I2 and 1-hydroxybenzotriazole [32]. The amide-bond activation protocol was then modified to tether the O6-(benzotriazol-1-yl) nucleosides onto a polymer support for high-throughput type of applications [33]. Interestingly, the amide bond activation when applied to the urea functionality of O6-benzyl-protected 2′-deoxyxanthosine did not yield the O2-(benzotriazol-1-yl) derivative but rather terminated in an isolable and synthetically useful phosphonium salt [34]. We also showed that guanosine and 2′-deoxyguanosine undergo facile reactions with BOP, and that O6-(benzotriazol-1-yl) guanine nucleosides are effective substrates for SNAr reactions as well [35]. In the combined course of these investigations we had ascertained plausible operative mechanisms of these amide-activation reactions, results that were later applied to a one-pot etherification protocol for purine nucleosides and pyrimidines [36].
Considering only the nucleoside modification literature, our BOP-mediated amide-activation methodology has found wide application [37,38,39,40,41,42,43]. Of specific interest to our current work was a report wherein some of our results on guanosine derivatives were reevaluated [44]. In addition to 2′,3′,5′-tri-O-(t-butyldimethylsilyl)guanosine, which we had originally investigated, five other compounds were also studied: unprotected 2′-deoxyguanosine, its 2′,3′,5′-triacetyl and the 2′,3′-isopropylidene derivatives, and two 2′,3′-isopropylidiene 5′-carboxamides [44]. These products were subsequently tested in diazotization-halogenation reactions, en route to 2-chloro and 2-iodo adenosine analogues [44]. 2,6-Dihalopurine ribonucleosides are more readily accessible for this purpose, as compared to 2′-deoxy derivatives. Thus, we were intrigued by the fact that diazotization-halogenation reactions of O6-(benzotriazol-1yl)-2′-deoxyguanosine have not been reported. This prompted use to investigate the chlorination and bromination of this compound, and in this context we decided to evaluate the synthesis of cladribine and other cladribine analogues via such an approach.
In 2001, diazotization reactions leading to cladribine and other halo derivatives have been performed on unprotected 2,6-diaminopurine 2′-deoxyribonucleoside [45]. However, no substituents other than NH2 were introduced into the C6 position, and the synthesis of the precursor is not particularly convenient. For the present study, we anticipated the need for saccharide protection, and both acetyl and t-butyldimethysilyl (TBS) protecting groups were considered. Between these, TBS was selected because acetyl groups can be susceptible to cleavage with amines, which could be a complicating problem in the method development stage. Thus, nucleosides 1a and 1b were silylated to give the corresponding products 2a and 2b, which were converted to the O6-(benzotriazol-1-yl) guanosine derivatives 3a and 3b, respectively (Scheme 1).
Scheme 1.

Preparation of O6-(benzotriazol-1-yl) guanosine derivatives and the chlorination reaction.
We opted for diazotization-chlorination conditions using t-BuONO/TMSCl, a reagent combination initially introduced for nucleoside modification in 2003 [46]. We [47] and others [44] have previously used non-aqueous conditions for halogenation at the C2 position of purine nucleoside derivatives. Under these conditions, reactions of substrate 3a proceeded modestly. However, the obtained product was contaminated with ~30% of the C2 protio O6-(benzotriazol-1-yl)-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyinosine (Table 1, entries 1 and 2). With the combination of t-BuONO/TMSCl/(BnNEt3)+Cl−, no C2 protio product was observed, but only a low product yield was obtained (entry 3).
Table 1.
Evaluation of conditions for the diazotization/chlorination of O6-(benzotriazol-1-yl)-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyguanosine.
| Entry | Conditions | Scale | Yield |
|---|---|---|---|
| 1 | t-BuONO (2.6 equiv), TMSCl (2.6 equiv), CH2Cl2, −10 °C to 0 °C, 2 h | 73 μmol of 3a | 43% a |
| 2 | t-BuONO (10 equiv), TMSCl (5 equiv), CH2Cl2, 0 °C, 1 h | 73 μmol of 3a | 58% a |
| 3 | t-BuONO (10 equiv), TMSCl (0.4 equiv), (BnNEt3)+Cl− (10 equiv), CH2Cl2, −10 °C to 0 °C, 2 h | 49 μmol of 3a | 26% |
| 4 | t-BuONO (3.5 equiv), SbCl3 (1.4 equiv), ClCH2CH2Cl, −10 °C to −15 °C, 1.5 h | 49 μmol of 3a | 35% |
| 5 | t-BuONO (10 equiv), (BnNEt3)+Cl− (10 equiv), CH2Cl2, −78 °C and then rt, 1 h | 49 μmol of 3a | 39% |
| 6 | t-BuONO (20 equiv), SbCl3 (0.4 equiv), (BnNEt3)+Cl− (20 equiv), CH2Cl2, −10 °C to 0 °C, 5 h | 49 μmol of 3a | 39% |
| 7 | t-BuONO (10 equiv), (BnNEt3)+Cl− (10 equiv), (Me3Si)2NH (10 equiv), CH2Cl2, −10 °C to 0 °C, 4 h | 49 μmol of 3a | 35% |
| 8 | t-BuONO (3.5 equiv), SbCl3 (1.4 equiv), CH2Cl2, −10 °C, 2 h | 49 μmol of 3a | 39% b |
| 9 | t-BuONO (3.5 equiv), SbCl3 (1.4 equiv), CH2Cl2, −10 °C to −15 °C, 2 h | 73 μmol of 3a | 51% |
| 10 | t-BuONO (3.5 equiv), SbCl3 (1.4 equiv), CH2Cl2, −10 °C, 2 h | 0.14 mmol of 3a | 60% |
| 11 | t-BuONO (3.5 equiv), SbCl3 (1.4 equiv), CH2Cl2, −10 °C to −15 °C, 2 h | 1.6 mmol of 3a | 65% |
| 12 | t-BuONO (3.5 equiv), SbCl3 (1.4 equiv), CH2Cl2, −10 °C to −15 °C, 3.5 h | 0.27 mmol of 3b | 61% |
| 13 | t-BuONO (10 equiv), TMSCl (5 equiv), CH2Cl2, 0 °C, 1 h | 2.69 mmol of 3b | 68% |
a Yield was calculated on the basis of the molecular weight of compound 4a. However, by 1H-NMR (500 MHz, CDCl3), the chromatographically homogenous product band was observed to contain a 2:1 ratio of compound 4a and the C2 protio O6-(benzotriazol-1-yl)-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyinosine. In addition to these, a minor uncharacterized nucleoside byproduct was also formed; b Compound 3a dissolved in CH2Cl2 was added to the mixture of reagents in CH2Cl2.
These results compelled us to consider other conditions. SbCl3 and SbBr3 have previously been used for the halogenation of nucleosides [48,49] Thus, the next series of experiments involved SbCl3, (BnNEt3)+Cl−, and combinations of these reagents (entries 4–11). Whereas most experiments yielded only 35%–39% of product 4a, a reasonable yield improvement was observed in entry 9. It was noted that efficient filtration of the reaction mixture after workup is critical to obtaining a good product recovery due to the pasty nature of the mixture (see the Experimental Section). On larger scales, better product recoveries were observed (entries 10 and 11). By comparison, diazotization/chlorination of the ribose derivative 3b proceeded well with both t-BuONO/SbCl3 (entry 12) and t-BuONO/TMSCl (entry 13), with the latter providing a better yield of compound 4b. No C2 protio product was apparent in the reactions of precursor 3b.
The next stage in the chemistry involved SNAr reactions at the C6 positions of substrates 4a and 4b. Cladribine and its ribose analogue were prepared by reactions with aqueous ammonia (see the Experimental Section for details). In order to prepare other analogues, reactions were conducted with 1.5 equiv each of methylamine, dimethylamine, pyrrolidine, piperidine, morpholine, and N,N,N′-trimethylethylenediamine (products, reaction times, and yields are shown in Figure 1).
Figure 1.

Structures of the products, reaction times, and yields obtained in the SNAr reactions.
Most reactions proceeded smoothly and in good to high yields. Methylamine (2 M in THF) was used for the synthesis of 6a and 6b, whereas a 40% aqueous solution of dimethylamine was used for the synthesis of 7a and 7b. As we have previously shown, water is not generally detrimental to reactions of these benzotriazolyl purine nucleosides [35]. In reactions of 4a and 4b with N,N,N′-tri-methylethylenediamine, yields were lower. In each ~10%–15% of compounds 7a and 7b were isolated as byproducts. The source of dimethylamine is currently unknown but its origin can be linked to N,N,N′-trimethylethylenediamine.
Finally, for biological testing, desilylation was performed. Because we did not anticipate decomposition of starting materials or products, we only tested the use of KF (2 equiv/silyl group) in MeOH at 80 °C (Scheme 2). The results of the desilylation reactions are shown in Table 2.
Scheme 2.

Desilylation of the protected nucleosides.
Table 2.
Structures of the products, reaction times, and yields of the desilylated compounds.
| Entry | R | Reaction Times/KF Equiv | Yields |
|---|---|---|---|
| 1 | 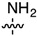 |
24 h with 4 equiv KF | 12a: 72% |
| 2 | 24 h with 6 equiv KF | 12b: 74% | |
| 3 | 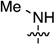 |
24 h with 4 equiv KF | 13a: 85% |
| 4 | 16 h with 6 equiv KF | 13b: 75% | |
| 5 | 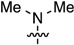 |
24 h with 4 equiv KF | 14a: 82% |
| 6 | 16 h with 6 equiv KF | 14b: 75% | |
| 7 | 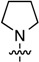 |
20 h with 4 equiv KF | 15a: 81% |
| 8 | 16 h with 6 equiv KF | 15b: 85% | |
| 9 | 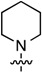 |
24 h with 4 equiv KF | 16a: 71% |
| 10 | 16 h with 6 equiv KF | 16b: 84% | |
| 11 | 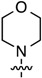 |
26 h with 4 equiv KF | 17a: 74% |
| 12 | 16 h with 6 equiv KF | 17b: 78% | |
| 13 | 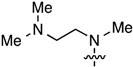 |
24 h with 4 equiv KF | 18a: 84% |
| 14 | 24 h with 6 equiv KF | 18b: 65% |
Because there is currently no known method for the diazotization/bromination of compound 3a, we investigated a route similar to that for chlorination. Results from these experiments are listed in Table 3. What is notable with the diazotization/bromination, in contrast to the chlorination, was that use of 3.5 equiv of t-BuONO led to incomplete conversion over 2 h at −10 to −15 °C. Addition of another 3.5 equiv of t-BuONO then led to complete conversion over an additional 1 h.
Table 3.
Diazotization-bromination of silyl-protected O6-(benzotriazol-1-yl) guanine nucleosides.
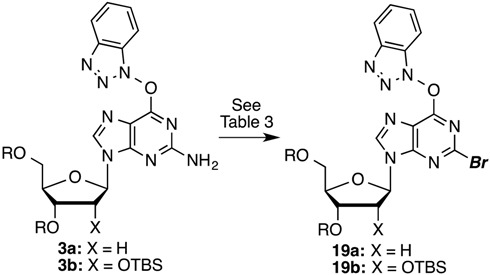
| Entry | Conditions | Scale | Yield |
|---|---|---|---|
| 1 | t-BuONO (7.0 equiv), SbBr3 (1.4 equiv), CH2Br2, −10 °C to −15 °C, 3 h | 73 μmol of 3a | 69% |
| 2 | t-BuONO (7.0 equiv), SbBr3 (1.4 equiv), CH2Br2, −10 °C to −15 °C, 3 h | 0.49 mmol of 3a | 63% |
| 3 | t-BuONO (7.0 equiv), SbBr3 (1.4 equiv), CH2Br2, −10 °C to −15 °C, 3 h | 0.49 mmol of 3b | 64% |
A similar conversion of 3b led to the ribose analogue 19b in a comparable 64% yield (Table 3, entry 3). Compound 19a was then converted to the bromo analogue of cladribine as shown in Scheme 3. SNAr displacement with aqueous NH3 proceeded in 83% yield producing compound 20a, which was finally desilylated with KF in anhydrous MeOH at 80 °C (28 h) to yield 2-bromo-2-deoxyadenosine (21a) in 66% yield. Corresponding conversions of the ribose analog 19b, via intermediate 20b, gave compound 21b. Yields for these conversions were comparable to the deoxyribose series.
Scheme 3.

Synthesis of 2-bromo-2′-deoxyadenosine.
