

Abstract
Loss of DNA mismatch repair (MMR) function leads to the development and progression of certain cancers. Currently, assays for DNA MMR activity involve the use of cell extracts and are technically challenging and costly. Here, we report a rapid, less labor-intensive method that can quantitatively measure MMR activity in live cells. A G–G or T–G mismatch was introduced into the ATG start codon of the enhanced green fluorescent protein (EGFP) gene. Repair of the G–G or T–G mismatch to G–C or T–A, respectively, in the heteroduplex plasmid generates a functional EGFP gene expression. The heteroduplex plasmid and a similarly constructed homoduplex plasmid were transfected in parallel into the same cell line and the number of green cells counted by flow cytometry. Relative EGFP expression was calculated as the total fluorescence intensity of cells transfected with the heteroduplex construct divided by that of cells transfected with the homoduplex construct. We have tested several cell lines from both MMR-deficient and MMR-proficient groups using this method, including a colon carcinoma cell line HCT116 with defective hMLH1 gene and a derivative complemented by transient transfection with hMLH1 cDNA. Results show that MMR-proficient cells have significantly higher EGFP expression than MMR-deficient cells, and that transient expression of hMLH1 alone can elevate MMR activity in HCT116 cells. This method is potentially useful in comparing and monitoring MMR activity in live cells under various growth conditions.
INTRODUCTION
The mismatch repair (MMR) system has been well studied in prokaryotes, especially in Escherichia coli (1–3). Nucleotides misincorporated during DNA replication that escape the proofreading activity of DNA polymerase can be recognized by the MutS protein. Binding of MutS to the mismatch site leads to recruitment of other proteins, including MutL and MutH, and triggers a series of enzymatic reactions resulting in the removal of the misincorporated nucleotides. The MMR system appears more complicated in humans than in E.coli (3–5). Multiple homologs of MutS and MutL have been identified and cloned in human. Three important MutS homologs are hMSH2, hMSH6 and hMSH3. hMSH2 can heterodimerize with either hMSH6 or hMSH3, forming two different protein complexes, designated hMutSα or hMutSβ, respectively (6–8). Several MutL homologs including hMLH1, hPMS1, hPMS2 and hMLH3 have been shown to form heterodimers, such as hMLH1-hPMS2 (hMutLα), hMLH1-hPMS1 (hMutLβ) and hMLH1-hMLH3 (9–12).
MMR defects have been strongly associated with certain types of cancer, especially hereditary non-polyposis colorectal cancer (HNPCC) and sporadic colorectal cancer (13,14). More than 70% of HNPCC patients have been found to have germline mutations in either the hMSH2 or the hMLH1 gene, and a small percentage have defective hPMS2 or hPMS1 gene (15). More recently, germline mutations of hMSH6 were identified in patients with hereditary colorectal cancer (16,17). Cancer cells from most HNPCC patients show a phenotype of replication error (RER+) or microsatellite instability (MSI+) with a high frequency of mutations in microsatellite sequences. While it is widely believed that MSI is a marker for defective MMR, a significant portion of patients with MSI+ cancers do not have mutations of known MMR genes (13). In fact, several studies have shown that the most common mechanism causing MSI in sporadic colon cancer is transcriptional silencing of the hMLH1 gene by methylation of the hMLH1 gene promoter (18–21). A limited number of studies have also reported changes of expression of MMR genes or MMR activity during cell cycle progression, by growth factor stimulation, or under different growth conditions (22–24). While these studies suggest that MMR activity may be regulated, it is not clear what physiological or pathological conditions might result in reduced MMR activity in various types of cells. Furthermore, few studies have addressed the significance of altered MMR activity in carcinogenesis. One roadblock in addressing these issues appears to be the lack of a simple and effective assay to compare and monitor MMR activity in various types of live cells under various physiological and pathological conditions.
In this study, we have devised an assay utilizing the reversion of a modified start codon of the enhanced green fluorescent protein (EGFP) gene to quantitatively measure MMR activity in live cells. This approach has previously been used for measuring other types of DNA repair (25,26). Our results indicate that this assay can effectively distinguish MMR− cells from MMR+ cells. Also, using this method, we show that ectopic expression of hMHL1 in HCT116 cells significantly elevates MMR activity.
