

Abstract
The ability of Serial Analysis of Gene Expression (SAGE) to provide a quantitative picture of global gene expression relies not only on the depth and accuracy of sequencing into the SAGE library, but also on the efficiency of each step required to generate the SAGE library from the starting mRNA material. The first critical step is the ligation of adaptors containing a Type IIS recognition sequence to the anchored 3′ end cDNA population that permits the release of short sequence tags (SSTs) from defined sites within the 3′ end of each transcript. Using an in vitro transcript as a template, we observed that only a small fraction of anchored 3′ end cDNA are successfully ligated with added SAGE adaptors under typical reaction conditions currently used in the SAGE protocol. Although the introduction of ∼500-fold molar excess of adaptor or the inclusion of 15% (w/v) PEG-8000 increased the yield of the adaptor-modified product, complete conversion to the desired adaptor:cDNA hetero-ligation product is not achieved. An alternative method of ligation, termed as directed ligation, is described which exploits a favourable mass-action condition created by the presence of NlaIII during ligation in combination with a novel SAGE adaptor containing a methylated base within the ligation site. Using this strategy, we were able to achieve near complete conversion of the anchored 3′ end cDNA into the desired adaptor-modified product. This new protocol therefore greatly increases the probability that a SST will be generated from every transcript, greatly enhancing the fidelity of SAGE. Directed ligation also provides a powerful means to achieve near-complete ligation of any appropriately designed adaptor to its respective target.
INTRODUCTION
The development of technologies aimed towards monitoring gene expression on a global scale has revolutionized the study of biology from a systems perspective (1). This perspective embraces the idea that the functional significance of gene products is not only related to their quantity in the cell, but also to how they interact and are strung together to form genetic and biochemical networks. Numerous technologies have been developed over the past decade, with the greatest attention being given to approaches based on either high-throughput sequencing or massively parallel analysis of the transcriptome (i.e. the set of all expressed genes weighted by transcript abundance) using array hybridization technology. The sequencing approach to monitoring gene expression on a global scale typically involves the creation of short representations of each transcript, such as expressed sequence tags (ESTs) or short sequence tags (SSTs) generated using Serial Analysis of Gene Expression (SAGE) technology (2,3). DNA microarray technology attempts to resolve the transcriptome by selectively binding and quantifying each transcript at one or more complementary registers of a high-density array (4–6). These technologies are now routinely used to identify families of genes—in many cases incompletely characterized or with previously unidentified functionality—which act in concert to define a given cell fate or outcome (7), and have been used to identify upstream sequence elements involved in directing the expression of these gene families. Although microarray technology offers an increasingly reliable and sensitive analysis of gene expression, its use is dependent on an a priori knowledge of genes, which are expressed under a given cell state, currently restricting application of the technology to the identification and quantification of these subsets of genes.
SAGE technology (2), in contrast, directly samples the entire transcriptome of an organism under a given cellular state through the generation of SSTs of 9–22 bp in length. Because a 9–10mer oligonucleotide can theoretically identify 49 (262 144) or 410 (1 048 576) unique sequences, the entire transcript population of any organism can potentially be repres ented (2,3). First, a cDNA copy of the mRNA population is digested with a restriction endonuclease (RE; e.g. NlaIII) and the most 3′ end restriction fragments of the digested population are purified. A short oligonucleotide adaptor that contains a unique primer sequence and a recognition sequence for a Type IIS RE is then ligated to the anchored cDNA. Because Type IIS REs are capable of cleaving DNA outside their recognition sequence (8), subsequent cleavage with a Type IIS RE (e.g. BsmFI) releases SSTs of equal length (2). A library of these SSTs is created through subsequent dimerization, amplification via the PCR, concatemerization and insertion into an appropriate vector. Finally, a representative population of clones is serially sequenced to identify and tally each SST. Since each SST is derived from a defined position within a particular cDNA, a given tag can be cross-referenced through organism- and/or tissue-specific genome databases to a particular gene to give a profile of global gene expression. An important advantage compared with microarray technology is that unreferenced SSTs that arise out of the SAGE analysis can be used to identify previously unknown genes and aid in the completion of genome annotations for the organism under study (2,3,9–17).
The ability of SAGE to provide an accurate measure of gene expression profiles is dependent upon the extent to which the distribution of transcript abundances inferred through the sequenced set of amplified SSTs fully reflects the real distribution of the abundances of associated transcripts in the original mRNA population. This fidelity depends upon the accuracy of the sequencing method used to identify the SSTs (18) and on the depth of sequencing applied to the SAGE library (19,20). Less appreciated, however, is the extent to which losses and processing artefacts in each of the 12 enzymatic and 10 purification steps—or 7 in the microSAGE protocol—used to convert the starting mRNA sample into a SAGE library (Table 1) can skew the sequencing results away from the real distribution. To illustrate, if 5 μg of mRNA (∼5 × 1012 molecules of average length 2 kb) are used as starting material for the SAGE protocol, a 50% average yield in each processing step would result in an overall yield of 0.000024% (i.e. 0.522), such that the final sample (∼1.2 × 106 molecules) would represent a minute fraction of the original. Such an overall yield would result in a form of sampling bias in SAGE analysis equivalent to the bias introduced by an insufficient depth of sequencing (19,20). Although inclusion of PCR steps in the SAGE protocol is intended to recover these losses, amplification after processing can only recover those ditags derived from targets that have survived the numerous enzymatic and purification steps. Clearly then, efforts to maximize yields and minimize artefacts introduced in each processing step are required to ensure the fidelity of SAGE.
Table 1. Outline of the enzymatic, purification and isolation steps involved in the SAGE and microSAGE protocols (http://www.sagenet.org/protocol/index.htm).
| Enzymatic steps | Purification and isolation steps | |
|---|---|---|
| MicroSAGE (SADE) | SAGE | |
| (1) mRNA preparation | Affinity purification | Precipitation, selection with biotinylated oligo(dT) |
| (2) cDNA synthesis | – | Phenol extraction, precipitation |
| (3) cleavage with anchoring enzyme (digest with NlaIII) | – | Phenol extraction, precipitation |
| (4) 3′ end cDNA isolation | – | Affinity purification |
| (5) Ligating adaptors to bound 3′ end cDNA | – | |
| (6) Release of cDNA tags (digest with BsmFI) | Phenol extraction, precipitation | |
| (7) Blunt-ending of cDNA tags | Phenol extraction, precipitation | |
| (8) Ligating tags to form ditags | – | |
| (9) PCR amplification of ditags | Phenol extraction, precipitation | |
| PAGE purification, gel extraction, precipitation | ||
| (10) Adaptor removal (NlaIII digestion) and purification of ditags | Phenol extraction, precipitation | |
| PAGE purification, gel extraction, precipitation | ||
| (11) Ligation of ditags to form concatamers | PAGE purification, size selection, gel extraction, precipitation | |
| (12) Insertion into vector | Phenol extraction, selection by host | |
Although a number of recent studies have resulted in the improvement of some of the purification steps in the SAGE protocol (13,15,21–28), little attention has been given to addressing the efficiencies of the enzymatic steps of the protocol. Given that the ability to generate a SAGE tag from a transcript is determined by the successful ligation of the SAGE adaptor to the anchored 3′ end cDNA population, the yield in this step is likely to contribute significantly to the overall fidelity of the SAGE protocol. Here we demonstrate, using adaptors 1A/B of the current SAGE protocol (version 1e; http://www.sagenet.org/protocol/index.htm), that the yield of this ligation step is generally low due to a strong propensity of the anchored 3′ end cDNA target to self-ligate. We then show that the addition of PEG-8000, traditionally used to favour the formation of linear ligation products (29–31), increases the yield of the desired adaptor-target heterodimer, but is unable to fully eliminate the formation of unwanted homodimer. Finally, we show that by using an alternative method of ligation, which we call ‘directed ligation’, a significant improvement in the SAGE protocol is achieved, increasing the efficiency of adaptor ligation and eliminating the irreversible formation of unwanted ligation products.