Notably, previously unknown compound 19a and 19b are relatively easily prepared, new bifunctional reactive nucleosides that can undergo SNAr reactions at the C6 and metal-mediated reactions at the C2 position. To the extent that C-Cl bonds can be activated by metal catalysts compounds 4a and 4b also offer this type of orthogonal reactivity. Results from the orthogonal reactivities of these new halo nucleosides will be reported in the future.
Results of Tests against HCL, TCL, CLL, and MDA-MB-231 Breast Cancer Cells
The newly synthesized compounds as well as cladribine and its bromo analogue were tested against HCL, TCL, and CLL. Data from these assays are shown in Table 4.
Table 4.
IC50 values (μM) of the cladribine and its analogues on HCL, TCL, and CLL a.
| Compound | HCL | TCL | CLL | |||
|---|---|---|---|---|---|---|
| ATP | 3H-Leu | ATP | 3H-Leu | ATP | 3H-Leu | |
| 12a | 0.065 | 0.090 | 0.020 | 0.039 | 0.004 | 0.011 |
| 12b | 0.81 | 0.85 | 4.81 | 9.40 | 1.33 | 0.88 |
| 13a | >33 | >33 | >33 | >33 | >33 | >33 |
| 13b | >32 | >32 | >32 | >32 | ND b | >32 |
| 14a | >32 | >32 | >32 | >32 | >32 | >32 |
| 14b | >30 | >30 | >30 | >30 | >30 | >30 |
| 15a | >29 | >29 | >29 | >29 | >29 | >29 |
| 15b | >28 | >28 | >28 | >28 | >28 | >28 |
| 16a | 11.5 | 16.3 | 9.6 | 9.0 | 4.3 | 5.4 |
| 16b | >27 | >27 | >27 | >27 | ND b | ND b |
| 17a | >28 | >28 | >28 | >28 | >28 | >28 |
| 17b | >27 | >27 | >27 | >27 | >27 | >27 |
| 18a | >27 | >27 | >27 | >27 | >27 | >27 |
| 18b | >26 | >26 | >26 | >26 | >26 | >26 |
| 21a | 0.76 | 0.96 | 0.13 | 0.16 | 0.13 | 0.14 |
| 21b | 1.8 | 7.2 | 8.6 | 10.2 | 2.0 | 4.0 |
a One patient per disease type; b ND = not determined.
From these data, across the entire series, cladribine (12a) was best. The bromo analogue of cladribine (21a) showed lower activities. A comparison of the ribose analogue of cladribine (12b) and the bromo analogue of cladribine (21a) is interesting. Both compounds 12b and 21a show comparable activities against HCL, but the latter shows higher activities against TCL and CLL. The bromo ribose derivative 21b showed activity but was inferior to compounds 12a, 12b, and 21a. In this series, the only other compound to demonstrate any activity was the piperidinyl derivative 16a.
The compounds were also tested against adenocarcinoma MDA-MB-231 breast cancer cells. These cells were treated with three-fold serial dilutions of the compounds ranging from 0–1.8 mM for 24 h. Viable cells were fixed with cold methanol and nuclei were stained with 0.4% propidium iodide (PI). Cell viability was calculated as the percentage of surviving cells after treatment as measured by differences in fluorescence units between treated and untreated wells (Figure 2).
Figure 2.

Effects of cladribine analogues on breast cancer cell viability. (A): Deoxyribose series and (B): Ribose series. Significant differences are described with * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001 compared to control.
IC50 values were obtained from dose response curve fittings using the non-linear regression function of GraphPad Prism® (La Jolla, CA, USA). Dashed horizontal line represents 50% cell viability. Columns represent means ± SEM of at least three independent experiments. Significant differences are described with * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001 compared to control.
From the IC50 values shown in Table 5, the two compounds that emerged as most promising were cladribine 12a and its ribose analogue 12b, followed by the bromo derivatives 21a and 21b, which were about 10 times less active.
Table 5.
IC50 values (mM) of the compounds synthesized on MDA-MB-231 breast cancer cells.
| 2′-Deoxyribose Series | Ribose Series | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 12a | 13a | 14a | 15a | 16a | 17a | 18a | 21a | 12b | 13b | 14b | 15b | 16b | 17b | 18b | 21b |
| 0.05 | 2.15 | 6.28 | 2.54 | 4.18 | 2.32 | 1.88 | 0.64 | 0.06 | 2.39 | 1.70 | 1.54 | 2.45 | 2.57 | 2.91 | 0.55 |
3. Experimental Section
3.1. General Considerations
Thin-layer chromatography was performed on 200 μm aluminum-foil-backed silica gel plates for the 2′-deoxynucleosides and on Merck 60F254 (Merck, Billerica, MA, USA) for the ribose analogues. Column chromatographic purifications were performed on 100–200 mesh silica gel. CH2Cl2 for the chlorination reactions was distilled over CaH2. Precursors 3a and 3b were prepared as described previously [35]. The yield of 3a was 71% on a 2.52 mmol scale and the yield of 3b was 71% on a 2.1 mmol scale. TMSCl was redistilled prior to use and all other commercially available compounds were used without further purification. 1H-NMR spectra were recorded at 500 MHz or at 400 MHz in the solvents indicated under the individual compound headings and are referenced to residual protonated solvent resonances. 13C-NMR spectra were recorded at 125 MHz or at 100 MHz in the solvents indicated under the individual compound headings and are referenced to the solvent resonances (Supplementary Materials). Chemical shifts (δ) are reported in parts per million (ppm), and coupling constants (J) are in hertz (Hz). Standard abbreviations are used to designate resonance multiplicities (s = singlet, d = doublet, t = triplet, dd = doublet of doublet, ddd = doublet of doublet of doublet, quint = quintet, m = multiplet, br = broad, app = apparent). The saccharide carbons of the nucleoside are numbered 1′ through 5′ starting at the anomeric carbon atom and proceeding via the carbon chain to the primary carbinol carbon atom. The purinyl proton is designated as H-8 and the saccharide protons are designated on the basis of the carbon atom they are attached to.
O6-(Benzotriazol-1-yl)-2-chloro-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (4a): A mixture of compound 3a (1.0 g, 1.63 mmol) and SbCl3 (520.6 mg, 2.28 mmol) in dry CH2Cl2 (16.3 mL) was cooled to −15 °C using dry ice and acetone, in a nitrogen atmosphere. t-BuONO (0.678 mL, 5.70 mmol) was added dropwise and the mixture was stirred at −10 to −15 °C for 3.5 h, at which time TLC indicated the reaction to be complete. The reaction mixture was poured into ice-cold, saturated aqueous NaHCO3 (25 mL) with stirring. The mixture was filtered using vacuum (note: use of vacuum for this filtration is critical for maximizing product recovery) and the residue was washed with CH2Cl2 (25 mL). The organic layer was separated and the aqueous layer was back extracted with CH2Cl2 (2 × 15 mL). The combined organic layer was washed with water (15 mL) and brine (15 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification of the crude material on a silica gel column sequentially eluted with hexanes, 5% EtOAc in hexanes, followed by 20% EtOAc in hexanes gave 0.67 g (65% yield) of compound 4a as a white foam. Rf (SiO2 and 30% EtOAc in hexanes) = 0.55. 1H-NMR (500 MHz, CDCl3): δ 8.47 (s, 1H, H-8), 8.09 (d, J = 8.3 Hz, 1H, Ar-H), 7.52–7.41 (m, 3H, Ar-H), 6.45 (t, J = 5.8 Hz, 1H, H-1′), 4.62 (m, 1H, H-3′), 4.01 (m, 1H, H-4′), 3.89 (dd, J = 3.4, 11.2 Hz, 1H, H-5′), 3.76 (dd, J = 2.4, 11.2 Hz, 1H, H-5′), 2.61 (app quint, Japp ~ 6.2 Hz, 1H, H-2′), 2.49–2.48 (app quint, Japp ~ 5.8 Hz, 1H, H-2′), 0.88 (s, 18H, t-Bu), 0.08 and 0.07 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 159.2, 154.9, 152.6, 144.4, 143.5, 129.0, 128.8, 125.0, 120.7, 119.1, 108.6, 88.4, 85.3, 71.6, 62.6, 41.6, 26.1, 25.9, 18.5, 18.1, −4.5, −4.7, −5.2, −5.3. HRMS (TOF) calcd for C28H43ClN7O4Si2 [M + H]+ 632.2598, found 632.2583.
2-Chloro-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyadenosine (5a): To a solution of compound 4a (126.0 mg, 0.2 mmol) in 1,2-DME (2 mL), 28%–30% aqueous ammonia (32 μL) was added, and the mixture was stirred at room temperature for 1.5 h. The mixture was diluted with EtOAc (5 mL) and washed with 5% aqueous NaCl (5 mL). The organic layer was separated and the aqueous layer was back extracted with EtOAc (5 mL). The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude material was chromatographed on a silica gel column by sequential elution with 20% EtOAc in hexanes followed by 5% MeOH in CH2Cl2 to afford 85.0 mg (82% yield) of compound 5a as a white solid. Rf (SiO2 and 10% MeOH in CH2Cl2) = 0.40. 1H-NMR (500 MHz, CDCl3): δ 8.12 (s, 1H, H-8), 6.54 (br s, 2H, NH2), 6.38 (t, J = 6.1 Hz, 1H, H-1′), 4.61 (m, 1H, H-3′), 3.99 (app q, Japp ~ 3.4 Hz, 1H, H-4′) 3.88 (dd, J = 3.9, 11.2 Hz, 1H, H-5′), 3.76 (dd, J = 3.0, 11.2 Hz, 1H, H-5′), 2.61 (app quint, Japp ~ 6.4 Hz, 1H, H-2′), 2.44–2.39 (ddd, J = 3.1, 6.3, 13.2 Hz, 1H, H-2′), 0.90 (s, 18H, t-Bu), 0.09 and 0.08 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 156.2, 154.0, 150.4, 139.5, 118.7, 87.9, 84.5, 71.7, 62.6, 41.3, 25.9, 25.7, 18.4, 18.0, −4.7, −4.8, −5.4, −5.5. HRMS (TOF) calcd for C22H40ClN5O3Si2Na [M + Na]+ 536.2250, found 536.2252.
3.2. General Procedure for the Synthesis of Cladribine Analogues
To a solution of compound 4a (126.0 mg, 0.2 mmol) in 1,2-DME (2 mL) the appropriate amine (1.5 equiv) was added, and the mixture was stirred at room temperature. The mixture was diluted with EtOAc (5 mL for compounds 6a, 9a, 11a, and 15 mL for compounds 7a, 8a, 10a) and washed 5% aqueous NaCl (5 mL for compounds 6a, 9a, 11a, and 15 mL for compounds 7a, 8a, 10a). The organic layer was separated and the aqueous layer was back extracted with EtOAc (5 mL for compounds 6a, 9a, 11a, and 15 mL for compounds 7a, 8a, 10a). The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude material was chromatographed on a silica gel column; see individual compound headings for details.
2-Chloro-N6-methyl-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyadenosine (6a): Compound 6a (85.0 mg, 80% yield) was obtained as a white, foamy solid after chromatography on a silica gel column by sequential elution with 5% EtOAc in hexanes and 15% EtOAc in hexanes. Rf (SiO2 and 30% EtOAc in hexanes) = 0.26. 1H-NMR (500 MHz, CDCl3): δ 8.02 (s, 1H, H-8), 6.37 (t, J = 6.4 Hz, 1H, H-1′), 6.17 (br s, 1H, NH), 4.60 (m, 1H, H-3′), 3.97 (app q, Japp ~ 3.4 Hz, 1H, H-4′), 3.87 (dd, J = 4.1, 11.2 Hz, 1H, H-5′), 3.75 (dd, J = 3.0, 11.2 Hz, 1H, H-5′), 2.61 (app quint, Japp ~ 6.4 Hz, 1H, H-2′), 2.42–2.37 (ddd, J = 2.8, 6.1, 12.7 Hz, 1H, H-2′), 0.90 (s, 18H, t-Bu), 0.09 and 0.08 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 155.9, 154.5, 149.2; 138.7, 119.1, 87.9, 84.4, 71.8, 62.7, 41.2, 27.5, 25.9, 25.7, 18.4, 18.0, −4.7, −4.8, −5.4, −5.5. HRMS (TOF) calcd for C23H42ClN5O3Si2Na [M + Na]+ 550.2407, found 550.2419.