MATERIALS AND METHODS
Modification of an EGFP expression plasmid
Two modifications were introduced to plasmid pEGFP-N1 (Clontech). First, a DNA fragment (230 bp) encompassing the SV40 replication origin was deleted from pEGFP-N1 by digesting the plasmid with StuI and SexAI, purifying the large fragment, filling in the ends with Klenow (New England Biolabs) and circularizing the large fragment with ligase. Second, two restriction enzyme sites, EcoRV and NruI, were engineered around the start codon ATG of EGFP gene using a PCR-based site-directed mutagenesis method (27). Briefly, two primers, 5′-ggtggcgaccgggatatcccgggccc-3′ and 5′-atggtgagcaatcgcgaggagctgttc-3′, were designed in such a way that they can anneal to the adjacent positions on opposite strands of pEGFP-N1. The underlined sequences are the introduced restriction sites, EcoRV and NruI, respectively. The PCR reaction contained 20 pmol of both primers, 59 ng pEGFP-N1, 50 nmol dNTPs and 2.5 U cloned Pfu polymerase (Strategene) in 1× cloned pfu buffer. After 20 cycles (94°C for 30 s, 72°C for 13 min), the PCR product was purified by phenol/chloroform extraction and ethanol precipitation, and then dissolved in 20 μl T4 ligase buffer with 400 U of T4 ligase (New England Biolabs). The ligation reaction was incubated at 16°C for 2 h and then heated at 65°C for 20 min to inactivate T4 ligase. DNA was precipitated by ethanol, re-dissolved in 20 μl DpnI buffer and digested with 20 U DpnI (New England Biolabs) for 2 h to destroy the methylated, wild-type DNA template. One microliter of the digestion mixture was used to transform an E.coli strain, JM110, and several transformed clones were expanded. The DNA prepared from these clones was first checked with restriction analyses and then sequenced. We selected one clone with the desired mutations and named it p95-1.
Construction of heteroduplex and homoduplex plasmids
The p95-1 (3 μg) was digested with ApaI (Life Technologies), whose site is within the multiple cloning site of pEGFP-N1, and NruI (New England Biolabs) (Figure 1). The larger fragment was purified from an agarose gel with NA45 membrane (Schleicher & Schuell Inc). Purified DNA was equally divided into three aliquots and subjected to three parallel ligations to generate a T–G or G–G mismatched heteroduplex plasmid, a homoduplex plasmid and a negative control. To construct the T–G heteroduplex plasmid, two oligonucleotides, EGFP3-11 (5′-cgggatccaccggtcgccaccatggtgagcaatcg-3′) and EGFP3-12 (5′-cgattgctcaccgtggtggcgaccggtggatcccgggcc-3′) were phosphorylated by T4 polynucleotide kinase. The phosphorylated oligonucleotides (20 pmol each) were then mixed together in 20 μl T4 ligase buffer, heated at 92°C for 3 min, and allowed to slowly cool down to room temperature, forming a double-stranded oligonucleotide with a T–G mismatch. Of the oligonucleotide mixture, 2 μl was mixed with 1 μg of p95-1 larger fragment, 5 μl ligase buffer, 42 μl H2O and 400 U T4 ligase, and incubated at 16°C for 10 min. The reaction was then further diluted by 4-fold with T4 ligase buffer and was kept at 16°C overnight. A G–G heteroduplex plasmid was constructed by replacing the oligonucleotide EGFP3-12 in the T–G mismatch construction with oligonucleotide EGFP3-15 (5′-cgattgctcacgatggtggcgaccggtggatcccgggcc-3′). The homoduplex plasmid was constructed in the same way as the heteroduplex plasmid except that the oligonucleotide EGFP3-12 was replaced with oligonucleotide EGFP3-13 (5′-cgattgctcaccatggtggcgaccggtggatcccgggcc-3′). For the negative control, the ligation reaction was carried out in the absence of any primer.
Figure 1.

Flow chart of protocol for the quantitation of DNA MMR in vivo. MMR efficiency as measured with relative EGFP expression was calculated using the equation of (M · IM − N · IN)/(C · IC − N · IN), where M, N or C is the percentage of green cells for heteroduplex, negative control or homoduplex transfection, respectively, and IM, IN or IC is the mean fluorescence intensity of green cells for M, N or C, respectively. PSAD stands for plasmid-safe ATP-dependent DNase.
After ligation, DNA was precipitated with ethanol. To destroy the residual parental p95-1 plasmid that might not be separated from the p95-1 larger fragment, we subjected the precipitated DNA to EcoRV digestion. Since the linear p95-1 large fragment and the circularized heteroduplex plasmid in the ligation mixture may undergo homologous recombination after being transfected into cells, we also digested the precipitated DNA with the plasmid-safe ATP-dependent DNase (PSAD) from Epicenter. Precipitated DNA from all three ligation reactions was resuspended in 50 μl PSAD buffer containing 20 U PSAD and 5 U EcoRV. After 30 min of digestion at 37°C, the reactions were heated at 80°C for another 30 min to inactivate the enzymes. Before transfection, a small aliquot of the reaction was subjected to electrophoresis in agrose gel to estimate DNA concentration.