MATERIALS AND METHODS
Enzymes and constructs
A 956 bp clone homologous to rat liver α transcription factor (GenBank ID: X65948) from rat brain with a polyadenylated 3′ end (58 bp), kindly provided by Dr Terry Snutch (Biotechnology Laboratory) in pBluescript SK− (Stratagene), was propagated in Escherichia coli DH5α (Invitrogen). Plasmids were isolated using the boiling miniprep method (32) from 3 ml Terrific broth (Sigma Aldrich) cultures in the presence of 100 μg/ml ampicillin (Sigma Aldrich) when required. Plasmids (∼20 μg each) were linearized with EcoRV and further purified using the Qiagen Qiaquick purification kit according to the manufacturer's protocol (Qiagen). Orientation and identification of the insert were verified by sequencing of 100 ng of the purified plasmid at the Nucleic Acids and Peptide Synthesis Unit, University of British Columbia. In vitro RNA transcripts in the sense orientation were generated from 1 μg of linearized plasmid using the T3 MEGAscript kit (Ambion) following the manufacturer's protocol and stored at −70°C in diethyl-pyrocarbonate (DEPC) treated H2O (Ambion). All reactions in this study were incubated using an Eppendorf Mixmaster programmed for 3 s mixtures at 1400 rpm every 15 min.
Preparation of 3′ end anchored cDNA
An aliquot of 5 μg (∼16 pmol) or 0.1 μg (∼0.3 pmol) of in vitro transcribed RNA was processed according to the regular SAGE protocol or the microSAGE protocol version 1e. Alternatively, in vitro transcribed RNA (0.6 μg or ∼1.9 pmol) was annealed to 3.0 mg oligo(dT)25 dynabeads (Dynal Biotech) in the presence of 600 U of SUPERase·In (Ambion). Annealed RNA was then processed according to the microSAGE protocol version 1e using components from a cDNA synthesis kit (Invitrogen) and scaled accordingly to a final volume of 600 μl with the following exception: after first strand synthesis, the reaction was cooled on ice, magnetized and 520 μl of the first strand reaction was replaced with 520 μl of a pre-chilled mixture of second strand synthesis reaction components and incubated for 16 h at 16°C. Anchored second strand products were then blunt-ended, washed and digested with NlaIII (New England Biolabs) as described. The resulting anchored 3′ end cDNAs (∼0.6 pmol/mg dynabeads) were stored at −20°C until ready for use.
Adaptors
Oligonucleotides corresponding to the adaptors and primers used in the SAGE and microSAGE protocols version 1e were obtained gel- or HPLC-purified (Qiagen) and are shown in Table 2. Stock concentrations (5 mM) of the following adaptors were prepared in 1 × NEB4 buffer (New England Biolabs) by mass dilutions: adaptor 1 (1A/1Bphos), adaptor 1m5C (1Am5C/1Bphos), adaptor 1m6A (1Am6A/1Bphos), adaptor 2 (2A/2Bphos), adaptor 2m5C (2Am5C/2Bphos) and adaptor 2m6A (2Am6A/2Bphos). Adaptors were annealed according to the annealing schedule described in the current SAGE protocols.
Table 2. List of oligonucleotides used in this study to form various SAGE adaptors.
| Oligo ID | Sequence (5′ → 3′) | MW (g/mol) |
|---|---|---|
| 1A | TTTGGATTTGCTGGTGCAGTACAACTAGGCTTAATAGGGACATG | 13657.06 |
| 1Am6Aa | TTTGGATTTGCTGGTGCAGTACAACTAGGCTTAATAGGGACAm6TG | 13670.95 |
| 1Am5Cb | TTTGGATTTGCTGGTGCAGTACAACTAGGCTTAATAGGGAC m5ATG | 13670.95 |
| 1Bphos | pTCCCTATTAAGCCTAGTTGTACTGCACCAGCAAATCC-NH2c | 11517.57 |
| 2A | TTTCTGCTCGAATTCAAGCTTCTAACGATGTACGGGGACATG | 12919.55 |
| 2Am6Aa | TTTCTGCTCGAATTCAAGCTTCTAACGATGTACGGGGACAm6TG | 12933.58 |
| 2Am5Cb | TTTCTGCTCGAATTCAAGCTTCTAACGATGTACGGGGAC m5ATG | 12933.58 |
| 2Bphos | pTCCCCGTACATCGTTAGAAGCTTGAATTCGAGCAG-NH2c | 11020.24 |
Oligonucleotides were obtained gel-purified and verified by mass spectrometry.
aAm6, N6-methyl-deoxyadenosine.
bCm5, 5-methyl-deoxycytosine.
cNH2, 3′ C7 amino spacer.
Standard ligation protocol used in SAGE
Ligation reactions using adaptor 1 at a final concentration of 80 nM were performed according to microSAGE protocol version 1e. Additional ligation reactions, scaled to a final volume of 10 μl (∼0.075 pmol cDNA per 125 μg dynabeads) and containing varying amounts of adaptor 1 (0.038–38 pmol), or supplemented with PEG-8000 [15% (w/v) final] using a final adaptor concentration of 1 μM were also performed. All reaction samples were incubated for 2 h at 16°C or 25°C.
Directed ligation
Titration of T4 DNA ligase activity with NlaIII
Stock ligase mixture containing T4 DNA ligase (5 Weiss U/μl; Fermentas) were prepared with various amounts of NlaIII (120 U/μl; New England Biolabs) in a final buffer composition of 15 mM Tris–HCl (pH 7.5), 0.1 mM EDTA, 1 mM DTT, 200 mM KCl, 0.5 mg/ml BSA and 50% glycerol, and stored at −70°C. Oligo(dT)25 dynabeads (125 μg) with anchored 3′end cDNA (0.075 pmol) were pre-incubated with adaptor 1, adaptor 1m5C or adaptor 1m6A (1 μM final) for 5 min at 37°C in 1× NEB4 buffer supplemented with 1 mM ATP and 100 ng/μl BSA in a volume of 9 μl. The reactions were initiated by adding 1 μl from one of the stock enzyme mixtures described above, overlaid with mineral oil, and incubated for 2 h at 37°C.
Directed ligation protocol for SAGE
A stock enzyme mixture containing NlaIII (25 U/μl final) and T4 DNA ligase (2.5 Weiss U/μl final) was prepared as described above. Oligo(dT)25 dynabeads (125 μg) with anchored 3′ end cDNA (0.075 pmol) were pre-incubated with 2.5 pmol of adaptor 1m6A for 5 min at 37°C in 1× NEB4 buffer supplemented with 100 ng/μl BSA and 1 mM ATP. After initiation with 1 μl of the stock enzyme mixture, reactions were spiked every 15 min with 2.5 pmol of adaptor 1m6A for a total incubation time of 1 h and a total addition of 10 pmol adaptor.
