

Abstract
Identification of operon structure is critical to understanding gene regulation and function, and pathogenesis, and for identifying targets towards the development of new antibiotics in bacteria. Recently, the complete genome sequences of a large number of important human bacterial pathogens have become available for computational analysis, including the major human Gram-positive pathogen Staphylococcus aureus. By annotating the predicted operon structure of the S.aureus genome, we hope to facilitate the exploration of the unique biology of this organism as well as the comparative genomics across a broad range of bacteria. We have integrated several operon prediction methods and developed a consensus approach to score the likelihood of each adjacent gene pair to be co-transcribed. Gene pairs were separated into distinct operons when scores were equal to or below an empirical threshold. Using this approach, we have generated a S.aureus genome map with scores annotated at the intersections of every adjacent gene pair. This approach predicted about 864 monocistronic transcripts and 533 polycistronic operons from the protein-encoding genes in the S.aureus strain Mu50 genome. When compared with a set of experimentally determined S.aureus operons from literature sources, this method successfully predicted at least 91% of gene pairs. At the transcription unit level, this approach correctly identified at least 92% of complete operons in this dataset. This consensus approach has enabled us to predict operons with high accuracy from a genome where limited experimental evidence for operon structure is available.
INTRODUCTION
Operons are a string of one or more genes co-transcribed as a unit. Understanding the organization of operons in a bacterial genome provides insight into both gene function and regulation. Very frequently an operon will contain a cluster of genes in a common pathway or mediating a common biological function. A classical example is the Escherichia coli lactose operon in which genes in lactose metabolism are clustered together and co-transcribed (1). Although some operons may contain genes unrelated by function, such as the E.coli rpsU-dnaG-rpoD operon containing three genes involved in translation, DNA replication and transcription, respectively, the early understanding that these genes were co-transcribed led to the hypothesis that this could allow for the cells to simultaneously coordinate regulation of these three macromolecular synthesis pathways (2). Linkage of genes in operons is certainly at the transcriptional level but operons can also contain signals for post-transcriptional regulation of gene expression (3) that can result in very large differences in the concentrations of gene products from adjacent, co-transcribed genes. Additionally, there can be anywhere from a single mRNA produced from one operon to extremely complicated patterns of transcription that defy a classical definition of an ‘operon’. Bacterial operons vary in length from simple monocistronic transcripts—as many as 70% of E.coli genes (4)—to very large operons encoding many ribosomal proteins (5). Knowledge of operon organization is becoming increasingly important in the search for novel antibacterial targets and for understanding the processes involved in bacterial pathogenesis. Directed or random methods to determine bacterial gene essentiality can create polar effects on downstream genes that must be deconvoluted for full understanding of gene function. Examples of these polar approaches include the use of shotgun antisense to discover essential bacterial genes (6), random transposon–promoter out methods (7) and all other gene replacement or knockout methods. Altogether, these facts point to the critical need to map operons in a targeted organism.
There have been recent attempts to predict operon structure, most of which have relied upon E.coli as the model organism. Databases such as RegulonDB have been developed as an exhaustive collection of operon organization and gene regulation (8). These efforts have facilitated our understanding of the gene regulatory network in E.coli; however, operon structures in most other organisms are poorly understood. With increasing genomic sequences becoming available, researchers ask the questions: how can we predict operons in less well characterized organisms from their genome sequences in the relative absence of extensive experimental data; how can we utilize the predicted information to guide future research and discovery in these organisms; how well will the predictions fit in with the existing and future experimental evidence? The Gram-positive bacterium, Staphylococcus aureus, a major human pathogen causing both community- and hospital-acquired infections, serves a good example of what we can learn about predicting operon organization from the genome sequence. In S.aureus there is limited experimental evidence to determine operons. Prediction of operon structures in S.aureus would also aid in drug target identification and antibiotic development.
Operon structure is typically not conserved during evolution (9), making operon prediction a non-trivial task. Several computational methods have been developed to predict operons and can be grouped into a few general categories. The first method is to predict operons by detecting promoters and transcription terminators. Although several programs have been developed to predict rho-independent transcription terminators (10–13), no efficient prokaryotic promoter-searching algorithm is available, even for the model organism E.coli (14). One approach to overcome this limit was to construct hidden Markov models (HMMs) based on known promoters and terminators, which enabled the prediction of operon structures. This method was reported to predict 60% of known operons in E.coli (15). However, this method is difficult to apply in organisms where promoters and terminators are not as well characterized. The second method is to use a probabilistic machine-learning approach to induce operon prediction models using a variety of data types including sequence data, gene expression data and functional annotation data. This method estimates the probability of any consecutive sequence of genes on the same strand to be an operon and yielded 67% accuracy in E.coli (16). With the generation of a large amount of microarray gene expression data, co-expression pattern has recently been used as a tool to improve operon prediction (17). Recently, Bockhorst et al. (18) developed a Bayesian network approach to operon prediction and showed the method was able to predict 78% of E.coli operons with 10% false positives. However, these methods again are only applicable to organisms in which vast amounts of experimental data are available. A third method is to predict operon organization by intergenic distances and functional relationships between adjacent genes based on the observation that genes within operons tend to have much shorter intergenic distances than genes at the borders of transcription units in E.coli (19). This method was reported to have a maximum of 88% accuracy in identification of adjacent gene pairs to be in an operon and found 75% of known transcription units in E.coli. This method has opened the possibility of operon predictions in bacterial genomes other than E.coli. A fourth method is to predict operons by conserved gene cluster analysis using a comparative genomics approach. Although this method has the advantage of high specificity (≥98%) in identifying co-transcribed gene pairs, it often fails to predict the whole operons and has low sensitivity (20–22). Increased understanding of the metabolic pathway networks contained in microbial genomes has allowed for novel computational algorithms. Zheng et al. (23) developed a computational pipeline to predict metabolism-related operons based on the fact that the genes in operons are sometimes involved in successive reactions in metabolic pathways. This algorithm has provided a method to putatively annotate unknown enzymes in microbial genomes in addition to prediction of operon structure. Despite its high prediction sensitivity (89%) and specificity (87%), the method is highly dependent on biochemical pathway knowledge and, thus, operons from genes involved in complex pathways or less-well known pathways are more difficult to be predicted. Furthermore, real operons may contain exceptions to this rule leading to incorrect exclusion.
Results of in silico predictions can frequently be improved by integration of various techniques. For example, Gelfand and co-workers have developed and validated an integrative approach to analyze transcription regulation sites by using comparative analysis of genes, functions, regulatory elements, etc., in bacterial genomes (24,25). Herein we describe a strategy to integrate various operon prediction methods, especially gene orientation analysis, intergenic distance analysis, conserved gene cluster analysis and terminator detections, and to score the confidence of likelihood of each adjacent gene pairs to be in the same operon. Operons are predicted by breaking apart low-scoring gene pairs with an empirical threshold. By using this approach, we have developed a computational pipeline to annotate operons in S.aureus genome. We also compare our results to a set of known S.aureus operons from the literature in an attempt to validate our method.
MATERIALS AND METHODS
Sequence data
The complete genome sequence of S.aureus Mu50 strain published by Kuroda et al. (26) was downloaded from NCBI (http://www.ncbi.nlm.nih.gov/genomes/MICROBES/Complete.html) and imported into the Elitra proprietary microbial database developed from Incyte PathoSeqTM. (The Elitra proprietary microbial database was developed and improved from the original Incyte PathoSeq™ database. In this database, unfinished genomic nucleotide sequences are imported as FASTA files and ORFs are annotated through Elitra's proprietary gene finding pipelines. Complete genome sequences from GenBank are directly imported with original annotations. Orthologs and paralogs from different genomes in the entire database are cross-referenced through Elitra's proprietary comparative genomics analysis pipeline. For more information on Elitra proprietary microbial database, please contact the authors.) The sequences of other bacterial genomes used for comparative genome analysis were also extracted from NCBI or TIGR and imported to the Elitra proprietary microbial database. The genome sequences from NCBI are: Aquifex aeolicus strain VF5, Borrelia burgdorferi strain B31, Bacillus halodurans C-125, Bacillus subtilis strain 168, Buchnera sp. APS, Clostridium acetobutylicum ATCC824, Caulobacter crescentus CB15, Campylobacter jejuni strain NCTC 11168, Chlamydia pneumoniae strain AR-39, Chlamydia trachomatis strain MoPn, Escherichia coli strain K-12, Haemophilus influenzae strain KW20, Helicobacter pylori strain 26695, Listeria monocytogenes, Listeria innocua, Lactococcus lactis strain IL1403, Mycoplasma genitalium isolate G37, Mycobacterium leprae strain TN, Mycoplasma pneumoniae strain M129, Mycoplasma pulmonis UAB CTIP, Mycobacterium tuberculosis CDC1551, Neisseria meningitides strain MC58, Pseudomonas aeruginosa strain PAO1, Pasteurella multocida Pm70, Rickettsia conorii Malish 7, Rickettsia prowazekii strain Madrid E, Sinorhizobium meliloti 1021, Streptococcus pneumoniae strain R6 hex, Streptococcus pyogenes strain M1 GAS, Salmonella typhi strain CT18, Salmonella typhimurium strain SGSC1412, Synechocystis sp. strain PCC 6803, Thermotoga maritime strain MSB8, Treponema pallidum strain Nichols, Ureaplasma urealyticum, Vibrio cholerae strain N16961, Xylella fastidiosa 9a5c and Yersinia pestis strainCO-92 Biovar Orientalis. The genome sequence of Enterococcus facaelis strain V583 was downloaded from TIGR.