Construction of nicked heteroduplex and homoduplex plasmids
The CMV promoter- EGFP gene cassette in pEGFP-N1 was subcloned into pGEM5Z(+) (Promega). This plasmid, pGEM5Z(+)-EGFP, was used to generate single-stranded circular DNA (ssDNA) containing the coding strand of EGFP with a helper phage (New England Biolab) according to the manufacturer's instruction. The ssDNA was annealed to BstXI-linearized pGEM5Z(+)-EGFP in 1.5-fold excess molar ratio to generate a homoduplex plasmid with a nick in the EGFP template strand located 692 bp 3′ upstream from the start codon. To generate a nicked heteroduplex plasmid with a G–G mismatch, we mutated the start codon in the EGFP template strand from 3′ TAC to 3′ TAG. Annealing of the ssDNA containing the coding strand of the wild-type EGFP with BstXI-linearized, mutated pGEM5Z(+)-EGFP produced a G–G mismatch at the start codon of EGFP with a nick located 694 bp 3′ upstream from the mismatch. After annealing, the mixture was digested with PSAD to degrade linearized DNA and ssDNA.
Cell lines and culture
The HCT116 human colon carcinoma cell line was kindly provided by Dr Michael Brattain and cultured in McCoy's 5A medium supplemented with bovine pancreas insulin, human transferrin, epidermal growth factor, pyruvate, vitamins, amino acids and antibiotics (28). LoVo, SW480 and HeLa were originally obtained from American Type Culture Collection. These four cell lines were adapted to McCoy's 5A medium supplemented with 10% fetal bovine serum (FBS), pyruvate, vitamins, amino acids and antibiotics (29). AA8, CHO EM9 and 46BR.1G1 cell lines were also cultured in this medium. HCT116+ch3 and HCT116+ch5 cell lines were kindly provided by Dr Minoru Koi. HCT116+ch3 was cultured in DMEM medium supplemented with 10% FBS and 325 μg/ml active G418 (Life Technologies). HCT116+ch5 was cultured in DMEM supplemented with 10% fetal calf serum and 6 μg/ml Blasticidin S (Calbiochem). Working cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 and routinely checked for mycoplasma contamination.
Transfection
Lipofectamine and lipofectamine PLUS reagents (Life Technologies) were used to transfect DNA into cultured cells. Cells were plated on 60 mm dishes at densities ranging from 0.4 × 106 to 1 × 106 cells/dish depending on the cell type. Transfections were performed the following day according to the protocols from the manufacturer. In some experiments, a red fluorescent protein (RFP) expression plasmid (1 μg), pDsRed1-N1 (Clontech), was co-transfected with the homoduplex or heteroduplex EGFP plasmid (0.75 μg) to ascertain that the transfection efficiency of the two kinds of EGFP plasmid was similar. The cells were trypsinized 24 h after transfection, and resuspended in PBS at a concentration of 0.5 × 106 cells/ml. In our preliminary experiments, 24 h incubation yielded higher relative EGFP expression in HeLa cells than a shorter incubation period such as 8, 12 or 16 h. The number of green cells and the intensity of the fluorescence were then determined by flow cytometry.
Flow cytometry
From each transfection, 30 000 cells were counted and their green fluorescence intensity at 530 ± 30 nm wavelength was measured using FACSCalibur (Becton Dickinson). The laser is a 15 mW, 488 nm, air-cooled argon-ion laser. The marker to identify positive cells was set in such a way that 0.2% cells transfected with the negative control DNA were considered as positive.
Calculation of relative EGFP expression and statistical analyses
MMR efficiency in each cell line was measured with relative EGFP expression. It was calculated according to the formula, relative EGFP expression = (M · IM − N · IN)/(C · IC − N · IN), where M, N and C are the percentages of green cells for heteroduplex, negative control and homoduplex transfection, respectively. IM, IN and IC are the mean fluorescence intensities of positive cells for M, N and C, respectively. N·IN was omitted in the calculation when it is negligible. When the red fluorescent protein expression plasmid was co-transfected with the heteroduplex or homoduplex plasmid, the percentage of red cells was used in the calculation to normalize transfection efficiency. Since the same amount of heteroduplex or homoduplex plasmid is transfected into each cell line, the maximal value of relative EGFP expression should be 100%. When comparing relative EGFP expression between two groups, we used one-tailed student t-test. When comparing multiple groups, we used ANOVA and Newman–Keuls test (30).