2-Chloro-N6,N6-dimethyl-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyadenosine (7a): Compound 7a (91.1 mg, 84% yield) was obtained as a colorless, thick gum after chromatography on a silica gel column by sequential elution with hexanes, 5% EtOAc in hexanes, and 20% EtOAc in hexanes. Rf (SiO2 and 30% EtOAc in hexanes) = 0.70. 1H-NMR (500 MHz, CDCl3): δ 7.93 (s, 1H, H-8), 6.36 (t, J = 6.3 Hz, 1H, H-1′), 4.58 (m, 1H, H-3′), 3.95 (m, 1H, H-4′), 3.83 (dd, J = 4.4, 11.2 Hz, 1H, H-5′), 3.73 (dd, J = 2.9, 11.2 Hz, 1H, H-5′), 3.50 (br s, 6H, N(CH3)2), 2.58 (app quint, Japp ~ 6.5 Hz, 1H, H-2′), 2.39–2.35 (m, 1H, H-2′), 0.88 (s, 18H, t-Bu), 0.08 and 0.06 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 155.2, 153.8, 151.4, 137.2, 119.6, 87.9, 84.4, 72.1, 62.9, 41.1, 26.1, 26.0, 25.9, 25.8, 18.6, 18.2, −4.5, −4.6, −5.2, −5.3. HRMS (TOF) calcd for C24H45ClN5O3Si2 [M + H]+ 542.2744, found 542.2749.
2-Chloro-6-(pyrrolidin-1-yl)-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (8a): Compound 8a (93.3 mg, 82% yield) was obtained as a colorless, thick gum after chromatography on a silica gel column by sequential elution with hexanes, 5% EtOAc in hexanes, and 20% EtOAc in hexanes. Rf (SiO2 and 30% EtOAc in hexanes) = 0.65. 1H-NMR (500 MHz, CDCl3): δ 7.92 (s, 1H, H-8), 6.37 (t, J = 6.6 Hz, 1H, H-1′), 4.59 (m, 1H, H-3′), 4.13 (br s, 2H, pyrrolidinyl NCH), 3.96 (m, 1H, H-4′), 3.83 (dd, J = 4.9, 11.2 Hz, 1H, H-5′), 3.74 (dd, J = 3.9, 11.2 Hz, 1H, H-5′), 3.79–3.73 (br m, 2H, pyrrolidinyl NCH), 2.61 (app quint, Japp ~ 6.5 Hz, 1H, H-2′), 2.39–2.35 (m, 1H, H-2′), 2.10–2.0 (br m, 2H, pyrrolidinyl CH), 1.96–1.85 (br m, 2H, pyrrolidinyl CH), 0.90 (s, 18H, t-Bu), 0.09 and 0.07 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 154.3, 153.5, 151.0, 137.8, 119.8, 88.1, 84.4, 72.3, 63.1, 49.1, 41.08, 26.4, 26.2, 25.9, 24.3, 18.6, 18.2, −4.5, −4.6, −5.2, −5.3. HRMS (TOF) calcd. for C26H47ClN5O3Si2 [M + H]+ 568.2900, found 568.2906.
2-Chloro-6-(piperidin-1-yl)-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (9a): Compound 9a (99.0 mg, 85% yield) was obtained as a white, foamy solid after chromatography on a silica gel column by sequential elution with 5% EtOAc in hexanes and 15% EtOAc in hexanes. Rf (SiO2 and 30% EtOAc in hexanes) = 0.65. 1H-NMR (500 MHz, CDCl3): δ 7.92 (s, 1H, H-8), 6.37 (t, J = 6.6 Hz, 1H, H-1′), 4.59 (m, 1H, H-3′), 4.45–3.73 (br m, 4H, piperidinyl N(CH2)2), 3.96 (app q, Japp ~ 3.6 Hz, 1H, H-4′), 3.83 (dd, J = 4.9, 11.2 Hz, 1H, H-5′), 3.74 (dd, J = 3.4, 11.2 Hz, 1H, H-5′), 2.59 (app quint, Japp ~ 6.3 Hz, 1H, H-2′), 2.40–2.35 (ddd, J = 3.3, 6.2, 13.3 Hz, 1H, H-2′), 1.68 (br m, 6H, piperidinyl CH), 0.90 (s, 18H, t-Bu), 0.09 and 0.07 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 153.8, 151.4, 136.7, 119.0, 109.9, 87.8, 84.2, 72.0, 62.8, 46.3, 40.9, 26.1, 25.9, 25.7, 24.6, 18.4, 18.0, −4.7, −4.8, −5.4, −5.5. HRMS (TOF) calcd for C27H49ClN5O3Si2 [M + H] + 582.3057 found 582.3074.
2-Chloro-6-(morpholin-4-yl)-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (10a): Compound 10a (97.4 mg, 83% yield) was obtained as a colorless, thick gum after chromatography on a silica gel column by sequential elution with hexanes, 5% EtOAc in hexanes, and 20% EtOAc in hexanes. Rf (SiO2 and 30% EtOAc in hexanes) = 0.68. 1H-NMR (500 MHz, CDCl3): δ 7.98 (s, 1H, H-8), 6.37 (t, J = 6.3 Hz, 1H, H-1′), 4.58 (m, 1H, H-3′), 4.55–3.98 (br m, 4H, morpholinyl N(CH2)2), 3.96 (m, 1H, H-4′), 3.84 (dd, J = 4.4, 11.2 Hz, 1H, H-5′), 3.78 (t, J = 4.6 Hz, 4H, morpholinyl CH2OCH2), 3.74 (dd, J = 2.9, 11.2 Hz, 1H, H-5′), 2.56 (app quint, Japp ~ 6.5 Hz, 1H, H-2′), 2.40–2.36 (m, 1H, H-2′), 0.89 (s, 18H, t-Bu), 0.08 and 0.07 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 154.0, 153.9, 151.8, 137.6, 119.3, 88.0, 84.4, 72.0, 67.1, 62.9, 43.5 (br), 41.3, 26.1, 26.0, 25.9, 18.6, 18.2, −4.5, −4.6, −5.2, −5.3. HRMS (TOF) calcd for C26H47ClN5O4Si2 [M + H]+ 584.2850, found 584.2855.
2-Chloro-6-[(2-dimethylamino)ethyl)(methyl)amino)]-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (11a): Compound 11a (77.0 mg, 64% yield) was obtained as a pale-yellow, thick gum after chromatography on a silica gel column by sequential elution with 10% EtOAc in hexanes, 20% EtOAc in hexanes, and 10% MeOH in CH2Cl2. Rf (SiO2 and 10% MeOH in CH2Cl2) = 0.10. 1H-NMR (500 MHz, CDCl3): δ 7.94 (s, 1H, H-8), 6.36 (t, J = 6.3 Hz, 1H, H-1′), 4.62 (m, 1H, H-3′), 4.28–3.84 (br m, 2H, NCH2), 3.99 (app q, Japp ~ 3.7 Hz, 1H, H-4′), 3.85 (dd, J = 4.4, 11.2 Hz, 1H, H-5′), 3.78 (dd, J = 3.4, 11.2 Hz, 1H, H-5′), 3.47 (br s, 3H, NCH3), 2.65–2.59 (m, 2H, NCH2 and 1H, H-2′), 2.41–2.37 (ddd, J = 4.2, 6.2, 13.1 Hz, 1H, H-2′), 2.34 (s, 6H, N(CH3)2), 0.92 and 0.91 (s, 18H, t-Bu), 0.11, 0.083, and 0.079 (3s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 154.9, 153.8, 151.4, 137.2, 119.4, 87.9, 84.4, 72.0, 62.9, 57.0, 48.6, 45.7, 41.0, 36.9, 26.0, 25.8, 18.4, 8.0, −4.6, −4.8, −5.4, −5.5. HRMS (TOF) calcd for C27H52ClN6O3Si2 [M + H]+ 599.3322 found 599.3313.
O6-(Benzotriazol-1-yl)-2-chloro-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (4b): To a solution of t-BuONO (2.78 g, 26.91 mmol) in CH2Cl2 (197 mL) was added TMSCl (1.46 g, 13.45 mmol) at 0 °C. To this mixture a solution of 3b (2 g, 2.69 mmol) in CH2Cl2 (197 mL) was added dropwise, and then the reaction mixture was stirred at 0 °C for 1 h at which time the reaction was completed as indicated by TLC. The reaction mixture was diluted with CH2Cl2 (200 mL), washed with saturated NaHCO3 (100 mL), H2O (100 mL), and brine (100 mL). The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. Purification of the crude material on a silica gel column sequentially eluted with hexanes, 5% EtOAc in hexanes, and 20% EtOAc in hexanes gave 1.4 g (68% yield) of compound 4b as a white foam. Rf (SiO2 and 30% EtOAc in hexanes) = 0.55. 1H-NMR (400 MHz, CDCl3): δ 8.56 (s, 1H, H-8), 8.14 (d, J = 8.0 Hz, 1H, Ar-H), 7.55–7.45 (m, 3H, Ar-H), 6.05 (d, J = 4.0 Hz, 1H, H-1′), 4.57 (t, J = 4.2 Hz, 1H, H-2′), 4.34 (t, J = 4.4 Hz, 1H, H-3′), 4.18–4.15 (m, 1H, H-4′), 4.08 (dd, J = 3.6, 11.6 Hz, 1H, H-5′), 3.83 (dd, J = 2.4, 11.6 Hz, 1H, H-5′), 0.96, 0.93, and 0.88 (3s, 27H, t-Bu), 0.15, 0.11, 0.02, and −0.11 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 159.0, 154.9, 152.6, 144.4, 143.4, 128.9, 128.7, 124.9, 120.6, 119.0, 108.5, 89.4, 85.3, 71.2, 62.0, 26.1, 25.8, 25.6, 18.5, 18.0, 17.9, −4.3, −4.6, −4.7, −4.9, −5.3, −5.4. HRMS (TOF) calcd for C34H57ClN7O5Si3 [M + H]+ 762.3412, found 762.3427.
2-Chloro-2′,3′,5′-tri-O-(t-butyldimethylsilyl)adenosine (5b): To a solution of compound 4b (500 mg, 0.655 mmol) in 1,2-DME (8 mL), 28%–30% aqueous ammonia (64 μL) was added and the mixture was stirred at room temperature for 1.5 h. The reaction mixture was diluted with EtOAc (25 mL) and washed 5% aqueous NaCl (25 mL). The organic layer was separated and the aqueous layer was back extracted with EtOAc (25 mL). The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude material was chromatographed on a silica gel column by sequential elution with 20% EtOAc in hexanes and 5% MeOH in EtOAc to afford 310 mg (75% yield) of compound 5b as an off-white solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.21. 1H-NMR (400 MHz, CDCl3): δ 8.12 (s, 1H, H-8), 6.17 (br s, 2H, NH2), 5.93 (d, J = 4.8 Hz, 1H, H-1′), 4.69 (t, J = 4.6 Hz, 1H, H-2′), 4.32 (t, J = 4.6 Hz, H-3′), 4.14–4.06 (m, 1H, H-4′), 4.06 (dd, J = 4.8, 11.6 Hz, 1H, H-5′), 3.80 (dd, J = 2.8, 11.2 Hz, 1H, H-5′), 0.94, 0.91, and 0.84 (3s, 27H, t-Bu), 0.14, 0.09, −0.01, and −0.17 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 156.1, 154.0, 150.7, 140.2, 118.9, 88.9, 85.4, 75.5, 71.7, 62.3, 26.0, 25.8, 25.7, 18.5, 18.0, 17.9, −4.3, −4.5, −4.7, −5.0, −5.3, −5.4. HRMS (TOF) calcd. for C28H55ClN5O4Si3 [M + H]+ 644.3245 found 644.3217.
3.3. General Procedure for the Synthesis of Ribose Analogues of Cladribine
To a solution of compound 4b (500 mg, 0.655 mmol) in 1,2-DME (8 mL) was added the appropriate amine (1.5 equiv) and the mixture was stirred at room temperature. The mixture was diluted with EtOAc (25 mL) and washed with 5% aqueous NaCl (15 mL). The organic layer was separated and the aqueous layer was back extracted with EtOAc (15 mL). The combined organic layers were dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude material was chromatographed on a silica gel column. See individual compound headings for details.