Besides S.aureus Mu50 strain, an additional seven S.aureus strains were also used in the analyses. The genomic sequences of these strains were either proprietary sequenced or downloaded from public sources as listed below and imported to the Elitra proprietary microbial database: Buttle strain and MRSA strain AS-5155 were sequenced by Incyte; MRSA strain 252 and MSSA strain 476 were downloaded from the Sanger Center (http://www.sanger.ac.uk/Projects/Microbes/); strain N315 was downloaded from NCBI; COL strain was downloaded from TIGR and strain NCTC8325 was downloaded from Advanced Center For Genome Technology at the University of Oklahoma (http://www.genome.ou.edu/).
Software
Rho-independent transcriptional terminators were identified using GCG Terminator software from the Wisconsin Package™ (10–11). All other work was performed using PERL scripts. The processed data were stored in an Oracle database.
Intergenic distance analysis
Genes in S.aureus were grouped by orientation relative to proximal 5′ and 3′ flanking genes. The intergenic distances between adjacent genes in the same orientations were calculated from the corresponding coordinates in the Mu50 strain. The intergenic distance between geneA and geneB was calculated in the following formula: distanceAB = geneB_start_position − geneA_end_position. The orientation and the distance of each gene pair were saved in Oracle.
Conserved gene cluster analysis
The deduced amino acid sequences from S.aureus and 39 other bacterial genomes were analyzed against one another using BLASTP (27,28). Paralogs of a gene in a genome were first detected if they were more similar to each other than to any genes from other genomes. Orthologs of a gene were identified as the reciprocal best BLAST hits between two genomes, after taking paralogs into account. Conserved gene clusters were identified as orthologs when gene orders between two genomes were conserved. Due to the combined requirement of reciprocal best hits and conservation of gene order, orthologs were not identified using clusters of orthologous groups (COGs) although we used COG functional categories to evaluate gene functions in predicted operons (see below). This allowed us to use conserved neighborhood as an additional criterion for orthology prediction and also to include orthologous groups that occur only in two genomes. The following is the procedure for identifying conserved gene clusters between S.aureus and 39 other genomes:
Step 1: conserved gene pairs between S.aureus and one of the 39 genomes were identified if two S.aureus genes (geneA, geneB) were adjacent and in the same orientation, and if their homologs (geneA′, geneB′) in the other genome were also adjacent and in the same orientation.
Step 2: all of the conserved gene pairs between S.aureus and the other organism were captured in a table with each row containing the gene IDs of one gene pair (geneA and geneB).
Step 3: repeat Step 1 and Step 2 to identify conserved gene pairs between S.aureus and every other organism.
- Step 4: use hashing function to cluster conserved gene pairs into conserved gene arrays. For every row from the above gene pair table, look up geneA and geneB in the cluster table (i.e. hash table):
- If the hash table entries for both genes are 0, these two genes belong to a new cluster together and they get assigned a new cluster number. The hash table entry is an offset into a cluster array that has the actual cluster value.
- If only one of the entries is 0, the gene corresponding to that entry gets a hash table entry equal to that of the other member.
- If both entries are non-zero and identical, it means the same gene pairs have been identified before by comparison with another organism. Skip this row.
- If both entries are non-zero and not identical, then replace all occurrences of the larger cluster value with the smaller one in the cluster array.
- Genes that have the same value in the hash table belong to the same conserved gene clusters. Order them by their relative coordinates in the S.aureus genome.
Searching for transcriptional terminators
The region encompassed by −20 to +200 nt around the stop codon of each gene was extracted from the S.aureus Mu50 genome. GCG Terminator software was used to search for rho-independent transcriptional terminators. Predicted terminators with S-value > 0 were extracted.
Scoring gene pairs and determination of operon boundaries
To determine whether two adjacent genes are likely to be in the same operon, we have established an empirical scoring scheme which assigns a numeric weight to the contribution of the individual analysis described above. We chose a scoring system that assigns a weight from 0 to 3, depending on the influence of each of these analyses. The exact weights and thresholds (e.g. intergenic distance) used to establish our scoring system were empirically derived based on our analysis of known operons in S.aureus and other organisms. The scoring system is described below:
Score = 0 if any of the following three criteria are met: the two adjacent genes are in different orientations; or the intergenic distance is >300 bp [in E.coli, the intergenic distance of genes in the same operons usually do not exceed 300 bp (15)]; or the intergenic distance is >100 bp and the number of conserved organisms = 0.
Score = 1 if a gene pair has an intergenic distance >60 bp, is conserved in less than 5 organisms analyzed and a predicted terminator exists in between.
Score = 3 if any of the following criteria are met: the gene pairs are conserved in at least 10 organisms; or the intergenic distance ≤30 bp; or if at least two of the following requirements were met: intergenic distance ≤50 bp; no predicted terminators; the number of organisms conserved greater than or equal to 5 but less than 10.
Score = 2 if a gene pair does not meet any of the above requirements.
To define operon boundaries, gene pairs were broken apart into two different operons if their scores were equal or below a user-defined threshold. We used empirical thresholds with scores of 0 or 1.
RESULTS AND DISCUSSION
Evaluation of individual operon prediction methods on S.aureus genome
A transcription unit containing one gene is defined as a monocistronic operon and a transcription unit containing multiple genes as a polycistronic operon. The ability to predict the operon structure of S.aureus could aid in better understanding pathogenesis and in identifying and understanding the role of novel antibiotic targets. Few operons in S.aureus have been experimentally determined and, therefore, are insufficient to build global operon prediction models. The currently available methods that may be still applicable to operon predictions in this organism are gene distance analysis, conserved gene cluster analysis and detection of promoters and terminators for transcription unit identifications. We evaluated each individual method in S.aureus before integrating them.
Binning genes by orientation
As the first step, genes in the S.aureus Mu50 genome were segregated into bins based on orientation relative to flanking genes. Consecutive genes in the same orientation were grouped into the same bin. A total of 670 bins from 2790 genes (including tRNA and rRNA genes) were collected. Among them, 273 bins contained a single gene, i.e. both 5′ and 3′ flanking genes were in opposite orientations. These 273 genes were simply assumed to be monocistronic operons and were not included in the downstream distance analysis or conserved gene cluster analyses. The remaining 397 bins had bin-sizes ranging from 2 to 63 genes with an average of 6.34 genes.
After the genes were binned by orientation, multi-gene bins were subjected to further analysis (Figure 1). Binning greatly simplified the operon annotation process because each bin could be taken as an entity and methods developed for an individual bin could be applied to other bins. The probability of each gene pair in each multi-gene bin to be in an operon was individually evaluated by intergenic distance analysis, conservation analysis and transcriptional terminator analysis.
Figure 1.

A flow chart of operon annotations in S.aureus by integrating various algorithms.
Intergenic distance analysis
Intergenic distances between genes within operons tend to be much shorter than distances for genes not within operons (15,19). Analysis of experimentally determined operons in E.coli revealed that the intergenic distances were mostly within 100 bp and generally not >300 bp (15). For the evaluation of intergenic distance analysis in operon prediction in S.aureus, a simple prediction scheme was used: gene pairs in the same bin were considered to be in the same putative operon provided their intergenic distance was below a predefined threshold. The optimal threshold for this determination was not obvious in view of the lack of experimental data on operon structure in S.aureus.
The overall intergenic distance distribution across all adjacent gene pairs was examined and compared to the overall intergenic distance distribution in E.coli to confirm whether the genome organizations in the two organisms were obviously different in terms of gene spacing. As shown in Figure 2, similar to E.coli, the intergenic distances of most of the adjacent gene pairs in the same orientation in S.aureus were between −20 and 60 bp around stop codons of the first genes with a clear peak at around 20 bp. Therefore, it is reasonable to conclude that the distribution of intergenic distances for co-transcribed genes in S.aureus might be similar to that of E.coli. In fact, most of the genomes analyzed had similar patterns (Figure 2), indicating that it may be feasible to use generic intergenic distance thresholds for operon prediction in a broad number of bacteria. This notion is confirmed by a recent study by Moreno-Hagelsieb and Collado-Vides (29). The only two genomes analyzed that had slightly different gene spacing distributions were B.subtilis and P.aeruginosa, both of which had a minor second peak around the 50 to 120 bp region. The role or significance of this minor second peak was not explored further.
Figure 2.

Comparisons of frequency distance distributions of adjacent gene pairs in the same orientations in various bacterial genomes.
Next, different distance thresholds for operon prediction in S.aureus were evaluated. Table 1 shows the operon prediction results from thresholds of 30, 50, 75, 100, 150 and 200 bp. By comparing the COG functional categories (30) and the operon information of other genomes from literature (if available), we inspected some of the putative operons especially focusing on those different operons between every two thresholds. While 30 or 50 bp thresholds appeared to be too stringent and 150 or 200 bp thresholds were too loose, in general thresholds of 75 or 100 bp seemed to balance false positives and false negatives better than the other thresholds (data not shown). Considering that multiple methods would be integrated for final operon predictions, these thresholds were not further refined.