Western blot
Cells recovered from the flow cytometer were rinsed twice with ice-cold PBS and lysed in 50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 0.02% sodium azide, 100 μg/ml phenylmethylsulfonyl fluoride and 1% Nonidet P-40. Equal amounts of proteins (50 μg of each extract) were separated by SDS–PAGE and transferred to a nitrocellulose membrane (Amersham Corp.). Membranes were blocked in TBST [100 mM Tris–HCl (pH 8.0), 150 mM NaCl, 0.05% Tween-20] containing 5% nonfat powder milk, and then incubated with mouse monoclonal anti-hMLH1 antibody (Oncogene Science) at a final concentration of 1.0 μg/ml. After three washes with TBST, the membrane was incubated with horseradish peroxidase (HRP)-conjugated secondary antibody at 1:1000 dilution (New England Biolabs) and washed again. Antigen–antibody complexes were detected by chemiluminescence according to the manufacturer's instructions (New England Biolabs). Following the same procedure, living colors DsRed monoclonal antibody (Clontech) was used to detect the red fluorescent protein, whereas living colors A.v. peptide antibody–HRP conjugate (Clontech) was used to detect green fluorescent protein.
RESULTS
Description of the method
The method used to measure DNA mismatch repair activity in vivo is shown schematically in Figure 1. In comparison with the parental plasmid pEGFP-N1, p95-1 does not have an SV40 replication origin, preventing its replication in cells expressing SV40 large T antigen. Moreover, it has two more restriction enzyme sites, EcoRV and NruI, flanking the start codon of EGFP. After digestion with ApaI and NruI, the larger fragment (gapped plasmid) was isolated by gel purification. The gapped plasmid was then incubated in three parallel ligation reactions. In the first reaction, the gapped plasmid was ligated to a heteroduplex oligonucleotide that is identical to the small ApaI/NruI DNA fragment except that it has a T–G mismatch and no EcoRV site. In the second reaction, the gapped plasmid was ligated to a homoduplex oligonucleotide that has no EcoRV site. No oligonucleotide was added to the third reaction. The ligation mixtures were then subjected to digestion by EcoRV and PSAD DNase. EcoRV linearized any residual parental plasmid p95-1. PSAD DNase was used to digest linear DNA molecules so that the possibility of homologous recombination after transfection between the gapped plasmid and the ligated, heteroduplex circular plasmid to generate wild-type EGFP sequence was eliminated.
To confirm the formation of circular homoduplex and heteroduplex plasmids, the ligation products were digested with NcoI restriction endonuclease. There are three NcoI restriction sites throughout p95-1, one of which encompasses the start codon of EGFP. After NcoI digestion and agarose gel electrophoresis, the homoduplex plasmid should yield 3 bands of 2285 bp, 1904 and 317 bp, while the heteroduplex plasmid should only yield 2 bands of 2602 bp and 1904 bp because NcoI cannot recognize and cut the mismatched site at the start codon. As shown in Figure 2A, NcoI digested the homoduplex plasmid into two larger fragments (2.3 and 1.9 kb), which were visible, and one smaller fragment (317 bp), which was not visible after photography. On the other hand, NcoI digestion of the ligation product of the gapped plasmid and the mismatched oligonucleotide only generated two fragments of 2.6 and 1.9 kb confirming the formation of the heteroduplex plasmid.
Figure 2.
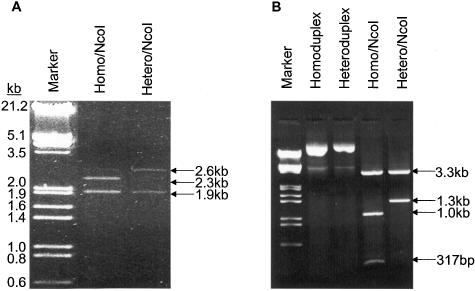
Restriction analysis of ligation products. (A) Digestion of un-nicked homoduplex and heteroduplex plasmids with NcoI. The ligation products was digested with NcoI endonuclease and then electrophoresed in 1% agarose gel. The homoduplex plasmid was digested into three fragments of 2.3, 1.9 and 317 bp. The ethidium bromide staining of the 317 bp fragment was too weak to be photographed. A faint band at 2.6 kb is likely the incompletely digested product that generates 2.3 and 317 bp fragments. The heteroduplex plasmid was digested into two fragments of 2.6 and 1.9 kb. (B) Digestion of nicked homoduplex and heteroduplex plasmids with NcoI. Homoduplex was digested into three fragments of 3.3, 1.0 and 317 bp, whereas the heteroduplex was digested into two fragments of 3.3 and 1.3 kb.
To compare DNA MMR activity measured with a circular heteroduplex plasmid versus a nicked, circular heteroduplex plasmid, we also constructed nicked, circular homoduplex and heteroduplex plasmids as described in the Materials and Methods. Similar to the intact, circular plasmids, digestion of the nicked, circular homoduplex plasmid with NcoI generated three expected fragments, 3.3, 1.0 and 317 bp, whereas digestion of the nicked, circular heteroduplex plasmid only generated two fragments, 3.3 and 1.3 kb (Figure 2B).