2-Chloro-N6-methyl-2′,3′,5′-tri-O-(t-butyldimethylsilyl)adenosine (6b): Compound 6b (340 mg, 80% yield) was obtained as a pale-yellow, thick gum after chromatography on a silica gel column by sequential elution with 10% EtOAc in hexanes, 20% EtOAc in hexanes, and 50% EtOAc in hexanes. Rf (SiO2 and EtOAc) = 0.20. 1H-NMR (400 MHz, CDCl3): δ 8.02 (s, 1H, H-8), 6.04 (br s, 1H, NH), 5.91 (d, J = 5.2 Hz, 1H, H-1′), 4.72 (t, J = 4.6 Hz, 1H, H-2′), 4.32 (t, J = 4.0 Hz, 1H, H-3′), 4.12–4.10 (m, 1H, H-4′), 4.06 (dd, J = 5.2, 11.6 Hz, 1H, H-5′), 3.79 (dd, J = 2.8, 11.2 Hz, 1H, H-5′), 3.17 (s, 3H, CH3), 0.94, 0.92, and 0.81 (3s, 27H, t-Bu), 0.13, 0.06, −0.02, and −0.19 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 155.9, 154.5, 139.3, 119.3, 88.8, 85.4, 75.3, 71.5, 65.2, 62.4, 26.0, 25.8, 25.7, 18.4, 18.0, 17.8, 14.0, 11.0, −4.4, −4.7, −5.1, −5.4, −5.7. HRMS (TOF) calcd for C29H57ClN5O4Si3 [M + H]+ 658.3401, found 658.3416.
2-Chloro-N6,N6-dimethyl- 2′,3′,5′-tri-O-(t-butyldimethylsilyl)adenosine (7b): Compound 7b (360 mg, 81% yield) was obtained as a pale-yellow, thick gum after chromatography on a silica gel column by sequential elution with 10% EtOAc in hexanes and 20% EtOAc in hexanes. Rf (SiO2 and 20% EtOAc in hexanes) = 0.40. 1H-NMR (400 MHz, CDCl3): δ 7.93 (s, 1H, H-8), 5.90 (d, J = 5.2 Hz, 1H, H-1′), 4.77 (t, J = 4.8 Hz, 1H, H-2′), 4.31 (t, J = 3.8 Hz, 1H, H-3′), 4.04–4.09 (m, 1H, H-4′), 4.06 (dd, J = 5.6, 11.2 Hz, 1H, H-5′), 3.78 (dd, J = 2.8, 11.2 Hz, 1H, H-5′), 3.58 (br s, 6H, CH3), 0.94, 0.92, and 0.82 (3s, 27H, t-Bu), 0.13, 0.10, −0.02, and −0.18 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 155.9, 153.7, 151.4, 137.9, 119.7, 88.8, 85.4, 74.8, 72.0, 62.5, 38.4, 26.0, 25.8, 25.7, 18.4, 18.0, 17.9, −4.3, −4.7, −5.0, −5.4. HRMS (TOF) calcd for C30H59ClN5O4Si3 [M + H]+ 672.3558, found 672.3561.
2-Chloro-6-(pyrrolidin-1-yl)-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (8b): Compound 8b (390 mg, 85% yield) was obtained as a pale-yellow, thick gum after chromatography on a silica gel column by sequential elution with 10% EtOAc in hexanes and 20% EtOAc in hexanes. Rf (SiO2 and 20% EtOAc in hexanes) = 0.30. 1H-NMR (400 MHz, CDCl3): δ 7.90 (s, 1H, H-8), 5.90 (d, J = 5.2 Hz, 1H, H-1′), 4.81 (t, J = 5.0 Hz, 1H, H-2′), 4.32 (t, J = 4.0 Hz, 1H, H-3′), 4.14–4.08 (m, 1H, H-4′ and br m, 2H, pyrrolidinyl NCH), 4.06 (dd, J = 5.6, 11.2 Hz, 1H, H-5′), 3.77–3.73 (m, 3H, H-5′ and pyrrolidinyl NCH), 2.04 (br m, 2H, pyrrolidinyl CH), 1.98 (br m, 2H, pyrrolidinyl CH), 0.93, 0.89, and 0.81 (3s, 27H, t-Bu), 0.12, 0.11, −0.03, and −0.20 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 154.0, 153.4, 151.0, 138.0, 119.9, 88.7, 85.5, 74.6, 72.2, 62.6, 48.9, 47.6, 26.0, 25.8, 25.7, 18.4, 18.1, 17.9, −4.4, −4.6, −4.7, −5.0, −5.3. HRMS (TOF) calcd for C32H61ClN5O4Si3 [M + H]+ 698.3714 found 698.3731.
2-Chloro-6-(piperidin-1-yl)-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (9b): Compound 9b (375 mg, 80% yield) was obtained as a pale-yellow, thick gum after chromatography on a silica gel column by sequential elution with 10% EtOAc in hexanes and 20% EtOAc in hexanes. Rf (SiO2 and 20% EtOAc in hexanes) = 0.50. 1H-NMR (400 MHz, CDCl3): δ 7.91 (s, 1H, H-8), 5.89 (d, J = 5.6 Hz, 1H, H-1′), 4.79 (t, J = 4.8 Hz, 1H, H-2′), 4.32 (t, J = 4.0 Hz, 1H, H-3′), 4.22 (br s, 4H, piperidinyl NCH2), 4.11–4.09 (m, 1H, H-4′), 4.06 (dd, J = 5.2, 10.8 Hz, 1H, H-5′), 3.77 (dd, J = 2.8, 10.8 Hz, 1H, H-5′), 1.69 (br m, 6H, piperidinyl CH2), 0.94, 0.90, and 0.84 (3s, 27H, t-Bu), 0.12, 0.10, −0.02, and −0.17 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 153.9, 153.8, 151.6, 137.6, 119.4, 88.8, 85.3, 74.7, 71.9, 62.5, 29.6, 25.8, 25.7, 24.6, 22.6, 18.4, 18.0, 17.9, 14.0, −4.3, −4.7, −5.0, −5.3. HRMS (TOF) calcd for C33H63ClN5O4Si3 [M + H]+ 712.3871, found 712.3875.
2-Chloro-6-(morpholin-4-yl)-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (10b): Compound 10b (430 mg, 90% yield) was obtained as a pale-yellow, thick gum after chromatography on a silica gel column by sequential elution with 10% EtOAc in hexanes and 20% EtOAc in hexanes. Rf (SiO2 and 20% EtOAc in hexanes) = 0.47. 1H-NMR (400 MHz, CDCl3): δ 7.98 (s, 1H, H-8), 5.92 (d, J = 4.8 Hz, 1H, H-1′), 4.73 (t, J = 4.8 Hz, 1H, H-2′), 4.31–4.20 (t, J = 4.2 Hz, 1H, H-3′and br m, 4H, morpholinyl N(CH2)2), 4.12–4.09 (m, 1H, H-4′), 4.06 (dd, J = 5.2, 11.2 Hz, 1H, H-5′), 3.83 (t, J = 5.0 Hz, 4H, morpholinyl CH2OCH2), 3.77 (dd, J = 3.2, 11.2 Hz, 1H, H-5′), 0.94, 0.92, and 0.83 (3s, 27H, t-Bu), 0.13, 0.10, −0.01, and −0.16 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 153.9, 153.7, 151.8, 138.1, 119.4, 88.8, 85.3,77.3,77.0,76.6,75.0, 71.8, 66.9, 62.3, 45.60, 29.6, 29.3, 26.0,25.8, 25.7, 22.6, 18.4, 18.0, 17.8,14.0, −4.3, −4.7, −5.0, −5.4. HRMS (TOF) calcd for C32H61ClN5O5Si3 [M + H]+ 714.3664 found 714.3668.
2-Chloro-6-[(2-dimethylamino)ethyl)(methyl)amino)]-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl] purine (11b): Compound 11b (280 mg, 58%) was obtained as a pale-yellow, thick gum after chromatography on a silica gel column by sequential elution with 10% MeOH in CH2Cl2, 20% MeOH in CH2Cl2, and 30% MeOH in CH2Cl2. Rf (SiO2 and 10% MeOH in CH2Cl2) = 0.10. 1H-NMR (500 MHz, CDCl3): δ 7.97 (s, 1H, H-8), 5.90 (d, J = 4.8 Hz, 1H, H-1′), 4.72 (t, J = 4.8 Hz, 1H, H-2′), 4.32 (t, J = 3.8 Hz, 1H, H-3′), 4.15–4.10 (m, 1H, H-4′ and br s, 2H, NCH2), 4.07 (dd, J = 5.2, 11.2 Hz, 1H, H-5′), 3.78 (dd, J = 2.8, 11.2 Hz, 1H, H-5′ and br m, 2H, NCH2), 2.59 (br s, 2H, NCH2), 2.32 (s, 6H, N(CH3)2), 0.94, 0.92, and 0.82 (3s, 27H, t-Bu), 0.13, 0.10, −0.01, and −0.15 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 154.8, 153.7, 151.4, 137.9, 119.5, 88.8, 85.2, 75.0, 71.9, 62.4, 45.7, 29.6, 26.0, 25.8, 25.7, 18.4, 18.0, 17.9, −4.3, −4.7, −5.0, −5.4. HRMS (TOF) calcd for C33H66ClN6O4Si3 [M + H]+ 729.4136, found 729.4157.
3.4. General Procedure for the Desilylation of Cladribine Analogues
To a 0.1 M solution of the silylated compound in anhydrous MeOH, KF (2 equiv/silyl group) was added. The mixture was heated at 80 °C for 20–26 h, cooled, and silica gel was added. The mixture was evaporated to dryness and the compound-impregnated silica gel was loaded onto a wet-packed silica gel column. The products were obtained by elution with appropriate solvents (see the individual compound headings for details).
2-Chloro-2′-deoxyadenosine (12a): Prepared from compound 5a (60.0 mg, 0.117 mmol) and KF (27.0 mg, 0.467 mmol) in MeOH (1.17 mL). Chromatography on a silica gel column sequentially eluted with 5% MeOH in EtOAc and 10% MeOH in EtOAc gave 24.0 mg (72% yield) of compound 12a as an off-white solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.13. 1H-NMR (500 MHz, CD3OD): δ 8.28 (s, 1H, H-8), 6.36 (t, J = 6.8 Hz, 1H, H-1′), 4.57 (m, 1H, H-3′), 4.04 (m, 1H, H-4′), 3.84 (dd, J = 2.9, 12.2 Hz, 1H, H-5′), 3.74 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 2.76 (app quint, Japp ~ 6.7 Hz, 1H, H-2′), 2.43–2.39 (ddd, J = 2.9, 5.9, 13.2 Hz, 1H, H-2′). 13C-NMR (125 MHz, CD3OD): δ 158.3, 155.3, 151.4, 141.9, 119.8, 89.9, 87.0, 73.0, 63.6, 41.6. HRMS (TOF) calcd for C10H12ClN5O3Na [M + Na]+ 308.0521, found 308.0523.
2-Chloro-N6-methyl-2′-deoxyadenosine (13a) [50]: Prepared from compound 6a (80.0 mg, 0.154 mmol) and KF (36.0 mg, 0.618 mmol) in MeOH (1.54 mL). Chromatography on a silica gel column sequentially eluted with 2.5% MeOH in EtOAc and 5% MeOH in EtOAc gave 39.0 mg (85% yield) of compound 13a as a white, foamy solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.21. 1H-NMR (500 MHz, CD3OD): δ 8.18 (s, 1H, H-8), 6.34 (t, J = 6.8 Hz, 1H, H-1′), 4.56 (m, 1H, H-3′), 4.04 (m, 1H, H-4′), 3.84 (dd, J = 2.4, 12.2 Hz, 1H, H-5′), 3.74 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 3.05 (br s, 3H, NCH3), 2.76 (app quint, Japp ~ 6.8 Hz, 1H, H-2′), 2.42–2.38 (ddd, J = 2.9, 5.8, 13.7 Hz, 1H, H-2′). 13C-NMR (125 MHz, CD3OD): δ 157.3, 155.5, 150.1, 141.2, 120.4, 89.9, 87.0, 73.0, 63.7, 41.6, 27.8. HRMS (TOF) calcd for C11H14ClN5O3Na [M + Na]+ 322.0677, found 322.0682.
2-Chloro-N6,N6-dimethyl-2′-deoxyadenosine (Cladribine, 14a): Prepared from compound 7a (80.0 mg, 0.147 mmol) and KF (34.3 mg, 0.590 mmol) in MeOH (1.4 mL). Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 38.0 mg (82% yield) of compound 14a as a white, foamy solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.29. 1H-NMR (500 MHz, CD3OD): δ 8.16 (s, 1H, H-8), 6.35 (t, J = 6.8 Hz, 1H, H-1′), 4.56 (m, 1H, H-3′), 4.04 (m, 1H, H-4′), 3.84 (dd, J = 2.9, 12.2 Hz, 1H, H-5′), 3.73 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 3.70–3.10 (br m, 6H, N(CH3)2), 2.74 (app quint, Japp ~ 6.8 Hz, 1H, H-2′), 2.41–2.36 (ddd, J = 2.9, 5.8, 13.2 Hz, 1H, H-2′). 13C-NMR (125 MHz, CD3OD): δ 156.4, 154.6, 152.2, 140.0, 120.72, 89.8, 86.9, 73.0, 63.7, 41.5, 39.0 (br). HRMS (TOF) calcd for C12H16ClN5O3Na [M + Na]+ 336.0834, found 336.0823.