Analysis of anchored ligation products
The reactions were heat-inactivated for 20 min at 65°C in 200 μl of 1× NEB4 supplemented with 100 ng/μl BSA, followed by two washes with the same buffer. Anchored ligation products were then cleaved off the dynabead support with 10 U DraI (New England Biolabs) in 30 μl of 1× NEB4 supplemented with BSA. After incubation for 1 h at 37°C, products were resolved via PAGE (6% PAGE; Owl Scientific) for 3 h at 12.5 V/cm. SYBR-Gold (Molecular Probes) stained gels were visualized using a CCD-based gel documentation system (Alpha Innotech) using a SYBR-green filter set (Molecular Probes) at a sub-saturating aperture setting and recorded as TIFF files. When required, densitometric analysis was performed using publicly available software (tnimage-3.3.7a; http://brneurosci.org/tnimage.html).
Preparation and PCR amplification of ditags
Adaptors 1 and 2 or adaptors 1m6A and 2m6A were ligated to anchored 3′ end cDNA derived from 100 ng of in vitro transcripts as described above using the standard microSAGE protocol version 1e or the directed ligation protocol. After ligation, the anchored products were processed according to microSAGE protocol version 1e to form ditags. Ditag ligation mixtures (3 μl) were brought up to a final volume of 20 μl with LoTE buffer (2 mM Tris–HCl, 0.2 mM EDTA, pH, 8.0). One microlitre aliquots of 1 : 20 and 1 : 200 dilutions of the ligation mixture in LoTE were then used as a template for PCR amplification with Platinum Pfx thermophilic DNA polymerase (Invitrogen) supplemented with 0.5× PCRX enhancer solution and 0.1 mM MgSO4 according to the manufacturer's protocol in a final volume of 50 μl. PCR amplification was performed in the presence or absence of template on an Eppendorf Mastercycler (Eppendorf) using primer 1 and primer 2 as described in the microSAGE protocol. After activation for 1 min at 95°C, 26 cycles were performed according to the following schedule: 95°C, 30 s; 55°C, 1 min and 72°C, 1 min. Upon completion, a 10 μl aliquot was then resolved via 6% PAGE for 1 h at 12.5 V/cm and visualized as described above.
RESULTS AND DISCUSSION
The ability of SAGE to provide a truly quantitative picture of gene expression relies on the efficiency of each step required to generate the library of SSTs from the harvested mRNA starting material. Currently, two general approaches to generate SAGE libraries are utilized (Table 1), each customized towards the amount of starting material available to the researcher. The original SAGE protocol described by Velculescu et al. (2) uses 5 μg of mRNA (∼7.8 pmol mRNA of average length 2 kb) as starting material. After conversion into biotinylated cDNA, half of this sample is digested with the RE NlaIII, and the 3′ end fragments are affinity purified via streptavidin-linked dynabeads (2 mg) to generate anchored 3′ end cDNA (3.9 pmol/mg dynabeads). In contrast, the microSAGE protocol, a modification of the SADE (SAGE Analysis for Down-sized Extracts) protocol of Virlon et al. (33) and commercially available I-SAGE™ kit from Invitrogen, is designed to process the RNA from 5 × 104 to 2 × 106 cells or up to 100 ng (∼0.16 pmol mRNA of average length 2 kb) of starting mRNA. Oligo(dT)25 dynabeads (0.5 mg) are used as an affinity support to directly harvest polyadenylated RNA from the sample. The anchored oligo(dT)25 on the support is used to prime cDNA synthesis which is then digested with NlaIII to generate anchored 3′ end cDNA (0.31 pmol/mg dynabeads).
When our in vitro RNA material was used as the starting material, we found that the amount of anchored 3′ end cDNA recovered using the original SAGE protocol was similar to that obtained through the microSAGE protocol despite using 25-fold more starting material (data not shown). This observation is consistent with work by Virlon et al. (33) where 200-fold less anchored 3′ end cDNA was recovered from microdissected renal tubules using the SAGE protocol compared to those recovered from their SADE protocol, which used half the amount of starting material and employed Sau3A I as the anchoring enzyme. Although this material loss is largely due to the presence of four additional extraction and precipitation steps in the original SAGE protocol prior to adaptor ligation (Table 1), additional losses may arise from the presence of excess biotinylated oligo(dT)20 primer used to prime first strand synthesis. Any such primer that survives the extraction and precipitation steps will compete with binding to the streptavidin support. This primer contamination is most probably small, however, as batch purification of biotinylated cDNAs using Qiaex II silica beads did not improve yields significantly.
After synthesis of the anchored 3′ end cDNA library on either streptavidin-linked Dynabeads (i.e. SAGE) or oligo(dT)25 Dynabeads (i.e. microSAGE), further processing towards generation of the SAGE library is essentially the same under the two protocols (Table 1).
Self-ligation of the anchored 3′ end cDNA competes with ligation of the adaptor
Under standard microSAGE reaction conditions, we observe that the ligation of SAGE adaptors to the cohesive end of the anchored 3′ end cDNA consistently produces two products. In the presence of T4 DNA ligase and the standard 80 nM concentration of adaptor 1, a relatively small fraction (<5%) of the anchored 3′ end cDNA was found to ligate to adaptor 1 to form the desired adaptor-target cDNA hetero-ligation product (Figure 1). The bulk of the anchored cDNA underwent an undesired reaction to form a high molecular weight product (lane 3). Comparisons with the control reaction in which no T4 DNA ligase was added (lane 2), and with a ligation reaction performed in the presence of NlaIII indicated that this high molecular weight product is a homodimer of the anchored 3′ end cDNA. Identical experiments were also carried out on streptavidin anchored 3′ end cDNA samples prepared by the original SAGE protocol and gave essentially the same results. Lower loading densities of in vitro RNA onto oligo(dT)20 dynabeads or biotinylated cDNA onto streptavidin-linked dynabeads only marginally inhibited formation of the homodimer, suggesting that homodimer formation depends on both the distance of separation between anchored 3′ end cDNA molecules on the surface of a given dynabead (intermolecular) as well as between those anchored on adjacent dynabeads (intramolecular). Formation of the homodimer was also observed when other in vitro RNA transcripts were utilized to generate anchored 3′ end cDNA targets ranging from 132 to 355 bp in length. Thus, under the ligation conditions described, most of the desired hetero-ligation product is lost in favor of self-ligation of two anchored cDNA fragments.
Figure 1.
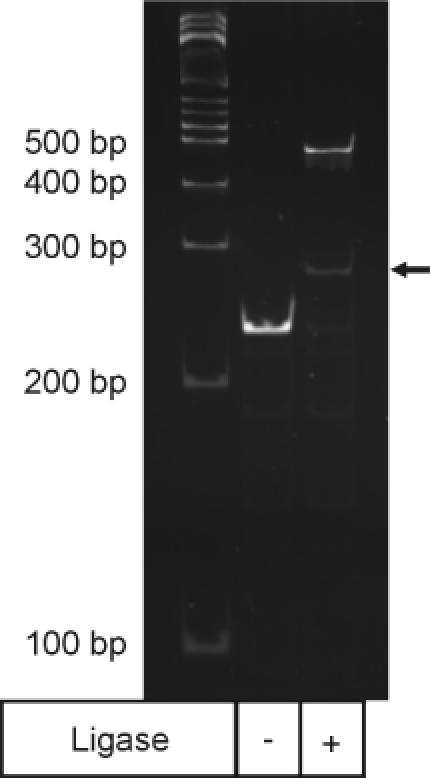
Ligation of SAGE adaptor 1A to anchored 3′ end cDNA. An aliquot of 100 ng of in vitro transcribed polyadenylated product was processed under the microSAGE protocol and split into half. Lane 2 shows a control reaction in which T4 DNA ligase was not added to the ligation mixture. Lane 3 shows the formation of a small amount of the hetero-ligation product indicated by the arrow as well as a high molecular weight band corresponding to twice the molecular weight of the unligated cDNA. Ligations were performed as described in Materials and Methods.