MMR+ cell lines showed significantly higher relative EGFP expression than MMR− cell lines
To determine whether the heteroduplex plasmid can be used to measure DNA MMR efficiency, we compared relative EGFP expression in two MMR deficient cell lines HCT116 and LoVo, and two MMR proficient cell lines HeLa and SW480. HCT116 cells harbor a nonsense mutation in exon 9 in hMLH1 gene (31–33). LoVo does not express hMSH2 protein (7,34). Nicked or un-nicked homoduplex or heteroduplex EGFP plasmid was co-transfected with RFP plasmid into the cells. Green and red fluorescent images of the transfected cells were taken before the cells were lifted and analyzed with flow cytometry. As shown in Figure 3A, EGFP was largely co-expressed with RFP in HeLa cells transfected with either homo- or heteroduplex EGFP plasmid. In contrast, many of RFP-expressing HCT116 cells did not show EGFP expression when transfected with the heteroduplex EGFP plasmid while the cells co-transfected with the homoduplex EGFP plasmid and RFP plasmid co-expressed the two fluorescent proteins. Similarly, flow cytometry analysis of HeLa and HCT116 cells after homoduplex or heteroduplex plasmid transfection showed that while the EGFP positive populations of HeLa cells transfected with either homo- or heteroduplex were very similar, the EGFP positive population of HCT116 cells transfected with the heteroduplex was considerably lower than that of HCT116 cells transfected with the homoduplex indicating a deficiency in MMR (Figure 3B). The percentage of gated EGFP- and RFP-positive cells and mean fluorescence intensity of EGFP-positive cells from the flow cytometry analysis in Figure 3B are presented in Table 1 for the calculation of relative EGFP expression. Repeated and independent analyses with flow cytometry showed that the relative EGFP expression of the two MMR+ cell lines, HeLa and SW480, was significantly (P < 0.001) higher than that of the two MMR− cell lines, HCT116 and LoVo (Figure 4A). To confirm that the lower number of green cells and reduced intensity of green fluorescence in MMR- cells was due to lower expression levels of EGFP, we quantified EGFP and RFP protein levels in HCT116 and HeLa cells that were transfected with nicked homoduplex or heteroduplex EGFP plasmid by western blotting. The EGFP level in the heteroduplex plasmid-transfected HCT116 cells was noticeably lower than that in the homoduplex plasmid-transfected HCT116 cells (Figure 4B). In contrast, the EGFP expression levels in the homoduplex and heteroduplex plasmid-transfected HeLa cells were very similar. Density analysis of the EGFP bands indicated that the EGFP expression from the heteroduplex plasmid was about a quarter of that from the homoduplex plasmid in HCT116 cells (Figure 4C).
Figure 3.
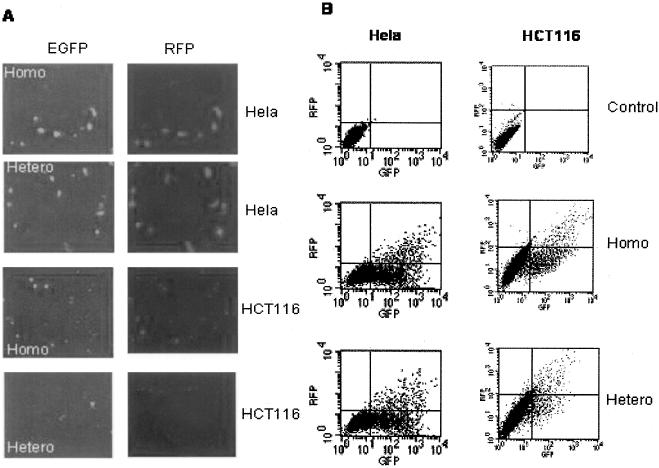
Heteroduplex EGFP plasmid is effectively repaired in HeLa cell, but not in HCT116 cell. (A) Fluorescent images of HeLa and HCT116 cells after co-transfection of nicked homo- or heteroduplex EGFP plasmid and RFP plasmid (pDsRed1-N1). (B). A typical flow cytometry data set of HeLa and HCT116 cells after co-transfection with nicked homo- or heteroduplex EGFP plasmid and RFP plasmid.
Table 1. Calculation of relative EGFP expression using flow cytometry data corresponding to Figure 3B.
| Quadrant | Control % Gated | Mean | Homoduplex % Gated | Mean | Heteroduplex % Gated | Mean | Relative EGFP expressiona | |
|---|---|---|---|---|---|---|---|---|
| HeLa | UL | 0.03 | 9.37 | 0.13 | 9.36 | 0.14 | 9.44 | 1829.99/1918.87 = 0.954 |
| UR | 0.02 | 32.19 | 4.46 | 751.36 | 3.73 | 672.30 | ||
| LL | 99.95 | 3.43 | 72.44 | 5.37 | 77.44 | 5.31 | ||
| LR | 0.00 | *** | 22.97 | 237.55 | 18.70 | 244.62 | ||
| HCT116 | UL | 0.03 | 6.35 | 0.19 | 10.81 | 0.36 | 11.77 | 234.69/1353.42 = 0.173 |
| UR | 0.01 | 793.45 | 1.64 | 766.25 | 1.59 | 155.75 | ||
| LL | 99.95 | 3.20 | 89.62 | 4.90 | 94.55 | 4.70 | ||
| LR | 0.01 | 20.82 | 8.54 | 142.87 | 3.50 | 60.00 |
aEGFP expression from homoduplex or heteroduplex plasmid transfection was calculated using the following equation.