2-Chloro-6-(pyrrolidin-1-yl)-9-(2-deoxy-β-d-ribofuranosyl)purine (15a): Prepared from compound 8a (60.0 mg, 0.105 mmol) and KF (24.5 mg, 0.422 mmol) in MeOH (1.0 mL). Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 29.1 mg (81% yield) of compound 15a as a white solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.19. 1H-NMR (500 MHz, CD3OD): δ 8.19 (s, 1H, H-8), 6.36 (t, J = 7.1 Hz, 1H, H-1′), 4.59 (m, 1H, H-3′), 4.14–4.07 (br m, 2H, pyrrolidinyl NCH), 4.04 (m, 1H, H-4′), 3.84 (dd, J = 3.4, 12.7 Hz, 1H, H-5′), 3.74 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 3.71–3.62 (br m, 2H, pyrrolidinyl NCH), 2.75 (app quint, Japp ~ 6.7 Hz, 1H, H-2′), 2.40–2.36 (ddd, J = 2.9, 5.8, 13.2 Hz, 1H, H-2′), 2.15–2.06 (br m, 2H, pyrrolidinyl CH), 2.04–1.92 (br m, 2H, pyrrolidinyl CH). 13C-NMR (125 MHz, CD3OD): δ 155.0, 154.7, 151.7, 140.6, 120.8, 89.9, 87.0, 73.0, 63.7, 50.2, 49.6, 41.6, 27.3, 25.2. HRMS (TOF) calcd for C14H19ClN5O3 [M + H]+ 340.1171, found 340.1149.
2-Chloro-6-(piperidin-1-yl)-9-(2-deoxy-β-d-ribofuranosyl)purine (16a): Prepared from compound 9a (70.0 mg, 0.120 mmol) and KF (28.0 mg, 0.481 mmol) in MeOH (1.20 mL). Chromatography on a silica gel column sequentially eluted with EtOAc and 2% MeOH in EtOAc gave 30.0 mg (71% yield) of compound 16a as a white, foamy solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.46. 1H-NMR (500 MHz, CD3OD): δ 8.15 (s, 1H, H-8), 6.34 (t, J = 6.6 Hz, 1H, H-1′), 4.55 (m, 1H, H-3′), 4.18 (br s, 4H, piperidinyl N(CH2)2), 4.03 (m, 1H, H-4′), 3.83 (dd, J = 2.4, 12.2 Hz, 1H, H-5′), 3.73 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 2.74 (app quint, Japp ~ 6.8 Hz, 1H, H-2′), 2.40–2.35 (ddd, J = 2.7, 5.9, 13.5 Hz, 1H, H-2′), 1.74 (br m, 2H, piperidinyl CH2), 1.65 (br m, 4H, piperidinyl CH2). 13C-NMR (125 MHz, CD3OD): δ 155.3, 154.9, 152.7, 139.6, 120.5, 89.8, 86.8, 73.0, 63.7, 47.8, 41.6, 27.3, 25.7. HRMS (TOF) calcd for C15H20ClN5O3Na [M + Na]+ 376.1147, found 376.1148.
2-Chloro-6-(morpholin-4-yl)-9-(2-deoxy-β-d-ribofuranosyl)purine (17a): Prepared from compound 10a (80.0 mg, 0.137 mmol) and KF (31.8 mg, 0.548 mmol) in MeOH (1.4 mL). Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 36.1 mg (74% yield) of compound 17a as a white, foamy solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.21. 1H-NMR (500 MHz, CD3OD): δ 8.22 (s, 1H, H-8), 6.37 (t, J = 7.1 Hz, 1H, H-1′), 4.56 (m, 1H, H-3′), 4.40–4.12 (br m, 4H, morpholinyl N(CH2)2), 4.04 (m, 1H, H-4′), 3.83 (dd, J = 2.9, 12.2 Hz, 1H, H-5′), 3.79 (t, J = 4.9 Hz, 4H, morpholinyl CH2OCH2), 3.74 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 2.74 (app quint, Japp ~ 6.7 Hz, 1H, H-2′), 2.41–2.37 (ddd, J = 2.9, 5.8, 13.2 Hz, 1H, H-2′). 13C-NMR (125 MHz, CD3OD): δ 155.2, 154.7, 152.7, 140.3, 120.6, 89.8, 86.8, 72.9, 68.0, 63.6, 47.0 (br), 41.5. HRMS (TOF) calcd for C14H19ClN5O4 [M + H]+ 356.1120, found 356.1107.
2-Chloro-6-[(2-dimethylamino)ethyl)(methyl)amino)]-9-(2-deoxy-β-d-ribofuranosyl)purine (18a): Prepared from compound 11a (70.0 mg, 0.117 mmol) and KF (27.0 mg, 0.467 mmol) in MeOH (1.17 mL). Chromatography on a silica gel column sequentially eluted with 10% MeOH in EtOAc and 20% MeOH in EtOAc gave 36.0 mg (84% yield) of compound 18a as a white, foamy solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.10. 1H-NMR (500 MHz, CD3OD): δ 8.14 (s, 1H, H-8), 6.35 (t, J = 6.9 Hz, 1H, H-1′), 4.56 (m, 1H, H-3′), 4.12 (br s, 2H, NCH2), 4.03 (m, 1H, H-4′), 3.83 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 3.74 (dd, J = 3.9, 12.2 Hz, 1H, H-5′), 3.48 (br s, 3H, NCH3), 2.76–2.71 (br m, 3H, NCH2 and 1H, H-2′), 2.42–2.41 (m, 1H, H-2′), 2.39 (s, 6H, N(CH3)2). 13C-NMR (125 MHz, CD3OD): δ 156.4, 154.7, 152.5, 140.1, 120.8, 89.8, 86.8, 73.0, 63.7, 57.5, 45.8, 41.6, 37.6, (one broadened resonance could not be identified). HRMS (TOF) calcd for C15H24ClN6O3 [M + H]+ 371.1598, found 371.1584.
3.5. General Procedure for the Desilylation of Ribose Cladribine Analogues
To a 0.1 M solution of the silylated compound in anhydrous MeOH, KF (2 equiv/silyl group) was added. The mixture was heated at 80 °C for 24 h, cooled, and silica gel was added. The mixture was evaporated to dryness and the compound-impregnated silica gel was loaded onto a wet-packed silica gel column. The products were obtained by elution with appropriate solvents (see the individual compound headings for details).
2-Chloroadenosine (12b): Prepared from compound 5b (100 mg, 0.155 mmol) and KF (54.0 mg, 0.93 mmol) in MeOH (1.55 mL). Chromatography on a silica gel column sequentially eluted with 5% MeOH in EtOAc and 10% MeOH in EtOAc gave 35.0 mg (74% yield) of compound 12b as an off-white solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.13. 1H-NMR (400 MHz, CD3OD): δ 8.27 (s, 1H, H-8), 5.92 (d, J = 6.0 Hz, 1H, H-1′), 4.72 (t, J = 5.6, 1H, H-2′), 4.32 (dd, J = 2.8, 5.2 Hz, 1H, H-3′), 4.14 (m, 1H, H-4′), 3.90 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.76 (dd, J = 2.8, 12.4 Hz, 1H, H-5′). 13C-NMR (100 MHz, DMSO-d6): δ 174.5, 156.7, 152.9, 150.3, 140.0, 118.1, 87.4, 85.7, 73.8, 70.3, 61.3, 25.3. HRMS (TOF) calcd for C10H13ClN5O4 [M + H]+ 302.0651, found 302.0627.
2-Chloro-N6-methyladenosine (13b): Prepared from compound 6b (100 mg, 0.152 mmol) and KF (52.9 mg, 0.912 mmol) in MeOH (1.52 mL). Chromatography on a silica gel column sequentially eluted with 2.5% MeOH in EtOAc and 5% MeOH in EtOAc gave 36.0 mg (75% yield) of compound 13b as a white, foamy solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.21. 1H-NMR (400 MHz, CD3OD): δ 8.20 (s, 1H, H-8), 5.90 (d, J = 6.0 Hz, 1H, H-1′), 4.68 (t, J = 5.6 Hz, 1H, H-2′), 4.31 (dd, J = 2,8, 4.8 Hz, 1H, H-3′), 4.14 (m, 1H, H-4′), 3.96 (dd, J = 2.4, 12.4 Hz, 1H, H-5′), 3.76 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.07 (br s, 3H, NCH3). 13C-NMR (100 MHz, DMSO-d6): δ 155.5, 153.2, 149.2, 139.7, 118.6, 87.3, 85.6, 73.6, 70.6, 70.3, 61.6, 61.3, 53.9, 27.1. HRMS (TOF) calcd for C11H15ClN5O4 [M + H]+ 316.0807, found 316.0808.
2-Chloro-N6,N6-dimethyladenosine (14b): Prepared from compound 7b (100 mg, 0.148 mmol) and KF (51.8 mg, 0.892 mmol) in MeOH (1.48 mL). Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 36.8 mg (75% yield) of compound 14b as an off-white, foamy solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.32. 1H-NMR (400 MHz, CD3OD): δ 8.17 (s, 1H, H-8), 5.91 (d, J = 6.4 Hz, 1H, H-1′), 4.67 (t, J = 5.4 Hz, 1H, H-2′), 4.31 (dd, J = 2.8, 4.8 Hz, 1H, H-3′), 4.14 (m, 1H, H-4′), 3.90 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.76 (dd, J = 2.4, 12.4 Hz, 1H, H-5′), 3.60–3.49 (br m, H, N(CH3)2). 13C-NMR (100 MHz, DMSO-d6): δ 154.4, 152.5, 151.1, 138.6, 118.5, 87.2, 85.6, 73.6, 70.2, 61.2, 37.5 (br). HRMS (TOF) calcd for C12H17ClN5O4 [M + H]+ 330.0964, found 330.0964.
2-Chloro-6-(pyrrolidin-1-yl)-9-(β-d-ribofuranosyl)purine (15b): Prepared from compound 8b (100 mg, 0.143 mmol) and KF (49.8 mg, 0.858 mmol) in MeOH (1.43 mL). Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 43.3 mg (85% yield) of compound 15b as a white solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.38. 1H-NMR (400 MHz, CD3OD): δ 8.18 (s, 1H, H-8), 5.91 (d, J = 6.4 Hz, 1H, H-1′), 4.67 (t, J = 5.4 Hz, 1H, H-2′), 4.32 (dd, J = 2.8, 4.8 Hz, 1H, H-3′), 4.14 (m, 1H, H-4′), 4.13–4.10 (br m, 2H, pyrrolidinyl NCH), 3.90 (dd, J = 2.8, 12.8 Hz, 1H, H-5′), 3.74 (dd, J = 2.4, 12.4 Hz, 1H, H-5′), 3.72–3.65 (br m, 2H, pyrrolidinyl NCH), 2.13–2.07 (br m, 2H, pyrrolidinyl CH), 2.05–1.95 (br m, 2H, pyrrolidinyl CH). 13C-NMR (100 MHz, DMSO-d6): δ 152.7, 152.7, 150.7, 139.1, 118.7, 87.2, 85.6, 73.7, 70.2, 61.2, 48.6, 47.3, 25.6, 23.6. HRMS (TOF) calcd for C14H19ClN5O4 [M + H]+ 356.1120, found 356.1127.
2-Chloro-6-(piperidin-1-yl)-9-(β-d-ribofuranosyl)purine (16b): Prepared from compound 9b (100 mg, 0.140 mmol) and KF (48.9 mg, 0.842 mmol) in MeOH (1.40 mL). Chromatography on a silica gel column sequentially eluted with EtOAc and 2% MeOH in EtOAc gave 43.6 mg (84% yield) of compound 16b as a white, foamy solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.41. 1H-NMR (400 MHz, CD3OD): δ 8.13 (s, 1H, H-8), 5.87 (d, J = 6.0 Hz, 1H, H-1′), 4.64 (t, J = 5.4 Hz, 1H, H-2′), 4.27 (dd, J = 2.8, 5.2 Hz, 1H, H-3′), 4.19 (br s, 4H, piperidinyl N(CH2)2), 4.11 (m, 1H, H-4′), 3.89 (dd, J = 2.8, 12.2 Hz, 1H, H-5′), 3.73 (dd, J = 2.4, 12.2 Hz, 1H, H-5′), 1.74 (br m, 2H, piperidinyl CH2), 1.64 (br m, 4H, piperidinyl CH2). 13C-NMR (100 MHz, DMSO-d6): δ 153.1, 152.6, 151.4, 138.5, 118.2, 87.2, 85.6, 73.6, 70.4, 70.2, 70.1, 61.2, 44.8 (br), 25.6, 25.2, 23.9, 14.0. HRMS (TOF) calcd for C15H21ClN5O4 [M + H]+ 370.1277, found 370.1302.