Table 1. Comparison of the putative operons in S.aureus at different intergenic distance thresholds using only the intergenic distance analysis method.
| Threshold (bp) | No. of total putative operons | Single-gene bins No. of monocistrons | Muti-gene bins No. of polycistrons | No. of polycistrons | No. of genes in largest putative operon |
|---|---|---|---|---|---|
| 30 | 1820 | 273 | 757 | 517 | 14 |
| 50 | 1718 | 273 | 632 | 540 | 25 |
| 75 | 1610 | 273 | 502 | 562 | 25 |
| 100 | 1541 | 273 | 428 | 567 | 25 |
| 150 | 1385 | 273 | 265 | 574 | 29 |
| 200 | 1237 | 273 | 113 | 578 | 46 |
Conserved gene cluster analysis
Several studies have found that clusters with conserved gene order across bacterial genomes have a high probability to be in the same operon (20–22). Taking advantage of the availability of the complete sequence for many bacterial genomes, conserved gene clusters in S.aureus were analyzed relative to every other available genome by comparing their sequences and genome locations. To reduce the rate of false positives, only gene pairs with ≤300 bp intergenic distances within the same multi-gene bins were analyzed. The numbers of conserved gene clusters between S.aureus and other genomes are shown in Table 2. A total of 39 bacterial genomes were used for this analysis: 11 Gram-positive, 23 Gram-negative and 5 other species. When only one genome was used, the number of conserved gene clusters predicted by this method was small, ranging from 27 to 245 clusters and the number of genes in the conserved gene clusters ranging from 2.8 to 27% of all the genes in the S.aureus Mu50 genome. The average cluster sizes range from 2.3 to 3 genes. These numbers are smaller than what would be expected for operons; however, when multiple genomes were used for comparison, these numbers increased with addition of each genome (Table 3), consistent with earlier studies (20,21). The increase in these numbers, however, was very slow and reached a plateau (Table 3). Thus, even if more genomes are added, the number of operons predicted is probably low (low coverage) and many of the predicted operons are probably still partial. This suggested that conserved gene cluster analysis alone is not sufficient for whole genome operon prediction and other methods should be combined for higher coverage and whole operon prediction.
Table 2. The number of conserved gene clusters in S.aureus when compared to other bacterial genomes.
| Organism compared | Type | No. of conserved gene clusters | No. of genes | % of S.aureus genes | Average cluster size | Largest cluster size |
|---|---|---|---|---|---|---|
| Aquifex aeolicus | G− | 27 | 76 | 2.8 | 2.8 | 10 |
| Bacillus halodurans | G+ | 245 | 714 | 26.3 | 2.9 | 23 |
| Bacillus subtilis | G+ | 244 | 722 | 26.6 | 3.0 | 23 |
| Borrelia burgdorferi | Spirochete | 39 | 114 | 4.2 | 2.9 | 22 |
| Buchnera sp. | G− | 40 | 119 | 4.4 | 3.0 | 14 |
| Campylobacter jejuni | G− | 42 | 121 | 4.5 | 2.9 | 19 |
| Caulobacter crescentus | G− | 75 | 190 | 7.0 | 2.5 | 14 |
| Chlamydia pneumoniae | G− | 40 | 101 | 3.7 | 2.5 | 8 |
| Chlamydia trachomatis | G− | 42 | 105 | 3.9 | 2.5 | 8 |
| Clostridium acetobutylicum | G+ | 138 | 384 | 14.1 | 2.8 | 23 |
| Enterococcus faecalis | G+ | 200 | 545 | 20.1 | 2.7 | 23 |
| Escherichia coli | G− | 98 | 249 | 9.2 | 2.5 | 14 |
| Haemophilus influenzae | G− | 74 | 191 | 7.0 | 2.6 | 11 |
| Helicobacter pylori | G− | 29 | 85 | 3.1 | 2.9 | 15 |
| Lactococcus lactis | G+ | 151 | 382 | 14.1 | 2.5 | 15 |
| Listeria innocua | G+ | 238 | 718 | 26.5 | 3.0 | 23 |
| Listeria monocytogenes | G+ | 243 | 732 | 27.0 | 3.0 | 23 |
| Mycobacterium leprae | G+ | 72 | 181 | 6.7 | 2.5 | 11 |
| Mycobacterium tuberculosis | G+ | 84 | 209 | 7.7 | 2.5 | 10 |
| Mycoplasma genitalium | Acid Fast | 49 | 139 | 5.1 | 2.8 | 19 |
| Mycoplasma pneumoniae | Acid Fast | 51 | 143 | 5.3 | 2.8 | 19 |
| Mycoplasma pulmonis | Acid Fast | 51 | 133 | 4.9 | 2.6 | 12 |
| Neisseria meningitidis | G− | 46 | 126 | 4.6 | 2.7 | 13 |
| Pasteurella multocida | G− | 73 | 185 | 6.8 | 2.5 | 14 |
| Pseudomonas aeruginosa | G− | 86 | 222 | 8.2 | 2.6 | 14 |
| Rickettsia conorii | G− | 35 | 97 | 3.6 | 2.8 | 14 |
| Rickettsia prowazekii | G− | 36 | 99 | 3.6 | 2.8 | 14 |
| Salmonella typhi | G− | 89 | 230 | 8.5 | 2.6 | 14 |
| Salmonella typhimurium | G− | 91 | 233 | 8.6 | 2.6 | 14 |
| Sinorhizobium meliloti | G− | 69 | 179 | 6.6 | 2.6 | 14 |
| Streptococcus pneumoniae | G+ | 155 | 369 | 13.6 | 2.6 | 14 |
| Streptococcus pyogenes | G+ | 137 | 396 | 14.6 | 2.7 | 28 |
| Synechocystis sp. | G− | 28 | 84 | 3.1 | 3.0 | 13 |
| Thermotoga maritima | G− | 70 | 192 | 7.1 | 2.7 | 23 |
| Treponema pallidum | Spirochete | 32 | 94 | 3.5 | 2.9 | 19 |
| Ureaplasma urealyticum | G− | 48 | 143 | 5.3 | 3.0 | 19 |
| Vibrio cholerae | G− | 80 | 203 | 7.5 | 2.6 | 14 |
| Xylella fastidiosa | G− | 60 | 163 | 6.0 | 2.7 | 14 |
| Yersinia pestis | G− | 89 | 230 | 8.5 | 2.6 | 14 |
Table 3. Dependence of the number of conserved gene clusters in S.aureus on the number of genomes to which S.aureus was compared.
| No. of genomes clustered | No. of conserved gene clusters | No. of genes | % of S.aureus genes | Average cluster size | Largest cluster size |
|---|---|---|---|---|---|
| 5 | 285 | 860 | 32 | 3 | 23 |
| 10 | 306 | 919 | 34 | 3 | 23 |
| 15 | 328 | 1047 | 39 | 3.2 | 29 |
| 20 | 332 | 1067 | 39 | 3.2 | 29 |
| 25 | 336 | 1079 | 40 | 3.2 | 29 |
| 30 | 344 | 1108 | 41 | 3.2 | 29 |
| 35 | 345 | 1112 | 41 | 3.2 | 29 |
| 39 | 345 | 1113 | 41 | 3.2 | 29 |
Detection of transcriptional terminators
The third method evaluated was the detection of signals at transcription unit boundaries. We extracted the first 70 bp of DNA sequence upstream of the start codons from the 273 S.aureus monocistronic operons determined by orientation as described earlier. The MEME program from Wisconsin Package™ was used in an effort to find conserved motifs in the promoter regions of these genes (31), followed by a MotifSearch program to search motifs from a 70 bp upstream region of each gene in the multi-gene bins using the profiles generated by MEME (32). Canonical −10 and −35 promoter elements were not detected (data not shown), probably due to the inherently high A + T content of the S.aureus genome. Instead, we focused on detection of transcription terminators only.
Rho-independent transcription terminators are distinct secondary structures in nascent RNAs. Putative terminators from all available bacterial genomes have been grouped by structure into five classes: L-shaped, I-shaped, V-shaped, U-shaped and X-shaped (13). About 85.1% of transcription terminators detected in S.aureus are L-shaped (13), a stem–loop followed by a U-trail. Taking advantage of this knowledge, the GCG Terminator software in the Wisconsin Package™ that searches for GC-rich dyad symmetry near a U-rich region (10–11), may be suitable to detect S.aureus transcription terminators. Nucleotide sequences −20 to 200 bp downstream of each gene's stop codon were extracted and imported into the software to identify putative transcriptional terminators. As a result, the software was able to detect 995 genes containing terminator-like secondary structures. About 36% of S.aureus genes have secondary structures predicted by GCG Terminator to be rho-independent transcription terminators, only slightly lower than what was reported by using GeSTer software (41%) (13), suggesting GCG Terminator software was comparable with GeSTer software for S.aureus transcription terminator detection. Most of the putative terminators were near stop codons, while a significant number of them were further than 50 bp downstream of stop codons (Figure 3). Although the GCG Terminator program might not detect all putative rho-independent transcription terminators, it is also likely that the majority of genes that lack detectable terminators are probably part of operons or use rho-factor-dependent transcriptional terminators. However, one would expect few rho-dependent terminators in S.aureus because deletion of its rho gene has no apparent effect on growth or virulence (33).