![]()
Relative EGFP expression = EGFP expression from heteroduplex/EGFP expression from homoduplex.
Figure 4.
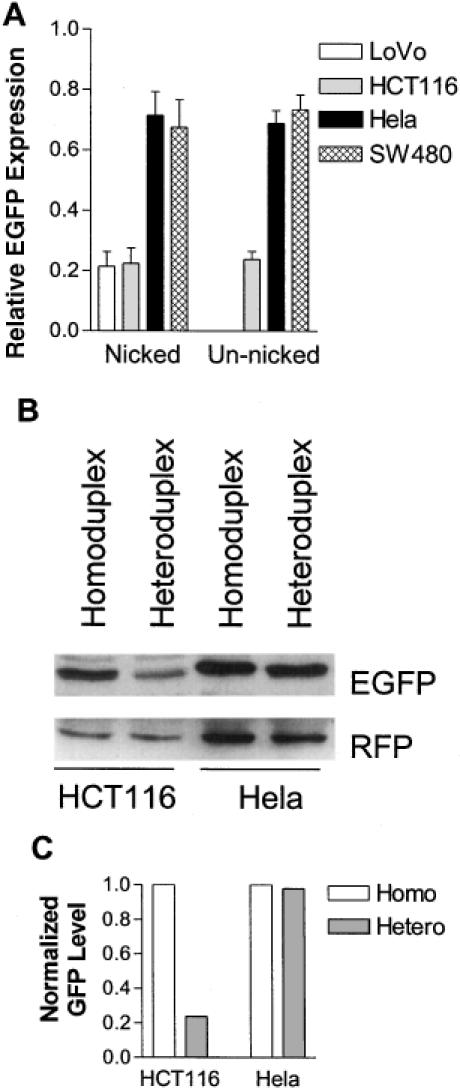
MMR+ cell lines showed significantly higher relative EGFP expression than MMR− cell lines. (A) Nicked or un-nicked homo- or heteroduplex (with a G–G mismatch) plasmids were transfected into the cell lines depicted. Percentage of green cells and their mean intensity from each transfection were obtained from flow cytometry, and relative EGFP expression was calculated as described in Materials and Methods. Each column represents mean ± SEM from five independent measurements for HCT116 and HeLa, and from three independent measurements for LoVo and SW480. (B) HCT116 and HeLa cells were co-transfected with nicked homoduplex or heteroduplex EGFP plasmid and RFP plasmid. Twenty-four hours later, the cells were harvested and western blot was done as described in Materials and Methods. (C) The density of EGFP bands was measured with Image-Pro Plus software (Media Cybernetics), normalized with that of the corresponding RFP bands, and plotted in an arbitrary unit.
Relative EGFP expression from a nicked heteroduplex was similar to that from an un-nicked heteroduplex in live cells
Since a single-strand nick was shown to be necessary for strand-specific repair of a mismatched base pair in nuclear extracts of human cells in vitro (35), we tested whether this is also the case in live cells. A homoduplex or heteroduplex EGFP plasmid with a single-strand nick was constructed such that the nick was placed in the template strand of EGFP. Preferential repair of the G–G mismatch in the template strand will result in the correct expression of EGFP and should consequently increase the relative EGFP expression. However, the presence of the nick had no significant effect on relative EGFP expression in MMR-proficient HeLa and SW480 cells, and MMR-deficient HCT116 cells (Figure 4A). It is possible that the nicked plasmid is efficiently ligated prior to the action of the MMR enzymes. To test this hypothesis, we measured relative EGFP expression in cell lines with reduced levels of either DNA ligase I (46BR.1G1 cell) or DNA ligase IIIα (CHO EM9 cells) activity. As shown in Figure 5, the relative EGFP expression was significantly higher in the CHO EM9 cells that have reduced levels of DNA ligase IIIα protein and activity (36). This suggests that the transfected nicked plasmid DNA is efficiently ligated by DNA ligase IIIα.
Figure 5.