2-Chloro-6-(morpholin-4-yl)-9-(β-d-ribofuranosyl)purine (17b): Prepared from compound 10b (100 mg, 0.139 mmol) and KF (48.7 mg, 0.839 mmol) in MeOH (1.40 mL). Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 40.6 mg (78% yield) of compound 17b as a white, foamy solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.21. 1H-NMR (400 MHz, CD3OD): δ 8.21 (s, 1H, H-8), 5.92 (d, J = 6.0 Hz, 1H, H-1′), 4.66 (t, J = 5.6 Hz, 1H, H-2′), 4.32 (dd, J = 2.8, 4.8 Hz, 1H, H-3′), 4.30–4.24 (br m, 4H, morpholinyl N(CH2)2), 4.14 (m, 1H, H-4′), 3.90 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.80 (t, J = 4.8 Hz, 4H, morpholinyl CH2OCH2), 3.76 (dd, J = 2.8, 12.4 Hz, 1H, H-5′). 13C-NMR (100 MHz, DMSO-d6): δ 153.3, 152.5, 151.6, 139.0, 118.3, 87.3, 85.6, 85.4, 73.7, 70.4, 70.2, 66.0, 61.4, 61.1, 48.5, 45.0 (br). HRMS (TOF) calcd for C14H19ClN5O5 [M + H]+ 372.1069, found 372.1056.
2-Chloro-6-[(2-dimethylamino)ethyl)(methyl)amino)]-9-(β-d-ribofuranosyl)purine (18b): Prepared from compound 11b (100 mg, 0.137 mmol) and KF (47.7 mg, 0.822 mmol) in MeOH (1.37 mL). Chromatography on a silica gel column sequentially eluted with 10% MeOH in EtOAc and 20% MeOH in EtOAc gave 34.5 mg (65% yield) of compound 18b as a white, foamy solid. Rf (SiO2 and 15% MeOH in EtOAc) = 0.10. 1H-NMR (400 MHz, CD3OD): δ 8.19 (s, 1H, H-8), 5.92 (d, J = 6 Hz, 1H, H-1′), 4.67 (t, 1H, J = 5.6 Hz, 1H, H-2′), 4.34 (dd, J = 3.2, 5.2 Hz 1H, H-3′), 4.14 (d, J = 2.4 Hz, 3H, H-4′), 3.90 (dd, J = 2.4, 12.4 Hz, 1H, H-5′), 3.76 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.49 (br s, 3H, NCH3), 2.83 (t, 2H, NCH2, J = 6.8 Hz ), 2.47 (s, 6H, N(CH3)2). 13C-NMR (100 MHz, CD3OD): δ 156.1, 154.7, 152.3, 140.5, 120.7, 90.8, 87.7, 75.3, 72.3, 63.2, 57.0, 49.6, 49.4, 49.2, 49.0, 48.7, 48.5, 48.3, 45.5, 37.4. HRMS (TOF) calcd for C15H24ClN6O4 [M + H]+ 387.1542, found 387.1513.
O6-(Benzotriazol-1-yl)-2-bromo-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (19a): A mixture of compound 3a (300.0 mg, 0.489 mmol) and SbBr3 (247.7 mg, 0.685 mmol) in dry CH2Br2 (4.9 mL) was cooled to −15 °C using dry ice and acetone, in a nitrogen atmosphere. t-BuONO (203.8 μL, 1.713 mmol) was added dropwise and the mixture was stirred at −10 °C to −15 °C for 2 h. Because TLC indicated the presence of starting material, another aliquot of t-BuONO (203.8 μL, 1.713 mmol) was added and the reaction proceeded for 1 h at −15 °C, at which time TLC indicated the reaction to be complete. The reaction mixture was poured into ice-cold, saturated aqueous NaHCO3 (5 mL) with stirring. The mixture was filtered using vacuum (note: use of vacuum for this filtration is critical for maximizing product recovery) and the residue was washed with CH2Cl2 (5 mL). The organic layer was separated and the aqueous layer was back extracted with CH2Cl2 (2 × 5 mL). The combined organic layer was washed with water (5 mL) and brine (5 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification of the crude material on a silica gel column sequentially eluted with hexanes, 5% EtOAc in hexanes, and 30% EtOAc in hexanes gave 208.5 mg (63% yield) of compound 19a as a white foam. Rf (SiO2 and 30% EtOAc in hexanes) = 0.60. 1H-NMR (500 MHz, CDCl3): δ 8.45 (s, 1H, H-8), 8.13 (d, J = 8.3 Hz, 1H, Ar-H), 7.56–7.45 (m, 3H, Ar-H), 6.47 (t, J = 5.9 Hz, 1H, H-1′), 4.63 (m, 1H, H-3′), 4.03 (m, 1H, H-4′), 3.90 (dd, J = 3.4, 11.2 Hz, 1H, H-5′), 3.78 (dd, J = 1.5, 11.2 Hz, 1H, H-5′), 2.61 (app quint, Japp ~ 6.2 Hz, 1H, H-2′), 2.49 (app quint, Japp ~ 5.8 Hz, 1H, H-2′), 0.91 (s, 18H, t-Bu), 0.11 and 0.09 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 158.7, 154.9, 144.3, 143.6, 142.6, 129.1, 128.9, 125.1, 120.8, 119.5, 108.7, 88.5, 85.4, 71.7, 62.7, 41.8, 26.2, 25.9, 18.6, 18.2, −4.4, −4.6, −5.2, −5.3. HRMS (TOF) calcd for C28H43BrN7O4Si2 [M + H]+ 676.2093, found 676.2078.
2-Bromo-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyadenosine (20a): To a solution of compound 19a (135.3 mg, 0.20 mmol) in 1,2-DME (2 mL), 28%–30% aqueous ammonia (48.6 μL) was added, and the mixture was stirred at room temperature for 45 min. The mixture was diluted with EtOAc (15 mL) and washed with 5% aqueous NaCl (10 mL). The organic layer was separated and the aqueous layer was back extracted with EtOAc (15 mL). The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude material was chromatographed on a silica gel column by sequential elution with hexanes, 20% EtOAc in hexanes and 40% EtOAc in hexanes to afford 93.1 mg (83% yield) of compound 20a as a white foam. Rf (SiO2 and EtOAc) = 0.50. 1H-NMR (500 MHz, CDCl3): δ 8.08 (s, 1H, H-8), 6.63 (br s, 2H, Ar-NH2), 6.37 (t, J = 6.1 Hz, 1H, H-1′), 4.61 (m, 1H, H-3′), 3.98 (m, 1H, H-4′), 3.88 (dd, J = 3.9, 11.2 Hz, 1H, H-5′), 3.75 (dd, J = 2.4, 11.2 Hz, 1H, H-5′), 2.61 (app quint, Japp ~ 6.3 Hz, 1H, H-2′), 2.43–2.38 (m, 1H, H-2′), 0.90 (s, 18H, t-Bu), 0.09 and 0.08 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 156.3, 150.4, 144.9, 139.6, 119.3, 88.2, 84.7, 71.9, 62.9, 41.4, 26.1, 25.9, 18.6, 18.2, −4.4, −4.6, −5.2, −5.3. HRMS (TOF) calcd for C22H41BrN5O3Si2 [M + H]+ 558.1926, found 558.1902.
2-Bromo-2′-deoxyadenosine (21a): As described in the general desilylation procedures, compound 21a was prepared from compound 20a (80.0 mg, 0.143 mmol) and KF (33.2 mg, 0.572 mmol) in MeOH (1.4 mL), over 28 h. Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 31.1 mg (66% yield) of compound 21a as a white/off-white solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.40. 1H-NMR (500 MHz, CD3OD): δ 8.25 (s, 1H, H-8), 6.36 (t, J = 6.8 Hz, 1H, H-1′), 4.57 (m, 1H, H-3′), 4.04 (m, 1H, H-4′), 3.84 (dd, J = 2.9, 12.2 Hz, 1H, H-5′), 3.74 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 2.76 (app quint, Japp ~ 6.7 Hz, 1H, H-2′), 2.43–2.39 (ddd, J = 2.9, 5.9, 13.2 Hz, 1H, H-2′). 13C-NMR (125 MHz, CD3OD): δ 158.1, 151.4, 145.9, 141.7, 120.3, 89.9, 86.9, 72.9, 63.7, 41.6. HRMS (TOF) calcd for C10H12BrN5O3Na [M + Na]+ 352.0016, found 352.0021.
O6-(Benzotriazol-1-yl)-2-bromo-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (19b): A mixture of compound 3b (500.0 mg, 0.67 mmol) and SbBr3 (340.8 mg, 0.94 mmol) in dry CH2Br2 (8.3 mL) was cooled to −15 °C using dry ice and acetone, in a nitrogen atmosphere. t-BuONO (280 μL, 2.352 mmol) was added dropwise and the mixture was stirred at −10 °C to −15 °C for 2 h. Because TLC indicated the presence of starting material, another aliquot of t-BuONO (280 μL, 2.352 mmol) was added and the reaction was allowed to progress for 1 h at −15 °C, at which time TLC indicated the reaction to be complete. The reaction mixture was poured into ice-cold, saturated aqueous NaHCO3 (10 mL) with stirring. The mixture was filtered using vacuum (note: use of vacuum for this filtration is critical for maximizing product recovery) and the residue was washed with CH2Cl2 (15 mL). The organic layer was separated and the aqueous layer was back extracted with CH2Cl2 (2 × 15 mL). The combined organic layer was washed with water (10 mL) and brine (10 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification of the crude material on a silica gel column sequentially eluted with hexanes, 5% EtOAc in hexanes, and 30% EtOAc in hexanes gave 350 mg (64% yield) of compound 19b as a white foam. Rf (SiO2 and 30% EtOAc in hexanes) = 0.80. 1H-NMR (400 MHz, CDCl3): δ 8.54 (s, 1H, H-8), 8.15 (d, J = 8.4 Hz, 1H, Ar-H), 7.58–7.45 (m, 3H, Ar-H), 6.05 (d, J = 4.0 Hz, 1H, H-1′), 4.57 (t, J = 4.2 Hz, 1H, H-2′), 4.33 (t, J = 4.6 Hz, 1H, H-3′), 4.17 (m, 1H, H-4′), 4.08 (dd, J = 3.6, 11.6 Hz, 1H, H-5′), 3.83 (dd, J = 2.4, 11.6 Hz, 1H, H-5′), 0.96, 0.93, and 0.85 (3s, 27H, t-Bu), 0.16, 0.11, −0.03, and −0.09 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 158.5, 154.8, 144.2, 143.4, 142.5, 128.8, 128.7, 124.8, 120.6, 119.4, 108.5, 89.5, 85.2, 76.1, 71.1, 61.9, 29.6, 26.1, 25.8, 25.6, 18.5, 18.0, 17.9, −4.2, −4.6, −4.7, −4.9, −5.3, −5.4. HRMS (TOF) calcd for C34H57BrN7O5Si3 [M + H]+ 806.2907, found 806.2912.
2-Bromo-2′,3′,5′-tri-O-(t-butyldimethylsilyl)-adenosine (20b): To a solution of compound 19b (200 mg, 0.247 mmol) in 1,2-DME (4 mL), 28%–30% aqueous ammonia (60 μL) was added, and the mixture was stirred at room temperature for 45 min. The mixture was diluted with EtOAc (20 mL) and washed with 5% aqueous NaCl (20 mL). The organic layer was separated and the aqueous layer was back extracted with EtOAc (25 mL). The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude material was chromatographed on a silica gel column by sequential elution with hexanes, 20% EtOAc in hexanes, and 40% EtOAc in hexanes to afford 140 mg (82% yield) of compound 20b as a white foam. Rf (SiO2 and EtOAc) = 0.55. 1H-NMR (400 MHz, CDCl3): δ 8.10 (s, 1H, H-8), 5.92 (d, J = 4.8 Hz, 1H, H-1′), 5.77 (br s, 2H, Ar-NH2), 4.69 (t, J = 4.6 Hz, 1H, H-2′), 4.32 (t, J = 4.2 Hz, 1H, H-3′), 4.13 (m, 1H, H-4′), 4.06 (dd, J = 4.8, 11.2 Hz, 1H, H-5′), 3.80 (dd, J = 2.8, 11.2 Hz, 1H, H-5′), 0.95, 0.93, and 0.83 (3s, 27H, t-Bu), 0.14, 0.11, −0.00, and −0.15 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 155.8, 150.5, 144.6, 140.0, 119.4, 110.0, 89.0, 85.3, 75.4, 71.7, 62.3, 26.0, 25.8, 25.7, 18.4, 18.0, 17.9, −4.3, −4.7, −5.0, −5.3, −5.4. HRMS (TOF) calcd for C28H55BrN5O4Si3 [M + H]+ 688.2740, found 688.2728.