Figure 3.

Distance distribution of S.aureus Rho-independent transcription terminators predicted by GCG Terminator software from Wisconsin Package™.
Scoring gene pairs and prediction of operons using a consensus approach
Each operon prediction method evaluated above has advantages and disadvantages. The intergenic distance analysis approach provides the best coverage on genome-wide operon prediction. It has high specificity when the distance cut-off length is short. With the increase of distance threshold, operon prediction sensitivity increases but its specificity decreases. The intergenic distance analysis approach, however, is dependent on the quality of genome annotation. Missed open reading frames (ORFs) in apparently long intergenic regions would result in false prediction of operons. Additionally, accurate prediction of the ORF start position continues to be a challenge in computational biology; this may significantly impact operon prediction. The conserved gene cluster analysis approach, on the other hand, has high specificity with low sensitivity, even if more bacterial genomes are used for analysis. Transcriptional promoters/terminators are natural signals of operon boundaries. Terminators can be used as flags for potential operon boundaries at the 3′ end despite the fact that we were not successful in detecting promoter sequences computationally in S.aureus and the existence of internal promoters/terminators within operons. To maximize the use of these methods, one could seed the process with the intergenic distance analysis by choosing an empirical distance threshold, extend truncated operons by conserved gene clusters, and break down chimerical operons by transcriptional terminators and promoters, if available. Another slightly different approach is to score the confidence of each adjacent gene pairs to be in the same operon by their orientation, intergenic distance, conservation across genomes and existence of terminators, and then determine operon boundaries by separating gene pairs with a score equal or lower than a user-defined threshold. We chose to use the latter strategy for operon prediction.
Scoring gene pairs
The confidence of a gene pair to be present in the same operon was scored using an empirical 0–3 scale. A score of 0 indicates that the two genes of a pair are very likely to be in two separate operons. A gene pair with a score of 1 indicates that these two genes might be in the same operon, but with low confidence. The intersections of such gene pairs are also potential operon boundaries. A gene pair with a score of 2 indicates that the two genes are probably in the same operon. A gene pair with a score of 3 indicates that the two genes are most likely in the same operon. The 0–3 scoring scheme can be translated to be ‘no confidence’, ‘little confidence’, ‘some confidence’ and ‘high confidence’ for two adjacent genes to be in the same operon. A score of 0 can also be considered to be ‘high confidence’ that two adjacent genes are in distinct operons. Empirical criteria (see Materials and Methods) for each score were derived based on our earlier analysis of individual operon prediction algorithms and the features of each algorithm reported in literature. The criteria were set based on the following principle: stringent on score 0 (operon boundaries) and score 3 (operon interior), less stringent on score 1 (potential operon boundaries) and score 2 (probable operon). Gene pairs with different orientations were given a score of 0. Gene pairs with the same orientation were also given a score of 0 provided they were >100 bp apart and not conserved in any of the 39 genomes to which S.aureus was compared. Though there may be certain exceptions, very few co-transcribed gene pairs that meet a 0 score criteria were expected.
An S.aureus genome map with scores annotated to the intersections of every adjacent gene pair was built. S.aureus strain Mu50 has 2790 genes including tRNA and rRNA genes and thus 2790 gene pairs. Based on the above scheme, the distribution of gene-pair scores in our genome map is shown in Table 4. A total of 51% of gene pairs scored 0 and 40% scored 3. This means that ∼91% of gene pairs had scores assigned under stringent criteria with high confidence to be in distinct operons or in the same operons. The predicted operons were validated in the next section.
Table 4. Distribution of gene-pair scores in S.aureus Mu50 genome. A score of 0 indicates that the two genes of a pair are very likely to be in two distinct operons.
| Gene-pair score | No. of gene pairs | % of gene pairs |
|---|---|---|
| 0 | 1410 | 51 |
| 1 | 62 | 2 |
| 2 | 210 | 8 |
| 3 | 1108 | 40 |
| Total | 2790 |
A score of 1 indicates that the two genes might be in the same operon, but with low confidence. A score of 2 indicates that the two genes are probably in the same operon. A score of 3 indicates that the two genes are most likely in the same operon.
Operon prediction
With assignment of gene-pair scores to the S.aureus whole genome, operon boundaries were determined by breaking apart gene pairs whose scores were equal to or below a user-defined threshold. Based on the definitions of these scores, scores of 0 or 1 were determined to be reasonable thresholds. Table 5 displays the operon prediction results using 0 or 1 as the maximum threshold. The criteria for predicting rRNA or tRNA transcription units might be different from protein-encoding genes so those were not included in the results. When a score of 0 was used as the threshold to break apart gene pairs, a total of 864 monocistrons and 533 polycistrons were generated, of which 80% of gene pairs in the 533 polycistrons have a score of 3. The complete list of predicted operons is available in the supplementary material. About 60% of the polycistrons have an average score of 3, indicating that all of the gene pairs in these polycistrons are of high confidence. Only 53 polycistrons had one or more gene pairs with a score of 1. Thus these 53 operons were split into two or more operons when a score of 1 was used as the threshold. This resulted in additional 62 operons.
Table 5. S.aureus operons generated at different thresholds using gene-pair scoring scheme.
| Threshold = 0 | Threshold ≤1 | |
|---|---|---|
| No. of total predicted operons | 1397 | 1459 |
| No. of monocistronic operons | 864 | 921 |
| No. of polycistronic operons | 533 | 538 |
| No. of polycistronic operons with minimum gene-pair score = 1 | 53 | 0 |
| No. of polycistronic operons with minimum gene-pair score = 2 | 160 | 162 |
| No. of polycistronic operons with every gene-pair score = 3 | 320 | 376 |
Transcription units for rRNA and tRNA genes were not included.
Given the lack of sufficient published experimental evidence in S.aureus, we preferred to err on the inclusive side for operon predictions, thus using a score of 0 as the threshold. Gene pairs with scores of 1 to 3, however, were flagged to indicate their confidence levels and to assist with experimental design. In this way, the largest transcription unit within a specified region may be predicted as well as other potential smaller transcription units in the same region.
The distribution of predicted operon size is shown in Table 6. For polycistronic operons, ∼97% have less than 10 genes or 88% have no more than 5 genes. The average size of polycistronic clusters was 3.47 genes. Using a score of 0 as the threshold, the largest predicted operon was the ribosomal superoperon containing 29 genes: rpsJ, rplC, rplD, rplW, rplB, rpsS, rplV, rpsC, rplP, rpmC, rpsQ, rplN, rplX, rplE, rpsN, rpsH, rplF, rplR, rpsE, rpmD, rplO, secY, adk, infA, rpmJ, rpsM, rpsK, rpoA, rplQ. A similar gene order exists in many bacterial genomes with minor differences. Experimental evidence exists in B.subtilis for the corresponding ribosomal superoperon containing 30 genes (34). The B.subtilis genome has an additional map gene encoding methionine aminopeptidase located between the adk and infA genes. In S.aureus, the map gene has been shuffled out of the operon to another location in the genome and forms a predicted monocistronic operon. According to our scoring scheme, gene pairs in the ribosomal superoperon in S.aureus are all scored 3 except the adk–infA pair. This pair was scored 1 due to a large intergenic distance (193 bp), existence of transcriptional terminators and conservation in only a small number of genomes.
Table 6. Size of S.aureus predicted operons at threshold of score 0. The average operon size of polycistrons was 3.47 genes.
| Predicted operon size | Number of predicted operons |
|---|---|
| 1 | 864 |
| 2 | 253 |
| 3 | 117 |
| 4 | 63 |
| 5 | 34 |
| 6 | 20 |
| 7 | 17 |
| 8 | 4 |
| 9 | 8 |
| 10 | 4 |
| 11 | 2 |
| 12 | 3 |
| 13 | 4 |
| 16 | 2 |
| 17 | 1 |
| 29 | 1 |
| Total | 1397 |
By integrating the various methods, operons that otherwise would be missed by using only one method should be predicted. For an example, dnaA–dnaN has 278 bp intergenic distance, much further than most of the gene pairs of known operons. In E.coli, these two genes are 5 bp apart and are in the same operon (35,36). We predicted dnaA and dnaN to be in the same operon in S.aureus with high confidence despite the large intergenic distance because this gene pair was conserved in 24 (out of 39) genomes to which S.aureus was compared. The fact that dnaA and dnaN were found to be in the same operon experimentally in B.subtilis despite an intergenic distance of 189 bp (37) suggests that our S.aureus prediction is likely accurate and highlights the differences between these two Gram-positive bacteria and the better-characterized E.coli. As another example, the genes SAV1419, murG and SAV1417 are predicted to be in the same operon with high confidence because of their short intergenic distance (12 and 17 bp, respectively) and lack of terminators even though the order of these genes is not conserved in any of the 39 other genomes examined (data not shown). The three-gene order, however, is conserved in all eight S.aureus strains examined (as listed in Materials and Methods). This operon would not be predicted by a comparative genomics or grouping by functions method alone.