The yield of the desired hetero-ligation product was found to depend on the amount of SAGE adaptor introduced into the ligation mixture, and increased with increasing adaptor concentration (Figure 2). However, even at very high concentrations of added adaptor (∼500:1, lane 10), formation of the unwanted cDNA self-ligation product remained significant, resulting in a loss of approximately half of the starting cDNA material. Under homogeneous reaction conditions (i.e. all reactants present in the solution phase), mass-action should favour the formation of two products, the desired adaptor–cDNA heterodimer and the adaptor–adaptor homodimer at these high concentrations of added adaptor. However, tethering of the target cDNA to the polystyrene surface of dynabeads creates a heterogeneous reaction environment. The distribution of ligation products may therefore be controlled by mass transfer effects that limit the concentration of adaptor in the solid–liquid interfacial region where the target cDNA is anchored and the reaction must take place. Consequently, adaptor–adaptor and cDNA–cDNA homodimers are produced preferentially, even in the presence of a large excess of the added adaptor.
Figure 2.
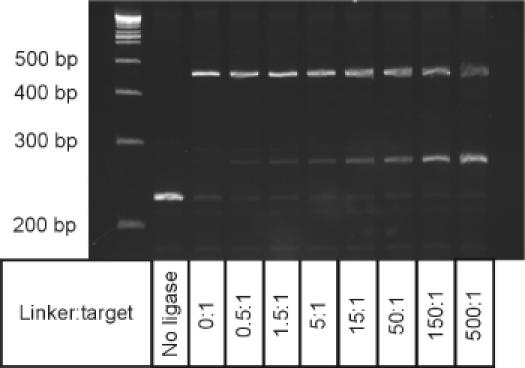
Influence of increasing adaptor:target molar ratios on the formation of adaptor–target heterodimer versus target homodimer. Increasing amounts of adaptor 1 (0–3.8 μM final) were introduced into standard ligation reactions containing 0.075 pmol anchored target in a final volume of 10 μl as described in Materials and Methods. In microSAGE, adaptors are introduced to a reaction mixture containing ∼0.08 pmol anchored target at a final concentration of 0.08 μM in a total volume of 20 μl, corresponding to adaptor:target ratio of approximately 20:1. The classic SAGE protocol introduces a final adaptor concentration of 0.8 μM to the ligation mixture containing ∼1.95 pmol anchored target in a total volume of 40 μl, corresponding to an adaptor: target ratio of ∼16:1.
Improving the yield of adaptor–cDNA heterodimer by increasing the adaptor concentration in the reaction mixture is impractical for large-scale SAGE projects. In addition to the high associated costs of preparing the adaptor, excess adaptor may have deleterious effects on subsequent steps in SAGE. High concentrations of adaptor promote the formation of a large number of adaptor dimers, which can interfere with subsequent PCR amplification steps or necessitate excessive washing of the anchored ligation product to remove unreacted adaptor and adaptor dimers. For this reason, some groups (33,34) have attempted to limit adaptor– dimer contamination of the ditag PCR mixture by reducing the concentration of adaptor used in the adaptor ligation step. However, our results show that lowering the added SAGE adaptor concentration below the standard concentration of 80 nM (i.e. lanes 4 and 5 of Figure 2) results in a significant reduction in the already low yield of the desired adaptor–cDNA hetero-ligation product. As the overall fidelity of SAGE to provide an accurate read of the distribution of transcript abundances will be affected by this sampling loss, there exists a need to develop cheaper and more effective methods to increase the yield of the desired hetero-ligation product by reducing or, better yet, eliminating the formation of self-ligation products.
Addition of macromolecular crowding agents increases the yield of adaptor modified anchored 3′ end cDNA
Other changes in reaction conditions that alter the distribution of ligation products were therefore explored to improve the yield of the desired hetero-ligation product. For example, lowering the reaction temperature can be used to slow the ligation reaction to a point where the rate of mass transfer of the adaptor to the solid–liquid interface no longer limits the formation of the hetero-ligation product. In this case, however, a significantly increased incubation time is required, extending the already lengthy process involved in producing a SAGE library. Varying the rate of mixing during the reaction to decrease the hydrodynamic boundary layer and increase the surface concentration of the free adaptor was explored, but led to only a marginal improvement in the yield of the hetero-ligation product.
Adding co-solutes that act as macromolecular crowding agents (i.e. compaction agents) has been shown to dramatically affect the thermodynamics of reaction mixtures, generally favouring the formation of products with compact conformations and for some proteins, linear rod-like aggregates (35,36). For ligation reactions, addition of 15% (w/v) of the neutral polymer polyethylene glycol (PEG) has been shown to enhance by up to 100-fold the formation of intermolecular ligation products (i.e. linear concatamers) during the ligation of cohesive or blunt-ended DNA fragments in the solution phase (30,31,37). The influence of increased concentrations of PEG-8000 on the formation of the desired hetero-ligation product was therefore examined (Figure 3). At the standard reaction temperature of 16°C and a fixed adaptor concentration of 1 μM (i.e. >10-fold than typically used in microSAGE), increasing the PEG-8000 concentration to 15% (w/v) (lane 3) significantly improved the yield over that obtained at 5% PEG-8000 (lane 2), such that the desired product represents slightly over half the total reaction product. This increase in hetero-ligation product yield in the presence of added PEG was also observed when the standard adaptor concentration (i.e. 80 nM) or a 10-fold higher concentration of anchored cDNA was used.
Figure 3.
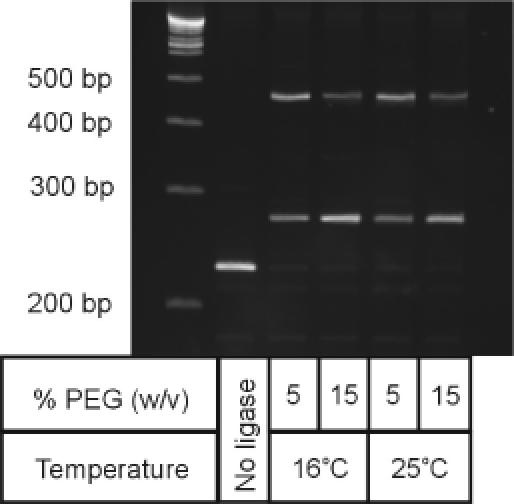
Influence of supplemental PEG-8000 and incubation temperature on the formation of adaptor–target heterodimer versus target homodimer. The standard ligation reaction in the microSAGE protocol is performed in the presence of 5% PEG-8000 (w/v) at 16°C for 2 h using a final adaptor concentration of 0.08 μM in a final volume of 20 μl. Ligation reactions shown were performed in a final volume of 10 μl as described in Materials and Methods using a adaptor concentration of 1 μM final in the presence or absence of PEG-8000 supplemented to a final concentration of 15% (w/v). The reactions were carried out for 2 h under the conditions indicated.