2-Bromoadenosine (21b): As described in the general desilylation procedures, compound 21b was prepared from compound 20b (100 mg, 0.145 mmol) and KF (50.5 mg, 0.870 mmol) in MeOH (1.9 mL), over 24 h. Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc which gave 34.9 mg (69% yield) of compound 21b as a pale yellow solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.39. 1H-NMR (400 MHz, CD3OD): δ 8.25 (s, 1H, H-8), 5.92 (d, J = 6.4 Hz, 1H, H-1′), 4.67 (d, J = 5.6 Hz, 1H, H-2′), 4.32 (dd, J = 2.8, 4.8 Hz, 1H, H-3′), 4.15 (m, 1H, H-4′), 3.90 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.76 (dd, J = 2.8, 12.4 Hz, 1H, H-5′). 13C-NMR (100 MHz, CD3OD): δ 156.5, 150.2, 144.1, 139.8, 118.4, 87.2, 85.7, 73.6, 70.3, 61.3. HRMS (TOF) calcd for C10H13BrN5O4 [M + H]+ 346.0145, found 346.0154.
3.6. Protocols for Tests against HCL, TCL, and CCL
Blood was obtained in sodium heparin tubes from patients as part of protocols with consent forms approved by the investigators review board (IRB) of the National Cancer Institute. The blood was diluted 1:1 with PBS without calcium or magnesium, layered over 15 mL Ficoll in 50 mL tubes, and centrifuged to obtain mononuclear cells. Patients with high lymphocytosis had leukemic cells >80%–90% pure after Ficoll. The cells were viably frozen in 7.5% DMSO in leucine-poor media (LPM, 88% leucine-free RPMI, 2% RPMI, and 10% FBS) in cryovials and stored under liquid nitrogen. LPM also contained penicillin, streptomycin, glutamine, gentamycin, and doxycycline. To assay, thawed cells were washed, suspended in LPM, added to 96-well round-bottom plates (15 μL/well), and treated with 15 μL of purine analogues diluted in LPM. The aliquots were incubated 3 days, then treated with 10 μL of either ATP (CellTiter-Glo, Promega, Madison, WI, USA) or {3H}-leucine (Perkin-Elmer, Waltham, MA, USA) diluted in leucine-free RPMI. After 30 min of ATP, the plate is read for bioluminescence. After 6 h of {3H}-leucine, the cells were liberated by freeze-thaw, harvested on to glass-fiber filters, counted either by a beta scintillation counter. The number of cells cultured in 30 μL aliquots for ATP assay was 20,000, 20,000, and 100,000 for HCL, TCL, and CLL, respectively. The cell number for {3H}-leucine assay was 60,000, 60,000 and 200,000 for HCL, TCL, and CLL, respectively. HCL and TCL cells were pulsed with 1 μCi of {3H}-leucine, while CLL cells were pulsed with 1.5–2 μCi/well. The IC50 was the calculated concentration needed for 50% inhibition, defined as the ATP uptake or {3H}-leucine incorporation corresponding to halfway between that of control (cells with LPM alone) and that of cycloheximide 10 μg/mL. Reported IC50 values were the means of 3 triplicate experiments.
3.7. Protocols for Tests against Breast Cancer Cells
MDA-MB-231 breast cancer (BC) cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), and were cultured at 37 °C in 5% CO2 using culture medium recommended by the supplier. Each tested compound was dissolved in sterile DMSO, then diluted (4.8–57.7 mM) in sterile 1X PBS for a final DMSO concentration of 0.1%. Then, 2 × 105 cells/well, were seeded and cultured for 24 h as described [51,52]. Afterwards, the media was changed to 5% FBS for 1 h and cells were treated in duplicate with dilutions of each treatment (0–1.8 mM) for 24 h. Cells were fixed (cold methanol), and nuclei stained (0.4% propidium iodide, (PI)) (Sigma-Aldrich, St Louis, MO, USA), and measured using a GloMax® Microplate Reader (Promega, Madison, WI, USA). Cell viability was calculated as percent of surviving cells after treatment relative to vehicle wells. The IC50 was obtained from dose response curve fittings using the non-linear regression function of GraphPad Prism® version 6.0b for Mac (GraphPad Software, San Diego, CA, USA) [53].
4. Conclusions
We have demonstrated, for the first time, the synthesis of O6-(benzotriazol-1-yl)-2-chloro-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (4a) by diazotization-chlorination of O6-(benzotriazol-1-yl)-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyguanosine (3a) with t-BuONO and SbCl3 in CH2Cl2. This procedure afforded better yields than the chlorination using t-BuONO and Me3SiCl. This compound and its ribose analogue, O6-(benzotriazol-1-yl)-2-chloro-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine, both undergo smooth reactions with ammonia, and primary, and secondary amines to produce cladribine (12a), its N-modified analogues (13a–18a), and the corresponding ribose derivatives (12b–18b), after a simple desilylation with KF in MeOH. These compounds were tested against HCL, TCL, and CLL, but none of the new compounds was more active than cladribine itself. The bromo as well as ribose analogues of cladribine displayed activity but the bromo analogue of cladribine was more active against TCL and CLL as compared to both the ribose equivalent and the bromo ribose analogue of cladribine. The compound containing both the bromine atom and a ribose ring was least active among the compounds possessing a primary amino group at the C6 position. Thus, it appears that a free amino group at this location is critical to the activity of cladribine. Interestingly, the C6 piperidinyl analog of cladribine showed low activity. Tests against MDA-MB-231 breast cancer cells showed that only cladribine and its ribose analogue showed some activity. The bromo analogues were about 10 times less active and all others showed no potential. Despite the lack of a major improvement in the activity of cladribine, or the identification of new compounds with activity against breast cancer, this work has provided a route to four doubly functionalizable nucleoside derivatives. The orthogonal reactivities of these compounds, i.e., SNAr at the C6 position and metal catalysis at the C2 position, can be used for development of novel nucleoside analogues. We anticipate pursuing further work along these lines in the future.
Acknowledgments
This work was supported by National Science Foundation Grant CHE-1265687 to MKL, in part by National Institutes of Health Grant SC3GM111171 to MMMM from the National Institute on General Medical Sciences, and Title V PPOHA US Department of Education P031M105050 to Universidad Central del Caribe. Infrastructural support at CCNY was provided by National Institute on Minority Health and Health Disparities Grant G12MD007603, while that at UCC was provided by G12MD007583. Siva Subrahmanyam Relangi, Venkateshwarlu Gurram, Muralidharan Kaliyaperumal, Somesh Sharma, and Narender Pottabathini gratefully acknowledge support from GVKBIO and Siva Subrahmanyam Relangi thanks Anna Venkateswara Rao (Koneru Lakshmaiah University, Guntur (Andhra Pradesh), India) for his support.
Supplementary Materials
Supplementary can be accessed at: http://www.mdpi.com/1420-3049/20/10/18437/s1.
Author Contributions
Sakilam Satishkumar, Prasanna K. Vuram, and Siva Subrahmanyam Relangi had equal contributions to the synthesis portion of this work. Sakilam Satishkumar and Prasanna K. Vuram (City College of New York) performed optimization of the synthetic procedures, executed syntheses of the deoxyribose series, performed spectroscopic analyses of the compounds, and produced a part of the experimental section. Messrs. Relangi and Venkateshwarlu Gurram (GVKBIO) executed syntheses of the ribose series, performed spectroscopic analyses of the compounds, and produced a part of the experimental section. Hong Zhou and Robert J. Kreitman (National Cancer Institute) tested the compounds against HCL, TCL, and CLL. Michelle M. Martínez Montemayor tested the compounds against breast cancer cell lines. Lijia Yang (City College of New York) performed HRMS analysis of all compounds synthesized in Lakshman’s laboratories. Narender Pottabathini (GVKBIO) was responsible for the oversight of the work performed in his laboratories. Mahesh K. Lakshman (City College of New York) conceived and designed the research, assisted with data analysis, and wrote a significant portion of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Limited amounts of compounds 12a–18a and 12b–18b may be available from the authors.
References and Notes
- 1.Venner H. Synthese der den natürlichen entsprechenden 2-desoxy-nucleoside des adenins, guanins, und hypoxanthins. Chem. Ber. 1960;63:100–110. doi: 10.1002/cber.19600930123. [DOI] [Google Scholar]
- 2.Carson D.A., Kaye J., Seegmiller J.E. Lymphospecific toxicity in adenosine deaminase deficiency and purine nucleoside phosphorylase deficiency: Possible role of nucleosidase kinase(s) Proc. Natl. Acad. Sci. USA. 1977;74:5677–5681. doi: 10.1073/pnas.74.12.5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carson D.A., Wasson D.B., Kaye J., Ullman B., Martin D.W., Jr., Robins R.K., Montgomery J.A. Deoxycytidine kinase-mediated toxicity of deoxyadenosine analogs toward malignant human lymphoblasts in vitro and toward murine L1210 leukemia in vivo. Proc. Natl. Acad. Sci. USA. 1980;77:6865–6869. doi: 10.1073/pnas.77.11.6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang L., Karlsson A., Arnér E.S.J., Eriksson S. Substrate specificity of mitochondrial 2′-deoxyguanosine kinase. Efficient phosphorylation of 2-chlorodeoxyadenosine. J. Biol. Chem. 1993;268:22847–22852. [PubMed] [Google Scholar]
- 5.Sjöberg A.H., Wang L., Eriksson S. Substrate specificity of human recombinant mitochondrial deoxyguanosine kinase with cytostatic and antiviral purine and pyrimidine analogs. Mol. Pharmacol. 1998;53:270–273. doi: 10.1124/mol.53.2.270. [DOI] [PubMed] [Google Scholar]
- 6.Genini D., Adachi S., Chao Q., Rose D.W., Carrera C.J., Cottam H.B., Carson D.A., Leoni L.M. Deoxyadenosine analogs induce programmed cell death in chronic lymphocytic leukemia cells by damaging the DNA and by directly affecting the mitochondria. Blood. 2000;96:3537–3543. [PubMed] [Google Scholar]
- 7.Marzo I., Pérez-Galán P., Giraldo P., Rubio-Félix D., Anel A., Naval J. Cladribine induces apoptosis in human leukaemia cells by caspase-dependent and -independent pathways acting on mitochondria. Biochem. J. 2001;359:537–546. doi: 10.1042/bj3590537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klöpfer A., Hasenjäger A., Belka C., Schulze-Osthoff K., Dörken B., Daniel P.T. Adenine deoxynucleotides fludarabine and cladribine induce apoptosis in a CD95/Fas receptor, FADD and caspase-8-independent manner by activation of the mitochondrial cell death pathway. Oncogene. 2004;23:9408–9418. doi: 10.1038/sj.onc.1207975. [DOI] [PubMed] [Google Scholar]
- 9.Goodman G.R., Burian C., Koziol J.A., Saven A. Extended follow-up of patients with hairy cell leukemia after treatment with cladribine. J. Clin. Oncol. 2003;21:891–896. doi: 10.1200/JCO.2003.05.093. [DOI] [PubMed] [Google Scholar]
- 10.Sigal D.S., Miller H.J., Schram E.D., Saven A. Beyond hairy cell: The activity of cladribine in other hematologic malignancies. Blood. 2010;116:2884–2896. doi: 10.1182/blood-2010-02-246140. [DOI] [PubMed] [Google Scholar]
- 11.The US National Institutes of Health Clinical Trials. [(accessed on 18 June 2015)]. Available online: https://www.clinicaltrials.gov/ct2/results?term=cladribine&Search=Search.