Evaluation of operon prediction results from the consensus approach
Validation of operon prediction
To validate the method, S.aureus experimentally determined operons were collected from publications by searching PubMed for ‘Staphylococcus aureus [AND] operons’ and related logical search terms, and then compared the published operon structures with our predictions. A sample totaling 40 unique operon results was collected from the literature. Due to strain variations, however, genes from 4 of the 40 operons were not found in strain Mu50 or were dispersed in various locations of the Mu50 chromosome. These four operons were thus excluded in our validation process. The remaining 36 operons were used to validate both the gene pair scoring scheme and the operon prediction (Table 7).
Table 7. S.aureus operon prediction validation results.
| No. | Operon name | Operon structure From experiments | Reference | Validation results |
|---|---|---|---|---|
| 1 | agr | agrBDCA | (40) | Predicted, exact |
| 2 | alr | orf1,orf2,orf3,dpj,alr,orf6,pemK | (47) | Predicted, exact |
| 3 | cap | capABCDEFGHIJKLMNOP | (38,39) | Predicted, exact |
| 4 | clp2161 | clp2161 | (48) | Predicted, exact |
| 5 | odh | csb22 (odh),csb22-1(nhaC S carnosus) | (48) | Predicted, exact |
| 6 | csb | csb28 (yhxD) | (48) | Predicted, exact |
| 7 | csb | csb29 (bmrU cotranscribed with bmr) | (48) | Predicted, exact |
| 8 | ctsR | ctsR, yacH, yacI,clp2392 | (48) | Predicted, exact |
| 9 | czr | czrAB | (49) | Predicted, exact |
| 10 | ddh | ddh | (50) | Predicted, exact |
| 11 | femAB | femA,femB | (51) | Predicted, exact |
| 12 | fhu | fhuC, fhuB, fhuD | (52) | Predicted, exact |
| 13 | glmM | orf1,orf2,glmM | (53) | Predicted, exact |
| 14 | hld | hld | (40) | Predicted, exact |
| 15 | hsp60 | hsp10(groES), hsp60(groEL) | (54,55) | Predicted, exact |
| 16 | lac | lacABCDFEG | (56) | Predicted, exact |
| 17 | mnh | mnhABCDEFG | (41) | Predicted, exact |
| 18 | nrd | nrdI,nrdE,nrdF | (46) | Predicted, exact |
| 19 | nrd | nrdD,nrdG | (46) | Predicted, exact |
| 20 | pheT | pheS, pheT | (57) | Predicted, exact |
| 21 | sar | sar | (58) | Predicted, exact |
| 22 | sigB | rsbU,rsbV,rsbW, sigB | (59–61) | Predicted, exact |
| 23 | sir | sirABC | (62) | Predicted, exact |
| 24 | spa | spa | (63) | Predicted, exact |
| 25 | ssp | sspA,sspB,sspC | (64) | Predicted, exact |
| 26 | sst | sstABCD | (65) | Predicted, exact |
| 27 | yurI | csb10(yurI),csb10-1(yurX),csb10-2 (yurW),csb10-3 (yurV),csb10-4(yurU) | (48) | Predicted, exact |
| 28 | dlt | dltABCD | (66,67) | Predicted, inclusive |
| 29 | gln | pr,glnR,glnA | (68,69) | Predicted, inclusive |
| 30 | hsp70 | hrc37, hsp20, hsp70, hsp40, orf35 | (54) | Predicted, inclusive |
| 31 | lrg | lrgA,lrgB | (70) | Predicted, inclusive |
| 32 | lyt | lytS,lytR | (70) | Predicted, inclusive |
| 33 | yckG | csb4 (yckG), csb4-1(yckF) | (48) | Predicted, inclusive |
| 34 | egc | seo,sem,sei,Ψent1,Ψent2,sen,seg | (71) | split |
| 35 | spl | splA, splB, splC, splD, splE, splF | (72) | split |
| 36 | tca | tcaR,tcaA,tcaB | (73) | split |
The meanings of the validation results are as follow. ‘Predicted, exact’: the published operon is exactly the same as predicted by the consensus method described in this paper. ‘Predicted, inclusive’: the published operon is predicted by the consensus method, but there are additional genes in the predicted operon. ‘split’: The published operon is divided into two or more predicted operons by the consensus method.
As shown in Table 8, the gene pairs from these 36 known operons including the pairs at the operon boundaries were compiled and compared to the gene pair scores. If a boundary was shared by two known operons adjacent to each other, this boundary pair was only counted once. A total of 156 gene pairs were collected, of which, 91% (142/156) pairs were successfully predicted. Breaking down the numbers into two categories, operon boundary and operon interior, 89% (62/70) of operon boundaries and 93% (80/86) of gene pairs inside known operons matched the prediction. If assigned gene pair scores, the ratio of the number of gene pairs with confidence scores 3, 2 and 1 was 70:9:1 when these gene pairs matched the prediction. About 97% of gene pairs with a score 3 were in the same operon when compared to known operons.
Table 8. Summary of the validation of the gene-pair scoring scheme in S.aureus.
| Predicted gene pair score | Predicted | Not predicted | Total | ||
|---|---|---|---|---|---|
| No. of pairs | % of pairs | No. of pairs | % of pairs | ||
| 0 | 62 | 89 | 8 | 11 | 70 |
| 1 | 1 | 50 | 1 | 50 | 2 |
| 2 | 9 | 75 | 3 | 25 | 12 |
| 3 | 70 | 97 | 2 | 3 | 72 |
| Total | 142 | 91 | 14 | 9 | 156 |
Gene pairs from the 36 known operons (in Table 7) were compiled including the pairs at the operon boundaries and compared to the predicted gene-pair scores.
When each individual operon was considered as a whole, 33 of the 36 operons (92%) were predicted and only 3 operons (8%) were split. Also, among these 33 predicted operons, 27 operons (82%) were identical to the prediction including both boundaries, while 6 operons had additional gene(s) in the prediction (Table 7, see below).
Among the 26 operons identical to the prediction, the largest was the 16-gene operon capABCDEFGHIJKLMNOP, whose products are involved in capsular polysaccharide biosynthesis (38,39). A more complicated situation was seen with the agrBDCA operon (40). The genes agrD and agrC were originally not predicted to be in the same operon based on ORFs in the Mu50 genome. The intergenic distance between these two genes is 198 bp and the gene order is not conserved in the 39 genomes examined. However, when we applied our operon prediction method to the genome sequence of the RN4220 strain in which operon experimental data were derived (40), the agrBDCA gene cluster was predicted to be a single operon. The distance between agrD and agrC in RN4220 is 25 bp. In Mu50, the agrC gene encodes a protein of 371 amino acids while it encodes a protein of 430 amino acids in RN4220. A similar analysis was performed on six more S.aureus genomes: Buttle strain, MRSA strain AS-5155, COL strain, MRSA strain 252, MSSA strain 476 and strain N315. Similar to strain Mu50, the agrC gene from strain N315 encodes a 371 amino-acid protein and is 198 bp away from agrD. However, agrC encodes a 430 amino-acid protein and is <30 bp away from agrD in the remaining strains. The observed differences in operon predictions between RN4220 and Mu50 strains could be due to either agrC ORF mis-annotation in strain Mu50 or strain-dependent genetic polymorphisms. Inspection of the 5′ region sequence upstream of the agrC gene of strain Mu50 has identified an additional 168 bp nucleotide region in frame with the original agrC ORF. The newly predicted agrC ORF encodes a 427 amino-acid protein, extended 56 amino acids at the N-terminus from the original 371 amino acids. Although both the newly extended ORF and the original ORF lack canonical Shine-Dalgarno sequences, the additional 56 amino acids share some sequence similarity to the N-termini of the agrC protein from the other S.aureus strains and is initiated with an ATG codon.
Of the 6 known operons predicted by our method to contain additional gene(s), some of the extra gene pairs were flagged with a score 1 or 2. For example, two additional genes, SAV1307 and SAV1308, in front of glnR–glnA operon, are predicted to be part of that operon. The confidence score between SAV1308 and glnR was 1, indicating that the likelihood of these two genes to be in the glnR–glnA operon was low. In other cases, the extra genes were predicted to be in the same operons with higher confidence. For an example, an additional gene, SAV0931, was predicted to be in the same operon as dltABCD. SAV0931 is 16 bp upstream of dltABCD. This gene is only found in S.aureus. We have realized that many of the cited studies from the literature did not exclude the possibility of additional genes in the same operon as those they investigated. Thus it is possible that we have correctly predicted the entire operons in these cases. On the other hand, genetic variations of different S.aureus strains could also be a source of the differences between the prediction and the published experimental data.