Given that the activity of T4 DNA ligase is higher at 25°C than at the standard 16°C reaction temperature used in microSAGE (38–40), we examined if increasing the rate of the ligation reaction by increasing the incubation temperature could further favour formation of the desired intermolecular ligation product. Moreover, as it is known that there is a temperature dependence to the effects of macromolecular crowding by added PEG (41–43), we also examined the effect of reaction temperature on the distribution of ligation products in the absence and presence of supplemental concentrations of PEG-8000. We observed that the desired product yield was not improved by increasing the reaction temperature to 25°C (lanes 4 and 5) compared to the standard temperature of 16°C (lanes 2 and 3), indicating that rates of formation of the two reaction products show similar temperature dependences. This is in contrast to some reports on the influence of PEG in ligations in solution, where cohesive-end ligations were shown to be enhanced by an increase in temperature (31,41,42,44).
We conclude that PEG-8000 added in moderate concentrations to ligation reactions performed at 16°C can improve hetero-ligation product yields. However, a large excess (50:1 or greater) of added adaptor is required to achieve better than 50% yield. More importantly, complete conversion to the desired hetero-ligation product is not observed at any realistic adaptor concentration.
Product distribution can be directed through the introduction of a restriction enzyme into the ligation reaction—directed ligation chemistry
The inability to adjust ligation conditions such that the hetero-ligation product becomes the only significant reaction product suggests that surface-anchoring of the target cDNA presents kinetic or mass-transfer barriers that cannot be overcome by simple adjustments to the reaction conditions. As the primary problem lay in the inability to offset self-ligation of the target cDNA molecules on the surface, we sought a novel method to limit or prevent formation of this undesired ligation product. Although removal of the 5′-phosphate on the recessed 5′ ends of the anchored 3′ end cDNAs using an appropriate alkaline phosphatase could potentially provide a means to eliminate self-ligation of the anchored 3′ end cDNAs, the efficiency of dephosphorylation by such phosphatases is often much lower for 5′-phosphates on these sites. This, combined with the background nuclease activity of the enzyme that can catalyse digestion of 5′ overhangs, would significantly reduce the overall yield of defined ligation products. In addition, the ligation of SAGE adaptors to such modified targets would lead to the formation of a nicked adaptor–cDNA hetero-ligation product that is inappropriate for further SAGE processing without the introduction of an additional enzymatic step prior to PCR amplification. Another approach would be to use an adaptor with an unphosphorylated 5′ end that would prevent adaptor dimer formation and thereby enhance reactivity by maintaining a large excess of adaptor relative to target cDNA. However, this approach would also result in a nicked strand that requires additional enzymatic steps for processing in SAGE.
As an alternative approach, we considered the effect of adding different amounts of NlaIII to the reaction mixture, with the aim of establishing a more favourable product distribution based on the relative rates of ligation-product formation catalysed by T4 DNA ligase and ligation product cleavage by NlaIII. In the presence of both enzymes, ligation would proceed until a steady-state product profile is reached in the presence of ATP. We observed that the addition of various amounts of NlaIII to a standard ligation reaction containing the SAGE adaptor clearly influenced the product distribution of hetero- versus homo-ligation product (Figure 4A). Titration of a standard ligation reaction containing 0.25 U/μl T4 DNA ligase and 1 μM of adaptor 1 with increasing amounts of NlaIII in the absence of PEG-8000 resulted in a gradual decrease in the amount of the high molecular weight homodimer as well as the desired heterodimer, and a concomitant increase in the amount of unmodified target DNA.
Figure 4.
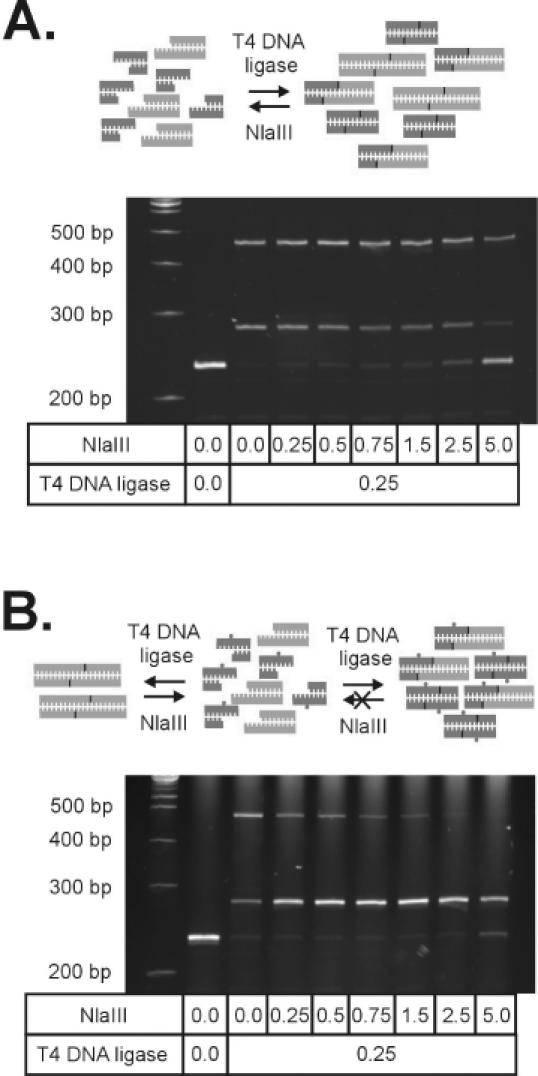
Outline of directed ligation. (A) Ligation of unmethylated adaptors (black) results in the formation of a mixture of adaptor homodimers, target homodimers and the adaptor–target heterodimer. In the presence of NlaIII, ligated products are converted back to their respective monomers. The final product distribution is determined by the relative rates of ligation by T4 DNA ligase and digestion by NlaIII. (B) In contrast, using an adaptor with a methylated base (N6-methyl-deoxy adenosine) within the site of ligation blocks digestion of the adaptor–target heterodimer, and product distribution is favoured towards the formation of the adaptor–target heterodimer. Titrations of T4 DNA ligase with increasing quantities of NlaIII were performed in the presence of 1 μM adaptor in a final volume of 10 μl for 2 h at 37°C as described in Materials and Methods.
Although we were unable to selectively enhance the formation of the hetero-ligation product relative to the undesired cDNA–cDNA homodimer, the results suggested that the competitive actions of NlaIII and T4 DNA ligase could provide an efficient route to complete conversion of the anchored 3′ end cDNA fragments into the desired hetero-ligation product if RE-catalysed digestion of the desired adaptor–cDNA heterodimer could be specifically inhibited. As NlaIII is one of a number of REs sensitive to the presence of a methylated base within its recognition sequence (Table 3), the introduction of a methylated base within the ligation site of the SAGE adaptor could potentially enable the selective inhibition of digestion of the desired ligation product. Through subsequent formation of a hemi-methylated site within the desired adaptor–3′ end cDNA hetero-ligation product, cleavage by NlaIII would be specifically inhibited. In contrast, all cDNA homodimers remain susceptible to NlaIII-catalysed digestion. Because the irreversible formation of the adaptor–cDNA heterodimer would direct formation of the desired product by rapidly digesting any self-ligated cDNA back to its unmodified state, we termed this technique ‘directed ligation’.