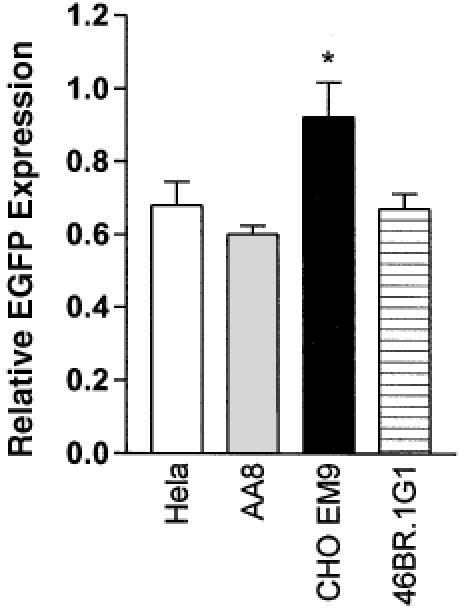
DNA ligase IIIα deficient cell line, CHO EM9, showed higher relative EGFP expression. Nicked homo- and heteroduplex plasmids were transfected into the cell lines depicted in the figure. CHO EM 9 was derived from the parental cell AA8, which is ligase IIIα-proficient. Relative EGFP expression was presented as mean ± SEM from three independent experiments. The asterisk indicates significant difference (P ≤ 0.05) from other mean values with student t tests.
The advantage of using the un-nicked circular heteroduplex plasmid DNA is that various kinds of mismatched heteroduplex oligonucleotides can be ligated into the gapped plasmid (see Figure 1) for MMR studies. To extend the results obtained with the G–G mismatched heteroduplex, we also measured relative EGFP expression from a T–G mismatch in an un-nicked circular plasmid in the four cell lines. The overall MMR efficiency of the T–G mismatch was lower than that of the G–G mismatch for all of the four cell lines (compare Figure 6A with Figure 4A). However, the ratios of relative EGFP expression on either G–G or T–G mismatch between MMR− cells and MMR+ cells remained basically the same. The mean ratios of HCT116's MMR efficiency to HeLa cells' MMR efficiency on G–G and T–G are 34.7 and 39.7%, respectively. Therefore, T–G and G–G mismatches can both be used to differentiate MMR+ and MMR− cells.
Figure 6.
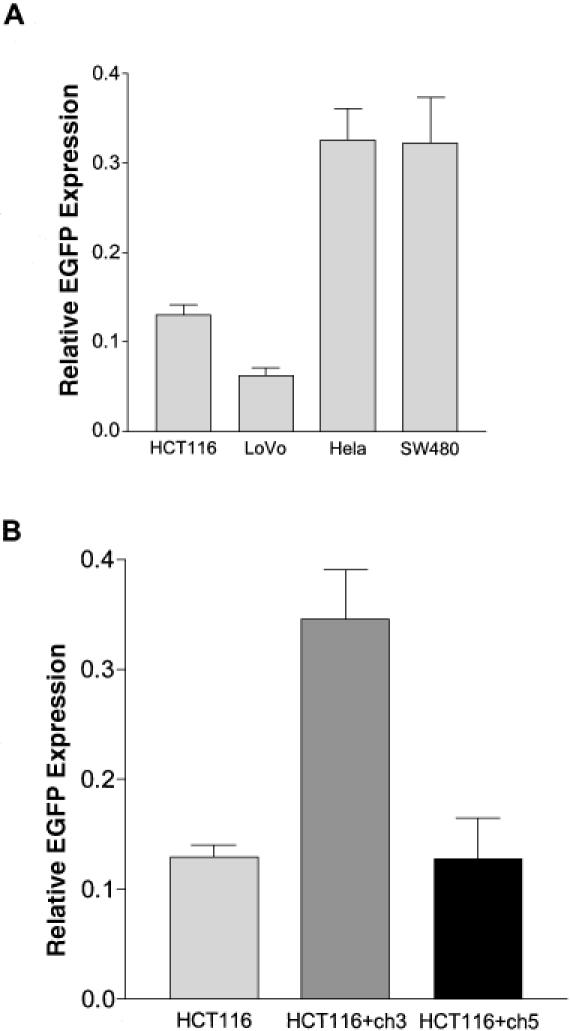
Relative EGFP expression from a T–G mismatch and the effect of chromosome 3 complementation on MMR in HCT116 cell. (A) The same amount of un-nicked heteroduplex (with a T–G mismatch), homoduplex, and negative control DNA were separately transfected into the depicted cell lines. Percentage of green cells and their mean intensity from each transfection were obtained from flow cytometry, and relative EGFP expression was calculated as described in Materials and Methods. Each column represents mean ± SEM from seven independent measurements for HCT116, HeLa and SW480, and six independent measurements for LoVo. According to Newman–Keuls multiple comparison test, the MMR efficiencies of HCT116 and LoVo are significantly different from those of HeLa or SW480 (P < 0.001), whereas the MMR efficiency between the two MMR- cell lines or between the two MMR+ cell lines is not significantly different from each other (P > 0.05). (B) Comparison of T–G MMR in HCT116, HCT116+ch3 and HCT116+ch5 cells. The relative EGFP expression in HCT116 is 0.13 ± 0.01 from 7 independent measurements. The relative EGFP expression in HCT116 + ch3 and HCT116 + ch5 is respectively 0.35 ± 0.05 and 0.13 ± 0.04 from 5 independent measurements. The data are presented as mean ± SEM. According to Newman–Keuls multiple comparison test, the MMR efficiency of HCT116 + ch3 is significantly higher from those of HCT116 and HCT116+ch5 (P < 0.01).