Comparison of the consensus approach to other individual methods
The consensus approach described herein has integrated an intergenic distance analysis method, a conserved gene cluster analysis method and a terminator prediction method for gene-pair scoring and operon prediction. Is this consensus method an improvement over the individual methods? Operon prediction by intergenic distances and functional relationships between adjacent genes was reported to have a maximum of 88% accuracy in identification of adjacent gene pairs to be in an operon and 75% of known transcription units in E.coli (19) while the promoter and terminator signal prediction method could predict 60% of known transcription units in E.coli (15). Ermolaeva et al. (20) performed conserved gene cluster analysis on 34 complete bacterial and archaeal genomes and predicted co-transcribed gene pairs with 30–50% sensitivity and 98% specificity in E.coli genome. The integrated consensus method has significantly increased the prediction accuracy to 91% for gene pair prediction and 92% for complete operon prediction.
To further analyze whether the difference of the prediction accuracy was an artifact of different validation datasets, the individual operon prediction methods were evaluated using the same set of 36 S.aureus known operons and the corresponding 156 gene pairs. Due to the difficulty in predicting promoters in S.aureus as described earlier, only the intergenic distance method and the conserved gene cluster method were compared to the consensus approach. As shown in Table 9, the intergenic distance method predicted 75% of the 36 known operons and 88% of the 156 gene pairs in S.aureus while the conserved gene cluster analysis method predicted 58% of the 36 operons and 64% of the 156 gene pairs. These results are consistent with those reported for these individual methods in E.coli and have provided additional evidence for the improvement in operon predictions made by the consensus approach.
Table 9. Comparison of S.aureus operon prediction results from the integrated consensus approach to other operon prediction results on the same dataset.
| Methods | Complete operons (total 36) | Gene pairs (total 156) | ||
|---|---|---|---|---|
| No. of operons predicted | % accuracy | No. of gene pairs predicted | % accuracy | |
| Integrated consensus approach | 33 | 92 | 142 | 91 |
| By intergenic distance only | 27 | 75 | 137 | 88 |
| By conserved gene clusters only | 21 | 58 | 100 | 64 |
Conservation of operons across organisms
A portion of the predicted S.aureus operons were compared to experimentally derived operons from other organisms in the literature or in the databases. In some cases, the operons are conserved across organisms. For an example, a 5.8 kb region in S.aureus Mu50 spans seven genes (mnhA, mnhB, mnhC, mnhD, mnhE, mnhF, mnhG) that encode the multi-subunit of Na+/H+ antiporter (41). Our method has predicted these seven genes to be in the same operon with high confidence. In B.subtilis, these seven genes were also demonstrated to be in the same operon (mrp operon) (42). In most cases, operons are not conserved as an entity, although a few gene pairs of an operon may be conserved across organisms. Gene loss and shuffling during evolution have resulted in operon rearrangement across organisms. One such example is the genes involved in the cell wall biosynthesis pathway. In E.coli, mraZ, mraW, ftsL, ftsI, murE, murF, mraY, murD, ftsW, murG, murC, ddlB, ftsQ, ftsA and ftsZ form a large operon (43). In S.aureus, although a similar operon structure has been predicted (SAV1177, SAV1178, SAV1179, ftsL, pbpA, mraY, murD, div1b, ftsA, ftsZ), several of the mur genes (murE, murF, murG, murC) have been rearranged and are present in different operons. These operons are located far apart in the S.aureus genome. Other mur gene operons, such as the murA, murB and murI operons, are also located far away from the above mur gene operons.
Another example that demonstrates that gene shuffling and gene loss during evolution impact operon conservation is the nrdIEF operon (Figure 4). In E.coli and S.typhimurium, both nrdAB and nrdIEF operons encode ribonucleoside-diphosphate reductase: nrdA and nrdE encode ribonucleoside-diphosphate reductase alpha subunit while nrdB and nrdF encode the ribonucleoside-diphosphate reductase beta subunit (44). The nrdI gene encodes an unknown protein. The nrdIEF operon is also conserved in S.aureus and B.subtilis (45,46). In some organisms such as M.genitalium and M.pneumoniae, the nrdF gene is shuffled to the beginning of the operon while it is rearranged to the other location of the genome in organisms such as M.leprae and M.tuberculosis (Figure 4). Some organisms such as H.influenzae and P.aeruginosa only have the nrdAB operon and the nrdIEF operon is lost. S.pneumoniae, on the other hand, has a nrdEF operon. But its nrdI gene has been shuffled to another location of the genome (Figure 4).
Figure 4.
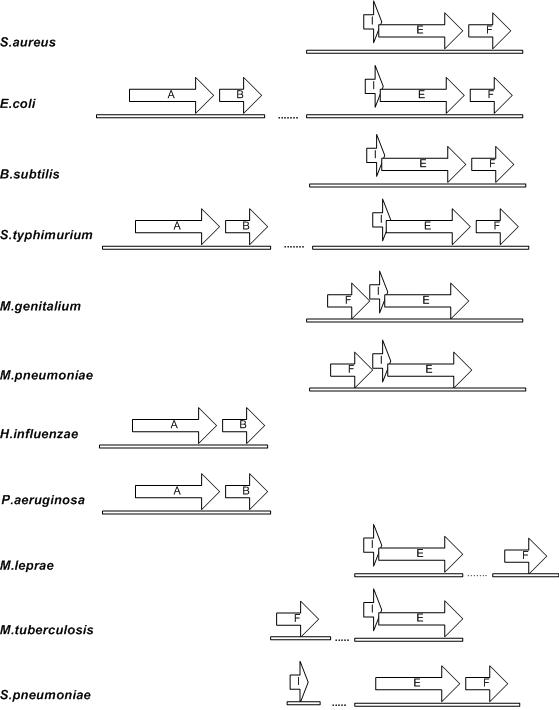
Comparison of nrdIEF operon across several bacterial organisms. Arrows represent the directions of the genes. A, nrdA; B, nrdB; I, nrdI; E, nrdE; F, nrdF. The double line represents the genome.
CONCLUSION
Understanding the organization of operons in S.aureus will aid in studying the complex array of S.aureus-specific pathogenesis determinants and in identifying and prioritizing novel antibiotic targets in this critically important pathogen. In this work, the complete S.aureus operon map was predicted through an integrated consensus algorithmic approach. This dataset will be very useful to support and interpret studies on this pathogen throughout the research community. It may be also used for comparative genomics studies with other bacterial organisms as more operon maps become available.
Various methods and algorithms have been devised to predict operons. By integrating gene orientation analysis, distance analysis, conserved gene cluster analysis and transcription terminator analysis, we have tried to maximize the operon prediction power using available algorithms. The consensus approach described above has several advantages. First, the overlapping and complementary strengths of each individual operon prediction method will serve to minimize the drawbacks from each method. For example, the impact of ‘mis-annotated’ start site of ORFs on the intergenic distance analysis approach may be overcome by conserved gene cluster analysis; on the other hand, the low-sensitivity of operon prediction by the conserved gene cluster analysis will be greatly enhanced by the intergenic distance approach. Second, the gene-pair scoring scheme allows the flexibility to err on the ‘inclusive’ or ‘exclusive’ side for operon prediction and aid with experimental designs. It should be noted that the scoring scale 0–3 is empirical and is based on our analysis of the S.aureus genome. We found this scale is both simple and sufficient to describe gene pairs for operon prediction. This scale could also be applied to other genomes and can be further refined or modified as needed. Nevertheless, the overall process and strategy described in this paper will still be valid even if the scoring scale is changed. Third, the consensus approach is simple to implement and easy to adapt. For example, additional operon prediction methods can be easily integrated into the pipeline with some alterations of the scoring parameters without changing the overall process. Fourth, this method does not rely on any experimental data, making this approach applicable to prediction of operons in genomes other than S.aureus where experimental training data may be even more limited. Based on currently available published experimental data, the method was able to successfully predict operon boundaries and gene pairs within operons at 89 and 93% accuracy in S.aureus, respectively. Whole operons were predicted with 92% accuracy. It is possible that this accuracy is an underestimation due to a couple of factors including the genetic variations of the strains used in experiments. For instance, distance between ORFs of actual pairs of operon members could be under selection in some strains and not in others. Additionally, experimental determination of operon structure is performed by several methods of varying levels of precision and accuracy. Rather than attempt to assess each of those, we have accepted the published conclusions of operon structure. Although the sample size used for validation was not very large due to limited literature information (36 operons and 156 gene pairs), the validation results still highlight the improvement made in operon predictions by integrating various algorithms. Additional experiments are currently underway to further validate the predicted operons.
A drawback of our method is the terminator detection software employed, which probably works well only on genomes whose rho-independent terminators are largely L-shaped (GC-rich stem–loop structure followed by U-trail) (13). Such genomes include S.aureus, B.subtilis, M.pulmonis, P.multocida, S.pneumoniae, S.pyogenes, U.urealyticum, etc. Using generic terminator prediction software such as GeSTer may improve the method (13). Another improvement may be to integrate promoter sequence detection in operon prediction since predicting promoter sequences in the AT-rich S.aureus genome was initially unsuccessful. When efficient promoter motif detection software and further scientific data on S.aureus promoters become available, the quality of the gene pair scores and the operon prediction may be further improved.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We acknowledge Dr Phil Youngman and Dr Jeff Winkelman for their thoughtful comments on this manuscript.