Table 3. List of methyl sensitive Type II restriction enzymes that generate overhangs suitable for directed ligation chemistry.
| Enzyme name | Recognition sequencewith cleavage siteab | Methylationsite and typec |
|---|---|---|
| AccI | GT↓MKAC | 5 (6) |
| AclI | AA↓CGTT | 3 (5) |
| AflIII | A↓CRYGT | 2 (4) |
| ApaI | GGGCC↓C | 4 (5) |
| BamHI | G↓GATCC | 5 (4) |
| BanII | GRGCY↓C | 4 (5) |
| BclI | T↓GATCA | 3 (6) |
| BglII | A↓GATCT | 5 (4) |
| BseCI | AT↓CGAT | 5 (6) |
| Bsp106I | AT↓CGAT | 5 (6) |
| BstNI | CC↓WGG | 2 (4) |
| BstYI | R↓GATCY | 5 (4) |
| Bsu15I | AT↓CGAT | 5 (6) |
| Cfr10I | R↓CCGGY | 2 (5) |
| Cfr13I | G↓GNCC | 4 (5) |
| Cfr9I | C↓CCGGG | 2 (4) |
| CfrI | Y↓GGCCR | 4 (5) |
| ClaI | AT↓CGAT | 5 (6) |
| DdeI | C↓TNAG | 1 (5) |
| DraIII | CACNNN↓GTG | 2 (6) |
| EaeI | Y↓GGCCR | 4 (5) |
| EcoO109I | RG↓GNCCY | 5 (5) |
| EcoRI | G↓AATTC | 3 (6) |
| EcoRII | ↓CCWGG | 2 (5) |
| EcoT38I | GRGCY↓C | 4 (5) |
| HapII | C↓CGG | 2 (5) |
| HhaI | GCG↓C | 2 (5) |
| HindIII | A↓AGCTT | 1 (6) |
| HinfI | G↓ANTC | 2 (6) |
| HinP1I | G↓CGC | 2 (5) |
| HpaII | C↓CGG | 2 (5) |
| Hpy188I | TCN↓GA | 5 (6) |
| HpyCH4IV | A↓CGT | 2 (5) |
| KpnI | GGTAC↓C | 4 (6) |
| MseI | T↓TAA | 4 (6) |
| MspI | C↓CGG | 1 (5) |
| MunI | C↓AATTG | 3 (6) |
| MvaI | CC↓WGG | 2 (4) |
| NgoMIV | G↓CCGGC | 2 (5) |
| NlaIII | CATG↓ | 2 (6) |
| NspI | RCATG↓Y | 2 (5) |
| PaeR7I | C↓TCGAG | 5 (6) |
| PspPI | G↓GNCC | 4 (5) |
| PstI | CTGCA↓G | 5 (6) |
| SacI | GAGCT↓C | 4 (5) |
| SalI | G↓TCGAC | 5 (6) |
| Sau3AI | ↓GATC | 4 (5) |
| Sau96I | G↓GNCC | 4 (5) |
| SinI | G↓GWCC | 4 (5) |
| Sse9I | ↓AATT | 2 (6) |
| TaqI | T↓CGA | 4 (6) |
| TfiI | G↓AWTC | 2 (6) |
| Tsp45I | ↓GTSAC | 4 (6) |
| TspRI | CASTGNN↓ | 1 (5) |
| Tth111I | GACN↓NNGTC | 2 (6) |
| VspI | AT↓TAAT | 5 (6) |
| XbaI | T↓CTAGA | 6 (6) |
| XcmI | CCANNNNN↓NNNNTGG | 3 (6) |
| XhoI | C↓TCGAG | 5 (6) |
| XhoII | R↓GATCY | 5 (4) |
This list was extracted from REBASE version 05/2003 (http://rebase.neb.com) and corresponds to commercially available enzymes which have well-characterized methylation sensitivities.
aRecognition sites listed 5′ to 3′ orientation with site of cleavage denoted by ‘↓’.
bDegenerate bases are indicated as follows: R, G or A; Y, C or T; M, A or C; K, G or T; S, G or C; W, A or T; B, not A (C or G or T); D, not C (A or G or T); H, not G (A or C or T); V, not T (A or C or G); and N, A or C or G or T.
cPosition of methylated base in recognition sequence with type of modification in parentheses as follows: 4, N4-methyl-2′-deoxy-cytosine; 5, 5-methyl-2′-deoxy-cytosine; and 6, N6-methyl-2′-deoxy-adenosine.
Near-complete conversion of anchored 3′ end cDNA to adaptor modified products via directed ligation chemistry
The principle of directed ligation chemistry was tested by designing SAGE adaptors with a methylated base (5-methyl-deoxy cytosine or N6-methyl-deoxy adenosine) within the ligation site. Each of these redesigned adaptors was then introduced into the ligation reaction performed in the presence of NlaIII. Substitution of the conventional SAGE adaptor with either modified adaptor had a dramatic effect in the overall distribution profile of ligation products (Figure 4B). When the methylated adaptor 1m6A was used, increasing amounts of NlaIII in the reaction mixture selectively reduced the amount of high molecular weight homodimer corresponding to the target cDNA self-ligation product, such that its formation was vanishingly small at a 20:1 NlaIII to ligase ratio. However, in contrast to the unmodified SAGE adaptor, the formation of the heterodimer increased dramatically as more NlaIII was introduced into the ligation mixture. As a result, very high yields of the desired adaptor–cDNA heterodimer were achieved when mixtures of 10:1 (lane 8) and 20:1 (lane 9) NlaIII to T4 DNA ligase were used. In both cases, the steady-state product distribution profiles shown in Figure 4B were obtained within the first 15 min of reaction. At 5% PEG-8000, a 10:1 enzyme mixture was sufficient enough to inhibit homodimer formation while promoting the formation of the desired heterodimer to the extent observed when PEG was excluded from the ligation mixture.
Although both the 5-methyl-deoxy cytosine and the N6-methyl-deoxy adenosine modified adaptors are extremely effective under directed ligation chemistry, the position of the 5-methyl-deoxy-cytosine base (i.e. adaptor 1m5C) within the recognition sequence of BsmFI would block the activity of this Type IIS enzyme (45), preventing the release of a SAGE tag from the transcript. We therefore employed the N6-methyl-deoxy adenosine modified adaptors for use in the SAGE protocol. Direct comparison of the efficiency of our modified SAGE protocol employing the SAGE 1m6A adaptor to that of the original microSAGE protocol demonstrated a remarkable increase in the yield of adaptor-modified anchored 3′ end cDNA (Figure 5). Under the directed ligation protocol, we achieved near complete conversion of the anchored 3′ end cDNA to the desired adaptor-target DNA hetero-ligation product (lane 9). This is in direct contrast to the <50% yield obtained in the microSAGE protocol when a >10-fold higher adaptor concentration was used (lane 2). We also verified that the N6-methyl-deoxy adenosine modified adaptors could be applied to downstream processing in SAGE by ligating both adaptors 1m6A and 2m6A to anchored 3′ end cDNA and subjecting the resulting ligation products to the remaining steps of the microSAGE protocol (Figure 6). PCR amplification of derived ditags demonstrated that more amplifiable template was present when the directed ligation protocol was employed (lanes 6 and 12) in place of the SAGE protocol employing a >10-fold excess of standard SAGE adaptors (lanes 3 and 9).
Figure 5.
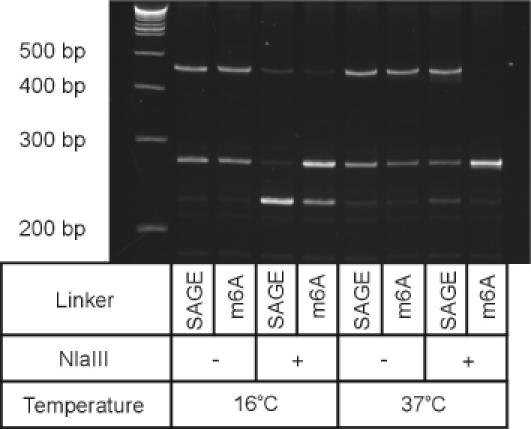
Comparison of ligation under the SAGE protocol versus under directed ligation chemistry. Ligation reactions were performed in 5% PEG-8000 (w/v) in the presence or absence of NlaIII, using standard SAGE adaptors (adaptor 1) or modified SAGE adaptors with an N6-methyl-deoxy-adenosine base (adaptor 1m6A) at a final concentration of 1 μM. The reactions were performed at a final reaction volume of 10 μl and incubated as described in Materials and Methods.