- 12.Ikehara M., Tada H. Studies of nucleosides and nucleotides. XVIV. Purine cyclonucleosides. I. 8,2′-Cyclonucleoside derived from 2-chloro-8-mercapto-9-β-d-xylofuranosyladenine. J. Am. Chem. Soc. 1965;87:606–610. doi: 10.1021/ja01081a038. [DOI] [PubMed] [Google Scholar]
- 13.Christensen L.F., Broom A.D., Robins M.J., Bloch A. Synthesis and biological activity of selected 2,6-disubstituted-(2-deoxy-α- and -β-d-erythro-pentofuranosyl)purines. J. Med. Chem. 1972;15:735–739. doi: 10.1021/jm00277a010. [DOI] [PubMed] [Google Scholar]
- 14.Kazimierczuk Z., Cottam H.B., Revankar G.R., Robins R.K. Synthesis of 2′-deoxytubercidin, 2′-deoxyadenosine, and related 2′-deoxynucleosides via a novel direct stereospecific sodium salt glycosylation procedure. J. Am. Chem. Soc. 1984;106:6379–6382. doi: 10.1021/ja00333a046. [DOI] [Google Scholar]
- 15.Lioux T., Gosselin G., Mathé C. Azido/tetrazole tautomerism in 2-azidoadenine β-d-pentofuranonucleoside derivatives. Eur. J. Org. Chem. 2003:3997–4002. doi: 10.1002/ejoc.200300274. [DOI] [PubMed] [Google Scholar]
- 16.Zhong M., Nowak I., Robins M.J. Regiospecific and highly stereoselective coupling of 6-(substituted-imidazol-1-yl)purines with 2-deoxy-3,5-di-O-(p-toluoyl)-α-d-erythro-pentofuranosyl chloride. Sodium-salt glycosylation in binary solvent mixtures: Improved synthesis of cladribine. J. Org. Chem. 2006;71:7773–7779. doi: 10.1021/jo061282+. [DOI] [PubMed] [Google Scholar]
- 17.Yang F., Zhu Y., Yu B. A dramatic concentration effect on the stereoselectivity of N-glycosylation for the synthesis of 2′-deoxy-β-ribonucleosides. Chem. Commun. 2012;48:7097–7099. doi: 10.1039/c2cc33155a. [DOI] [PubMed] [Google Scholar]
- 18.Henschke J.P., Zhang X., Huang X., Mei L., Chu G., Hu K., Wang Q., Zhu G., Wu M., Kuo C., Chen Y. A stereoselective process for the manufacture of a 2′-deoxy-β-d-ribonucleoside using the Vorbrüggen glycosylation. Org. Process Res. Dev. 2013;17:1419–1429. doi: 10.1021/op4002005. [DOI] [Google Scholar]
- 19.Xu S., Yao P., Chen G., Wang H. A new synthesis of 2-chloro-2′-deoxyadenosine (cladribine), CdA) Nucleosides Nucleotides Nucleic Acids. 2011;30:353–359. doi: 10.1080/15257770.2011.587701. [DOI] [PubMed] [Google Scholar]
- 20.Sakakibara N., Kakoh A., Maruyama T. First synthesis of [6-15N]-cladribine using ribonucleoside as a starting material. Heterocycles. 2012;85:171–182. [Google Scholar]
- 21.Mikhailopulo I.A., Zinchenko A.I., Kazimierczuk Z., Barai V.N., Bokut S.B., Kalinichenko E.N. Synthesis of 2-chloro-2′-deoxyadenosine by microbiological transglycosylation. Nucleosides Nucleotides. 1993;12:417–422. doi: 10.1080/07328319308017836. [DOI] [Google Scholar]
- 22.Barai V.N., Zinchenko A.I., Eroshevskaya L.A., Kalinichenko E.N., Kulak T.I., Mikhailopulo I.A. A universal biocatalyst for the preparation of base- and sugar-modified nucleosides via an enzymatic transglycosylation. Helv. Chim. Acta. 2002;85:1901–1908. doi: 10.1002/1522-2675(200207)85:7<1901::AID-HLCA1901>3.0.CO;2-C. [DOI] [Google Scholar]
- 23.Komatsu H., Araki T. Efficient chemo-enzymatic syntheses of pharmaceutically useful unnatural 2′-deoxynucleosides. Nucleosides Nucleotides Nucleic Acids. 2005;24:1127–1130. doi: 10.1081/NCN-200060154. [DOI] [PubMed] [Google Scholar]
- 24.Taran S.A., Verevkina K.N., Esikova T.Z., Feofanov S.A., Miroshnikov A.I. Synthesis of 2-chloro-2′-deoxyadenosine by microbiological transglycosylation using a recombinant Escherichia coli strain. Appl. Biochem. Microbiol. 2008;44:162–166. doi: 10.1134/S0003683808020063. [DOI] [PubMed] [Google Scholar]
- 25.Janeba Z., Francom P., Robins M.J. Efficient syntheses of 2-chloro-2′-deoxyadenosine (cladribine) from 2′-deoxyguanosine. J. Org. Chem. 2003;68:989–992. doi: 10.1021/jo020644k. [DOI] [PubMed] [Google Scholar]
- 26.Peng Y. A practical synthesis of 2-chloro-2′-deoxyadenosine (cladribine) from 2′-deoxyadenosine. J. Chem. Res. 2013;37:213–215. doi: 10.3184/174751913X13618878756705. [DOI] [Google Scholar]
- 27.Wan Z.-K., Wacharasindhu S., Binnun E., Mansour T. An efficient direct amination of cyclic amides and cyclic ureas. Org. Lett. 2006;8:2425–2428. doi: 10.1021/ol060815y. [DOI] [PubMed] [Google Scholar]
- 28.Wan Z.-K., Binnun E., Wilson D.P., Lee J. A highly facile and efficient one-step synthesis of N6-adenosine and N6-2′-deoxyadenosine derivatives. Org. Lett. 2005;7:5877–5880. doi: 10.1021/ol052424+. [DOI] [PubMed] [Google Scholar]
- 29.Bae S., Lakshman M.K. O6-(Benzotriazol-1-yl)inosine derivatives: Easily synthesized, reactive nucleosides. J. Am. Chem. Soc. 2007;129:782–789. doi: 10.1021/ja064682n. [DOI] [PubMed] [Google Scholar]
- 30.Wan Z.-K., Wacharasindhu S., Levins C.G., Lin M., Tabei K., Mansour T.S. The scope and mechanism of phosphonium-mediated SNAr reactions in heteocyclic amides and ureas. J. Org. Chem. 2007;72:10194–10210. doi: 10.1021/jo7020373. [DOI] [PubMed] [Google Scholar]
- 31.Ashton T.D., Scammells P.J. Microwave-assisted direct amination: Rapid access to multi-functionalized N6-substituted adenosines. Aust. J. Chem. 2008;61:49–58. doi: 10.1071/CH07340. [DOI] [Google Scholar]
- 32.Bae S., Lakshman M.K. Unusual deoxygenation and reactivity studies related to O6-(benzotriazol-1yl)inosine derivatives. J. Org. Chem. 2008;73:1311–1319. doi: 10.1021/jo7021795. [DOI] [PubMed] [Google Scholar]
- 33.Bae S., Lakshman M.K. A novel polymer supported approach to nucleoside modification. J. Org. Chem. 2008;73:3707–3713. doi: 10.1021/jo702558n. [DOI] [PubMed] [Google Scholar]
- 34.Bae S., Lakshman M.K. Synthetic utility of an isolable nucleoside phosphonium salt. Org. Lett. 2008;10:2203–2206. doi: 10.1021/ol8006106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lakshman M.K., Frank J. A simple method for C-6 modification of guanine nucleosides. Org. Bimol. Chem. 2009;7:2933–2940. doi: 10.1039/b905298d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kokatla H.P., Lakshman M.K. One-pot etherification of purine nucleosides and pyrimidines. Org. Lett. 2010;12:4478–4881. doi: 10.1021/ol101655h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lakshman M.K., Singh M.K., Parrish D., Balachandran R., Day B.W. Azide-tetrazole equilibrium of C-6 azidopurine nucleosides and their ligation reactions with alkynes. J. Org. Chem. 2010;75:2461–2473. doi: 10.1021/jo902342z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lakshman M.K., Kumar A., Balachandran R., Day B.W., Andrei G., Snoeck R., Balzarini J. Synthesis and biological properties of C-2 triazolylinosine derivatives. J. Org. Chem. 2012;77:5870–5883. doi: 10.1021/jo300628y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghosh S., Greenberg M.M. Synthesis of cross-linked DNA containing oxidized abasic site analogues. J. Org. Chem. 2014;79:5948–5957. doi: 10.1021/jo500944g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Devine S.M., May L.T., Scammells P.J. Design, synthesis, and evaluation of N6-substituted 2-aminoadenosine-5′-N-methylcarboxamides as A3 adenosine receptor agonists. Med. Chem Commun. 2014;5:192–196. doi: 10.1039/c3md00364g. [DOI] [PubMed] [Google Scholar]
- 41.Krüger S., Meier C. Synthesis of site-specific damaged DNA strands by 8-(acetylarylamino)-2′-deoxyguanosine adducts and effects on various DNA polymerases. Eur. J. Org. Chem. 2013;2013:1158–1169. doi: 10.1002/ejoc.201200984. [DOI] [Google Scholar]
- 42.Van der Henden van Noort G.J., Overkleeft H.S., van der Marel G.A., Filippov D.V. Synthesis of nucleotidylated poliovirus VPg proteins. J. Org. Chem. 2010;75:5733–5736. doi: 10.1021/jo100757t. [DOI] [PubMed] [Google Scholar]
- 43.Printz M., Richert C. Pyrenylmethyldeoxyadenosine: A 3′-cap for universal DNA hybridization probes. Chem. Eur. J. 2009;15:3390–3402. doi: 10.1002/chem.200801587. [DOI] [PubMed] [Google Scholar]
- 44.Devine S.M., Scammells P.J. Synthesis and utility of 2-halo-O6-(benzotriazol-1-yl)-functionalized purine nucleosides. Eur. J. Org. Chem. 2011:1092–1098. doi: 10.1002/ejoc.201001395. [DOI] [Google Scholar]
- 45.Sampath U., Bartlett L. ((Reliable Biopharmaceutical, Inc., St. Louis, MO, USA)). Process for the Production of 2-halo-6-aminopurine Derivatives. 6,252,061 B1. US Patent. 2001 Jun 26;
- 46.Francom P., Robins M.J. Nucleic acid related compounds. 118. Nonaqueous diazotization of aminopurine derivatives. Convenient access to 6-halo and 2,6-dihalopurine nucleosides and 2′-deoxynucleosides with acyl or silyl halides. J. Org. Chem. 2003;68:666–669. doi: 10.1021/jo020625a. [DOI] [PubMed] [Google Scholar]
- 47.Pottabathini N., Bae S., Pradhan P., Hahn H.-G., Mah H., Lakshman M.K. Synthesis and reactions of 2-chloro and 2-tosyloxy 2′-deoxyinosine derivatives. J. Org. Chem. 2005;70:7188–7195. doi: 10.1021/jo050847j. [DOI] [PubMed] [Google Scholar]
- 48.Robins M.J., Uznański B. Nucleic acids related compounds. 34. Non-aqueous diazotization with tert-butyl nitrite. Introduction of fluorine, chlorine, and bromine at C-2 of purine nucleosides. Can. J. Chem. 1981;59:2608–2611. doi: 10.1139/v81-375. [DOI] [Google Scholar]
- 49.Francom P., Janeba Z., Shibuya S., Robins M.J. Nucleic acid related compounds. 116. Nonaqueous diazotization of aminopurine nucleosides. Mechanistic considerations and efficient procedures with tert-butyl nitrite or sodium nitrite. J. Org. Chem. 2002;67:6788–6796. doi: 10.1021/jo0204101. [DOI] [PubMed] [Google Scholar]
- 50.Kazimierczuk Z., Vilpo J.A., Seela F. Base-modified nucleosides related to 2-chloro-2′-deoxyadenosine. Helv. Chim. Acta. 1992;75:2289–2297. doi: 10.1002/hlca.19920750715. [DOI] [Google Scholar]
- 51.Martínez-Montemayor M.M., Rosario-Acevedo R., Otero-Franqui E., Cubano L.A., Dharmawardhane S.F. Ganoderma lucidum (Reishi) inhibits cancer cell growth and expression of key molecules in inflammatory breast cancer. Nutr. Cancer. 2011;63:1085–1094. doi: 10.1080/01635581.2011.601845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suarez-Arroyo I.J., Rosario-Acevedo R., Aguilar-Perez A., Clemente P.L., Cubano L.A., Serrano J., Schneider R.J., Martinez-Montemayor M.M. Anti-tumor effects of Ganoderma lucidum (Reishi) in inflammatory breast cancer in in vivo and in vitro models. PLoS ONE. 2013;8:e57431. doi: 10.1371/journal.pone.0057431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.GraphPad Software. Available online: http://www.graphpad.com.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.