REFERENCES
- 1.Jacob F. and Monod,J. (1961) Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol., 3, 318–356. [DOI] [PubMed] [Google Scholar]
- 2.Burton Z.F., Gross,C.A., Watanabe,K. and Burgess,R.R. (1983) The operon that encodes the sigma subunit of RNA polymerase also encodes ribosomal protein S21 and DNA primase in E.coli K12. Cell, 32, 335–349. [DOI] [PubMed] [Google Scholar]
- 3.Little R., Fiil,N.P., Dennis,P.P. (1981) Transcriptional and post-transcriptional control of ribosomal protein and ribonucleic acid polymerase genes. J. Bacteriol., 147, 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blattner F.R., Plunkett,G.,III, Bloch,C.A., Perna,N.T., Burland,V., Riley,M., Collado-Vides,J., Glasner,J.D., Rode,C.K., Mayhew,G.F. et al. (1997) The complete genome sequence of Escherichia coli K-12. Science, 277, 1453–1474. [DOI] [PubMed] [Google Scholar]
- 5.Zurawski G., Zurawski,S.M. (1985) Structure of the Escherichia coli S10 ribosomal protein operon. Nucleic Acids Res., 13, 4521–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forsyth R.A., Haselbeck,R.J., Ohlsen,K.L., Yamamoto,R.T., Xu,H., Trawick,J.D., Wall,D., Wang,L., Brown-Driver,V., Froelich,J.M. et al. (2002) A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol. Microbiol., 43, 1387–1400. [DOI] [PubMed] [Google Scholar]
- 7.Judson N. and Mekalanos,J.J. (2000) TnAraOut, a transposon-based approach to identify and characterize essential bacterial genes. Nat. Biotechnol., 18, 740–745. [DOI] [PubMed] [Google Scholar]
- 8.Huerta A.M., Salgado,H., Thieffry,D. and Collado-Vides,J. (1998) RegulonDB: a database on transcriptional regulation in Escherichia coli. Nucleic Acids Res., 26, 55–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Itoh T., Takemoto,K., Mori,H. and Gojobori,T. (1999) Evolutionary instability of operon structures disclosed by sequence comparisons of complete microbial genomes. Mol. Biol. Evol., 16, 332–346. [DOI] [PubMed] [Google Scholar]
- 10.Brendel V. and Trifonov,E.N. (1984) A computer algorithm for testing potential prokaryotic terminators. Nucleic Acids Res., 12, 4411–4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brendel V. and Trifonov,E.N. (1984) Computer-aided mapping of DNA–protein interaction sites. Proceedings of the Ninth International CODATA Conference, Jerusalem, Israel, pp. 17–20, 115–118.
- 12.Ermolaeva M.D., Khalak,H.G., White,O., Smith,H.O. and Salzberg,S.L. (2000) Prediction of transcription terminators in bacterial genomes. J. Mol. Biol., 301, 27–33. [DOI] [PubMed] [Google Scholar]
- 13.Unniraman S., Prakash,R. and Nagaraja,V. (2002) Conserved economics of transcription termination in eubacteria. Nucleic Acids Res., 30, 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ozoline O.N., Deev,A.A. and Arkhipova,M.V. (1997) Non-canonical sequence elements in the promoter structure. Cluster analysis of promoters recognized by Escherichia coli RNA polymerase. Nucleic Acids Res., 23, 4703–4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yada T., Nakao,M., Totoki,Y. and Nakai,K. (1999) Modeling and predicting transcriptional units of Escherichia coli genes using hidden Markov models. Bioinformatics, 15, 987–993. [DOI] [PubMed] [Google Scholar]
- 16.Craven M., Page,D., Shavlik,J., Bockhorst,J. and Glasner,J. (2000) A probabilistic learning approach to whole-genome operon prediction. Proceedings of the Eighth International Conference on Intelligent Systems for Molecular Biology, La Jolla, CA, pp. 116–127. [PubMed]
- 17.Sabatti C., Rohlin,L., Oh,M. and Liao,J.C. (2002) Co-expression pattern from DNA microarray experiments as a tool for operon prediction. Nucleic Acids Res., 30, 2886–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bockhorst J., Craven,M., Page,D., Shavlik,J. and Glasner,J. (2003) A Bayesian network approach to operon prediction. Bioinformatics, 19, 1227–1135. [DOI] [PubMed] [Google Scholar]
- 19.Salgado H., Moreno-Hagelsieb,G., Smith,T.F. and Collado-Vides,J. (2000) Operons in Escherichia coli: genomic analyses and predictions. Proc. Natl Acad. Sci. USA, 97, 6652–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ermolaeva M., White,O. and Salzberg,S.L. (2001) Prediction of operons in microbial genomes. Nucleic Acids Res., 29, 1216–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolf Y.I., Rogozin,I.B., Kondrashov,A.S. and Koonin,E.V. (2001) Genome alignment, evolution of prokaryotic genome organization, and prediction of gene function using genomic context. Genome Res., 11, 356–372. [DOI] [PubMed] [Google Scholar]
- 22.Overbeek R., Fonstein,M., D'Souza,M., Pusch,G.D. and Maltsev,N. (1999) The use of gene clusters to infer functional coupling. Proc. Natl Acad. Sci. USA, 96, 2896–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng Y., Szustakowski,J.D., Fortnow,L., Roberts,R.J. and Kasif,S. (2002) Computational identification of operons in microbial genomes. Genome Res., 12, 1221–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mironov A.A., Koonin,E.V., Roytberg,M.A. and Gelfand,M.S. (1999) Computer analysis of transcription regulatory patterns in completely sequenced bacterial genomes. Nucleic Acids Res., 27, 2981–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vitreschak A.G, Rodionov,D.A., Mironov,A.A. and Gelfand,M.S. (2002) Regulation of riboflavin biosynthesis and transport genes in bacteria by transcriptional and translational attenuation. Nucleic Acids Res., 30, 3141–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuroda M., Ohta,T., Uchiyama,I., Baba,T., Yuzawa,H., Kobayashi,I., Cui,L., Oguchi,A., Aoki,K., Nagai,Y., Lian,J., Ito,T. et al. (2001) Whole genome sequencing of methicillin-resistant Staphylococcus aureus, the major hospital pathogen. Lancet, 357, 1225–1240. [DOI] [PubMed] [Google Scholar]
- 27.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 28.Altschul S.F., Madden,T.L., Schäffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moreno-Hagelsieb G and Collado-Vides,J (2002) A powerful non-homology method for the prediction of operons in prokaryotes. Bioinformatics, 18 (Suppl. 1), S329–S336. [DOI] [PubMed] [Google Scholar]
- 30.Tatusov R.L., Natale,D.A., Garkavtsev,I.V., Tatusova,T.A., Shankavaram,U.T., Rao,B.S., Kiryutin,B., Galperin,M.Y., Fedorova,N.D. and Koonin,E.V. (2001) The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res., 29, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bailey T.L. and Elkan,C. (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology, Stanford, CA, pp. 28–36. [PubMed]
- 32.Bailey T.L. and Gribskov,M. (1998) Combining evidence using p-values: application to sequence homology searches. Bioinformatics, 14, 48–54. [DOI] [PubMed] [Google Scholar]
- 33.Washburn R.S., Marra,A., Bryant,A.P., Rosenberg,M. and Gentry,D.R. (2001) rho is not essential for viability or virulence in Staphylococcus aureus. Antimicrob. Agents Chemother., 45, 1099–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X., Lindahl,L., Sha,Y. and Zengel,J.M. (1997) Analysis of the Bacillus subtilis S10 ribosomal protein gene cluster identifies two promoters that may be responsible for transcription of the entire 15-kilobase S10-spc-alpha cluster. J. Bacteriol., 179, 7046–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sako T. and Sakakibara,Y. (1980) Coordinate expression of Escherichia coli dnaA and dnaN genes. Mol. Gen. Genet., 179, 521–526. [DOI] [PubMed] [Google Scholar]
- 36.Perez-Roger I., Garcia-Sogo,M., Navarro-Avino,J.P., Lopez-Acedo,C., Marcian,F. and Armengod,M.E. (1991) Positive and negative regulatory elements in the dnaA–dnaN–recF operon of Escherichia coli. Biochimie, 73, 329–334. [DOI] [PubMed] [Google Scholar]