Figure 6.
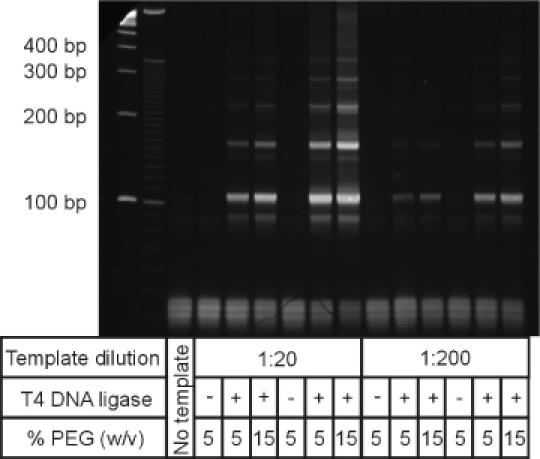
PCR amplification of ditags derived from adaptor-modified anchored 3′ end cDNA obtained using the microSAGE protocol or directed ligation chemistry. After ligation under the microSAGE protocol using ∼10-fold greater amount of adaptors 1 and 2 under standard conditions (lanes 3–5, 9–11) or using adaptors 1m6A and 2m6A using directed ligation chemistry (lanes 6–8, 12–14), tags were released with BsmFI, blunt-ended with Klenow and ligated to form ditags as described under the microSAGE protocol version 1e in the presence (lanes 5, 8, 11 and 14) or absence (lanes 4, 7, 10 and 13) of added PEG-8000 [15% (w/v) final]. After ligation, mixtures were diluted 1:20 (lanes 3–8) or 1:200 in LoTE (lanes 9–14) and 1 μl was used as a template for PCR amplification as described in Materials and Methods.
Thus, directed ligation chemistry appears to provide an extremely efficient route to ensuring complete conversion of anchored 3′ end cDNA to the desired adaptor modified product, and can thereby ameliorate the loss of sample due to self-ligation of the anchored cDNA.
CONCLUSIONS
As the cost of sequencing continues to decrease, SAGE technology will continue to evolve into a powerful, more accessible alternative to microarray technology for the study of gene expression on a global scale. However, for SAGE to provide a truly quantitative picture of global gene expression, it is clear that many of its processing steps require optimization in order to ensure the fidelity of SAGE prior to analysis of the SAGE tag library via sequencing.
We have demonstrated that the efficiency of one critical step in the SAGE protocol, namely the ligation of the SAGE adaptor that permits the use of a Type IIS RE to generate a SAGE tag, is compromised by the tendency for the anchored target to self-ligate. Although optimization of reaction conditions to improve ligation efficiency resulted in a significant improvement in the yield of the desired ligation product, complete conversion could not be achieved. We therefore developed a simple approach termed ‘directed ligation’, that provides near complete conversion into hetero-ligation products, thereby ensuring the fidelity of the transcriptome sample at this step in SAGE analysis.
Finally, given that the ligation of specifically designed adaptors is a fundamental step in many genomic technologies, it is likely that this self-ligation reaction problem is not unique to SAGE. Our directed ligation chemistry may therefore provide a means of improving a range of important functional genomics technologies.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Terry Snutch for the gift of the rat α-factor construct used in this study, as well as members of the laboratory of Dr Jim Kronstad for helpful discussions and use of reagents and equipment. A.P.S. is a Ph.D. candidate in the Individual Interdisciplinary Studies Graduate Program. C.A.H. holds a Senior Canada Research Chair in Interfacial Biotechnology. This researched was supported in part by a grant from the Canadian Institutes of Health Research.
REFERENCES
- 1.Ideker T., Thorsson,V., Ranish,J.A., Christmas,R., Buhler,J., Eng,J.K., Bumgarner,R., Goodlett,D.R., Aebersold,R. and Hood,L. (2001) Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science, 292, 929–934. [DOI] [PubMed] [Google Scholar]
- 2.Velculescu V.E., Zhang,L., Vogelstein,B. and Kinzler,K.W. (1995) Serial analysis of gene expression. Science, 270, 484–487. [DOI] [PubMed] [Google Scholar]
- 3.Adams M.D. (1996) Serial analysis of gene expression: ESTs get smaller. BioEssays, 18, 261–262. [DOI] [PubMed] [Google Scholar]
- 4.Schena M., Shalon,D., Davis,R.W. and Brown,P.O. (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science, 270, 467–470. [DOI] [PubMed] [Google Scholar]
- 5.Epstein C.B. and Butow,R.A. (2000) Microarray technology—enhanced versatility, persistent challenge. Curr. Opin. Biotechnol., 11, 36–41. [DOI] [PubMed] [Google Scholar]
- 6.Lipshutz R.J., Fodor,S.P., Gingeras,T.R. and Lockhart,D.J. (1999) High density synthetic oligonucleotide arrays. Nat. Genet., 21, 20–24. [DOI] [PubMed] [Google Scholar]
- 7.Lercher M.J., Urrutia,A.O. and Hurst,L.D. (2002) Clustering of housekeeping genes provides a unified model of gene order in the human genome. Nat. Genet., 31, 180–183. [DOI] [PubMed] [Google Scholar]
- 8.Szybalski W., Kim,S.C., Hasan,N. and Podhajska,A.J. (1991) Class-IIS restriction enzymes—a review. Gene, 100, 13–26. [DOI] [PubMed] [Google Scholar]
- 9.Saha S., Sparks,A.B., Rago,C., Akmaev,V., Wang,C.J., Vogelstein,B., Kinzler,K.W. and Velculescu,V.E. (2002) Using the transcriptome to annotate the genome. Nat. Biotechnol., 20, 508–512. [DOI] [PubMed] [Google Scholar]
- 10.van den Berg A., van der Leij,J. and Poppema,S. (1999) Serial analysis of gene expression: rapid RT–PCR analysis of unknown SAGE tags. Nucleic Acids Res., 27, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Velculescu V.E., Vogelstein,B. and Kinzler,K.W. (2000) Analysing uncharted transcriptomes with SAGE. Trends Genet., 16, 423–425. [DOI] [PubMed] [Google Scholar]
- 12.Velculescu V.E., Zhang,L., Zhou,W., Vogelstein,J., Basrai,M.A., Bassett,D.E.,Jr, Hieter,P., Vogelstein,B. and Kinzler,K.W. (1997) Characterization of the yeast transcriptome. Cell, 88, 243–251. [DOI] [PubMed] [Google Scholar]
- 13.Lee S., Chen,J., Zhou,G. and Wang,S.M. (2001) Generation of high-quantity and quality tag/ditag cDNAs for SAGE analysis. BioTechniques, 31, 348–350, 352–344. [DOI] [PubMed] [Google Scholar]
- 14.Caron H., van Schaik,B., van der Mee,M., Baas,F., Riggins,G., van Sluis,P., Hermus,M.C., van Asperen,R., Boon,K., Voute,P.A. et al. (2001) The human transcriptome map: clustering of highly expressed genes in chromosomal domains. Science, 291, 1289–1292. [DOI] [PubMed] [Google Scholar]
- 15.Munasinghe A., Patankar,S., Cook,B.P., Madden,S.L., Martin,R.K., Kyle,D.E., Shoaibi,A., Cummings,L.M. and Wirth,D.F. (2001) Serial analysis of gene expression (SAGE) in Plasmodium falciparum: application of the technique to A-T rich genomes. Mol. Biochem. Parasitol., 113, 23–34. [DOI] [PubMed] [Google Scholar]
- 16.Boheler K.R. and Stern,M.D. (2003) The new role of SAGE in gene discovery. Trends Biotechnol., 21, 55–57. [DOI] [PubMed] [Google Scholar]
- 17.Chen J., Sun,M., Lee,S., Zhou,G., Rowley,J.D. and Wang,S.M. (2002) Identifying novel transcripts and novel genes in the human genome by using novel SAGE tags. Proc. Natl Acad. Sci. USA, 99, 12257–12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colinge J. and Feger,G. (2001) Detecting the impact of sequencing errors on SAGE data. Bioinformatics, 17, 840–842. [DOI] [PubMed] [Google Scholar]
- 19.Stern M.D., Anisimov,S.V. and Boheler,K.R. (2003) Can transcriptome size be estimated from SAGE catalogs? Bioinformatics, 19, 443–448. [DOI] [PubMed] [Google Scholar]
- 20.Stollberg J., Urschitz,J., Urban,Z. and Boyd,C.D. (2000) A quantitative evaluation of SAGE. Genome Res., 10, 1241–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Powell J. (1998) Enhanced concatemer cloning—a modification to the SAGE (Serial Analysis of Gene Expression) technique. Nucleic Acids Res., 26, 3445–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Datson N.A., van der Perk-de Jong,J., van den Berg,M.P., de Kloet,E.R. and Vreugdenhil,E. (1999) MicroSAGE: a modified procedure for serial analysis of gene expression in limited amounts of tissue. Nucleic Acids Res., 27, 1300–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kenzelmann M. and Muhlemann,K. (1999) Substantially enhanced cloning efficiency of SAGE (Serial Analysis of Gene Expression) by adding a heating step to the original protocol. Nucleic Acids Res., 27, 917–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Angelastro J.M., Klimaschewski,L.P. and Vitolo,O.V. (2000) Improved NlaIII digestion of PAGE-purified 102 bp ditags by addition of a single purification step in both the SAGE and microSAGE protocols. Nucleic Acids Res., 28, e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ye S.Q., Zhang,L.Q., Zheng,F., Virgil,D. and Kwiterovich,P.O. (2000) miniSAGE: gene expression profiling using serial analysis of gene expression from 1 microg total RNA. Anal. Biochem., 287, 144–152. [DOI] [PubMed] [Google Scholar]
- 26.Margulies E.H., Kardia,S.L. and Innis,J.W. (2001) Identification and prevention of a GC content bias in SAGE libraries. Nucleic Acids Res., 29, e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mathupala S.P. and Sloan,A.E. (2002) ‘In-gel’ purified ditags direct synthesis of highly efficient SAGE Libraries. BMC Genomics, 3, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Damgaard Nielsen M., Millichip,M. and Josefsen,K. (2003) High-performance liquid chromatography purification of 26-bp serial analysis of gene expression ditags results in higher yields, longer concatemers, and substantial time savings. Anal. Biochem., 313, 128–132. [DOI] [PubMed] [Google Scholar]
- 29.Harrison B. and Zimmerman,S.B. (1984) Polymer-stimulated ligation: enhanced ligation of oligo- and polynucleotides by T4 RNA ligase in polymer solutions. Nucleic Acids Res., 12, 8235–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayashi K., Nakazawa,M., Ishizaki,Y., Hiraoka,N. and Obayashi,A. (1986) Regulation of inter- and intramolecular ligation with T4 DNA ligase in the presence of polyethylene glycol. Nucleic Acids Res., 14, 7617–7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pheiffer B.H. and Zimmerman,S.B. (1983) Polymer-stimulated ligation: enhanced blunt- or cohesive-end ligation of DNA or deoxyribooligonucleotides by T4 DNA ligase in polymer solutions. Nucleic Acids Res., 11, 7853–7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sambrook J. and Russell,D.W. (2001) Molecular Cloning: A Laboratory Manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 33.Virlon B., Cheval,L., Buhler,J.M., Billon,E., Doucet,A. and Elalouf,J.M. (1999) Serial microanalysis of renal transcriptomes. Proc. Natl Acad. Sci. USA, 96, 15286–15291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Angelastro J.M., Klimaschewski,L., Tang,S., Vitolo,O.V., Weissman,T.A., Donlin,L.T., Shelanski,M.L. and Greene,L.A. (2000) Identification of diverse nerve growth factor-regulated genes by serial analysis of gene expression (SAGE) profiling. Proc. Natl Acad. Sci. USA, 97, 10424–10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hall D. and Minton,A.P. (2003) Macromolecular crowding: qualitative and semiquantitative successes, quantitative challenges. Biochim. Biophys. Acta, 1649, 127–139. [DOI] [PubMed] [Google Scholar]
- 36.Zimmerman S.B. and Minton,A.P. (1993) Macromolecular crowding: biochemical, biophysical, and physiological consequences. Annu. Rev. Biophys. Biomol. Struct., 22, 27–65. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi K., Nakazawa,M., Ishizaki,Y. and Obayashi,A. (1985) Influence of monovalent cations on the activity of T4 DNA ligase in the presence of polyethylene glycol. Nucleic Acids Res., 13, 3261–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu D.Y. and Wallace,R.B. (1989) Specificity of the nick-closing activity of bacteriophage T4 DNA ligase. Gene, 76, 245–254. [DOI] [PubMed] [Google Scholar]
- 39.Pohl F.M., Thomae,R. and Karst,A. (1982) Temperature dependence of the activity of DNA-modifying enzymes: endonucleases and DNA ligase. Eur. J. Biochem., 123, 141–152. [DOI] [PubMed] [Google Scholar]
- 40.Faulhammer D., Lipton,R.J. and Landweber,L.F. (2000) Fidelity of enzymatic ligation for DNA computing. J. Comput. Biol., 7, 839–848. [DOI] [PubMed] [Google Scholar]
- 41.Louie D. and Serwer,P. (1991) Effects of temperature on excluded volume-promoted cyclization and concatemerization of cohesive-ended DNA longer than 0.04-Mb. Nucleic Acids Res., 19, 3047–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Louie D. and Serwer,P. (1994) Quantification of the effect of excluded-volume on double-stranded DNA. J. Mol. Biol., 242, 547–558. [DOI] [PubMed] [Google Scholar]
- 43.Murphy L.D. and Zimmerman,S.B. (1995) Condensation and cohesion of lambda-dna in cell-extracts and other media—implications for the structure and function of DNA in prokaryotes. Biophys. Chem., 57, 71–92. [DOI] [PubMed] [Google Scholar]
- 44.Hayashi K., Nakazawa,M., Ishizaki,Y., Hiraoka,N. and Obayashi,A. (1985) Stimulation of intermolecular ligation with E. coli DNA ligase by high concentrations of monovalent cations in polyethylene glycol solutions. Nucleic Acids Res., 13, 7979–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roberts R.J., Vincze,T., Posfai,J. and Macelis,D. (2003) REBASE: restriction enzymes and methyltransferases. Nucleic Acids Res., 31, 418–420. [DOI] [PMC free article] [PubMed] [Google Scholar]