- 37.Ogura Y., Imai,Y., Ogasawara,N. and Moriya,S. (2001) Autoregulation of the dnaA-dnaN operon and effects of DnaA protein levels on replication initiation in Bacillus subtilis. J. Bacteriol., 183, 3833–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sau S., Sun,J. and Lee,C.Y. (1997) Molecular characterization and transcriptional analysis of type 8 capsule genes in Staphylococcus aureus. J. Bacteriol., 179, 1614–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ouyang S., Sau,S. and Lee,C.Y. (1999) Promoter analysis of the cap8 operon, involved in type 8 capsular polysaccharide production in Staphylococcus aureus. J. Bacteriol., 181, 2492–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bischoff M., Entenza,J.M. and Giachino,P. (2001) Influence of a functional sigB operon on the global regulators sar and agr in Staphylococcus aureus. J. Bacteriol., 183, 5171–5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hiramatsu T., Kodama,K., Kuroda,T., Mizushima,T. and Tsuchiya,T. (1998) A putative multisubunit Na+/H+ antiporter from Staphylococcus aureus. J. Bacteriol., 180, 6642–6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ito M., Guffanti,A.A., Oudega,B. and Krulwich,T.A. (1999) mrp, a multigene, multifunctional locus in Bacillus subtilis with roles in resistance to cholate and to Na+ and in pH homeostasis. J. Bacteriol., 181, 2394–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hara H., Yasuda,S., Horiuchi,K. and Park,J.T. (1997) A promoter for the first nine genes of the Escherichia coli mra cluster of cell division and cell envelope biosynthesis genes, including ftsI and ftsW. J. Bacteriol., 179, 5802–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jordan A., Aragall,E., Gibert,I.And Barbe,J. (1996) Promoter identification and expression analysis of Salmonella typhimurium and Escherichia coli nrdEF operons encoding one of two class I ribonucelotide reductases present in both bacteria. Mol. Microbiol., 19, 777–790. [DOI] [PubMed] [Google Scholar]
- 45.Scotti C., Valbuzzi,A., Perego,M., Galizzi,A. and Albertini,A.M. (1996) The Bacillus subtilis genes for ribonucleotide reductase are similar to the genes for the second class I nrdE/nrdF enzymes of Enterobacteriaceae. Microbiology, 142, 2995–3004. [DOI] [PubMed] [Google Scholar]
- 46.Masalha M., Borovok,I., Schreiber,R., Aharonowitz,Y. and Cohen,G. (2001) Analysis of transcription of the Staphylococcus aureus aerobic class 1b and anaerobic class III ribonucleotide reductase genes in response to oxygen. J. Bacteriol., 183, 7260–7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kullik I., Jenni,R. and Berger-Bachi,B. (1998) Sequence of the putative alanine racemase operon in Staphylococcus aureus: insertional interruption of this operon reduces d-alanine substitution of lipoteichoic acid and autolysis. Gene, 219, 9–17. [DOI] [PubMed] [Google Scholar]
- 48.Gertz S., Engelmann,S., Schmid,R., Ziebandt,A., Tischer,K., Scharf,C., Hacker,J. and Hecker,M. (2000) Characterization of the sigma(B) regulon in Staphylococcus aureus. J. Bacteriol., 182, 6983–6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuroda M., Hayashi,H. and Ohta,T. (1999) Chromosome-determined zinc-responsible operon czr in Staphylococcus aureus strain 912. Microbiol. Immunol., 43, 115–125. [DOI] [PubMed] [Google Scholar]
- 50.Boyle-Vavra S., de Jonge,B.L., Ebert,C.C. and Daum,R.S. (1997) Overproduction of a 37-kilodalton cytoplasmic protein homologous to NAD+-linked d-lactate dehydrogenase associated with vancomycin resistance in Staphylococcus aureus. J. Bacteriol., 179, 6756–6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kopp U., Roos.M., Wecke,J. and Labischinski,H. (1996) Staphylococcal peptidoglycan interpeptide bridge biosynthesis: a novel antistaphylococcal target? Microb. Drug Resist., 2, 29–41. [DOI] [PubMed] [Google Scholar]
- 52.Cabrera G., Xiong,A., Uebel,M., Singh,V.K., Jayaswal,R.K. (2001) Molecular characterization of the iron-hydroxamate uptake system in Staphylococcus aureus. Appl. Environ. Microbiol., 67, 1001–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Glanzmann P., Gustafson,J., Komatsuzawa,H., Ohta,K. and Berger-Bachi,B. (1999) GlmM operon and methicillin-resistant glmM suppressor mutants in Staphylococcus aureus. Antimicrob. Agents Chemother., 43, 240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuroda M., Kobayashi,D., Honda,K., Hayashi,H. and Ohta,T. (1999) The hsp operons are repressed by the hrc37 of the hsp70 operon in Staphylococcus aureus. Microbiol. Immunol., 43, 19–27. [DOI] [PubMed] [Google Scholar]
- 55.Ohta T., Honda,K., Kuroda,M., Saito,K. and Hayashi,H. (1993) Molecular characterization of the gene operon of heat shock proteins HSP60 and HSP10 in methicillin-resistant Staphylococcus aureus. Biochem. Biophys. Res. Commun., 193, 730–737. [DOI] [PubMed] [Google Scholar]
- 56.Breidt F. Jr, Hengstenberg,W., Finkeldei,U. and Stewart,G.C. (1987) Identification of the genes for the lactose-specific components of the phosphotransferase system in the lac operon of Staphylococcus aureus. J. Biol. Chem., 262, 16444–16449. [PubMed] [Google Scholar]
- 57.Savopoulos J.W., Hibbs,M., Jones,E.J., Mensah,L., Richardson,C., Fosberry,A., Downes,R., Fox,S.G., Brown,J.R. and, Jenkins,O. (2001) Identification, cloning, and expression of a functional phenylalanyl-tRNA synthetase (pheRS) from Staphylococcus aureus. Protein Expr. Purif., 21, 470–484. [DOI] [PubMed] [Google Scholar]
- 58.Bischoff M., Entenza,J.M. and Giachino,P. (2001) Influence of a functional sigB operon on the global regulators sar and agr in Staphylococcus aureus. J. Bacteriol., 183, 5171–5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kies S., Otto,M., Vuong,C. and Gotz,F. (2001) Identification of the sigB operon in Staphylococcus epidermidis construction and characterization of a sigB deletion mutant. Infect Immun., 69, 7933–7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kullik I.I. and Giachino,P. (1997) The alternative sigma factor sigmaB in Staphylococcus aureus: regulation of the sigB operon in response to growth phase and heat shock. Arch. Microbiol., 167, 151–159. [DOI] [PubMed] [Google Scholar]
- 61.Wise A.A. and Price,C.W. (1995) Four additional genes in the sigB operon of Bacillus subtilis that control activity of the general stress factor sigma B in response to environmental signals. J. Bacteriol., 177, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Horsburgh M.J., Ingham,E. and Foster,S.J. (2001) In Staphylococcus aureus, fur is an interactive regulator with PerR, contributes to virulence, and is necessary for oxidative stress resistance through positive regulation of catalase and iron homeostasis. J. Bacteriol., 183, 468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uhlen M., Guss,B., Nilsson,B., Gatenbeck,S., Philipson,L. and Lindberg,M. (1984) Complete sequence of the staphylococcal gene encoding protein A. A gene evolved through multiple duplications. J. Biol. Chem., 259,1695–702. [PubMed] [Google Scholar]
- 64.Rice K., Peralta,R., Bast,D., de Azavedo,J. and McGavin,M.J. (2001) Description of staphylococcus serine protease (ssp) operon in Staphylococcus aureus and nonpolar inactivation of sspA-encoded serine protease. Infect. Immun., 69, 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morrissey J.A., Cockayne,A., Hill,P.J. and Williams,P. (2000) Molecular cloning and analysis of a putative siderophore ABC transporter from Staphylococcus aureus. Infect. Immun., 68, 6281–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakao A., Imai,S. and Takano,T. (2000) Transposon-mediated insertional mutagenesis of the D-alanyl-lipoteichoic acid (dlt) operon raises methicillin resistance in Staphylococcus aureus. Res. Microbiol., 151, 823–829. [DOI] [PubMed] [Google Scholar]
- 67.Peschel A., Otto,M., Jack,R.W., Kalbacher,H., Jung,G. and Gotz,F. (1999) Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J. Biol. Chem., 274, 8405–8410. [DOI] [PubMed] [Google Scholar]
- 68.Stranden A.M., Roos,M. and Berger-Bachi,B. (1996) Glutamine synthetase and heteroresistance in methicillin-resistant Staphylococcus aureus. Microb. Drug Resist., 2, 201–207. [DOI] [PubMed] [Google Scholar]
- 69.Gustafson J., Strassle,A., Hachler,H., Kayser,F.H. and Berger-Bachi,B. (1994) The femC locus of Staphylococcus aureus required for methicillin resistance includes the glutamine synthetase operon. J. Bacteriol., 176, 1460–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Groicher K.H., Firek,B.A., Fujimoto,D.F. and Bayles,K.W. (2000) The Staphylococcus aureus lrgAB operon modulates murein hydrolase activity and penicillin tolerance. J. Bacteriol., 182, 1794–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jarraud S., Peyrat,M.A., Lim,A., Tristan,A., Bes,M., Mougel,C., Etienne,J., Vandenesch,F., Bonneville,M. and Lina,G. (2001) egc, a highly prevalent operon of enterotoxin gene, forms a putative nursery of superantigens in Staphylococcus aureus. J. Immunol., 166, 669–677. [DOI] [PubMed] [Google Scholar]
- 72.Reed S.B., Wesson,C.A., Liou,L.E., Trumble,W.R., Schlievert,P.M., Bohach,G.A. and Bayles,K.W. (2001) Molecular characterization of a novel Staphylococcus aureus serine protease operon. Infect. Immun., 69, 1521–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brandenberger M., Tschierske,M., Giachino,P., Wada,A. and Berger-Bachi,B. (2000) Inactivation of a novel three-cistronic operon tcaR-tcaA-tcaB increases teicoplanin resistance in Staphylococcus aureus. Biochim. Biophys. Acta, 1523, 135–139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.