Complementation of MMR genes significantly elevated MMR activity in MMR− cells
HCT116 does not express functional hMLH1 protein and therefore has an impaired MMR system. Previous studies have shown that transfer of chromosome 3, which carries a copy of the normal hMLH1 gene, into HCT116 cells restores MMR activity to cell extracts (37). This effect is chromosome specific, since transfer of chromosome 2 or 5 did not elevate MMR activity in cell extracts. To confirm that MMR complementation can also be observed in vivo, we tested our method using HCT116+ch3 and HCT116+ch5 cells. As expected, the relative EGFP expression of HCT116+ch5 cells was similar to that of HCT116 cells (Figure 6B). In contrast, HCT116+ch3 had a significantly higher (P < 0.01) relative EGFP expression, which was similar to those of HeLa and SW480 cells as shown in Figure 6A. Thus, the presence of chromosome 3, but not chromosome 5, significantly elevates in vivo MMR activity in HCT116 cells.
Complementation of the MMR defect in HCT116 cells by the hMLH1 gene or cDNA alone has not been demonstrated because healthy HCT116 clones were not produced (38). To examine whether the expression of hMLH1 alone is sufficient to increase MMR activity in HCT116 cells, we transiently co-transfected HCT116 cells with an hMLH1 cDNA expression plasmid and either the homoduplex or G–G heteroduplex plasmid. Expression of hMLH1 protein was detected by western blotting 48 h after transfection (Figure 7A). Even though the transfection efficiency was <10%, we were able to detect a significant (P < 0.02) increase of MMR activity as reflected with an increase of relative EGFP expression in the hMLH1-transfected cells when compared with the MMR efficiency of the cells transfected with an empty vector, pRC/CMV (Figure 7B). Transfection of the hMLH1 expression plasmid into SW480 cells had no effect on the relative EGFP expression. These results indicate that our method can detect restored MMR activity by the expression of hMLH1 protein in HCT116 cells.
Figure 7.
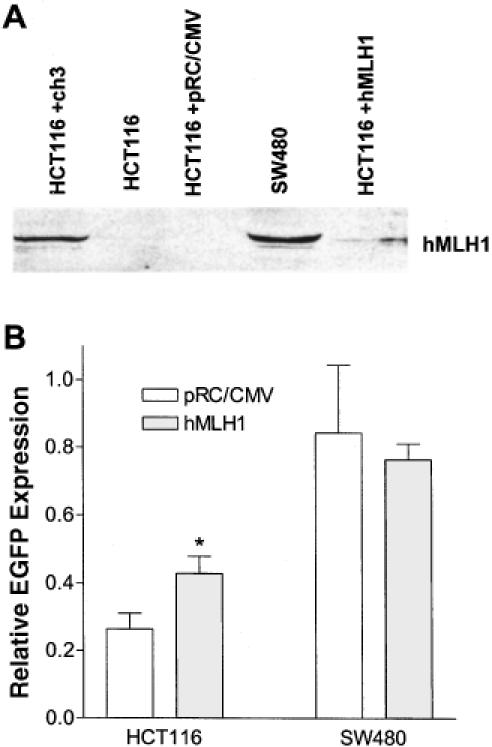
Ectopic expression of hMLH1 increased MMR efficiency in HCT116 cells. (A) Detection of hMLH1 protein with western blotting. HCT116 and SW480 were co-transfected with 1 μg pCMV-MLH1 or 0.6 μg pRC/CMV (control) and the homo- or heteroduplex (with a G–G mismatch) plasmid. Forty-eight hours later, the transfected cells were lifted and analyzed with flow cytometry. Cells recovered from the flow cytometer, along with HCT116+ch3 and SW480 cells, were lysed. Proteins were extracted, separated on 8% SDS-PAGE, and immunoblotted by a hMLH1 antibody. (B) Effect of hMLH1 transfection on G–G MMR in HCT116 and SW480 cells. Relative EGFP expression was calculated after flow cytometry analysis as described in the Materials and Methods. According to student t-test, HCT116 transfected with hMLH1 expression plasmid has a significant higher (P < 0.02) MMR efficiency (0.43 ± 0.05) than HCT116 transfected with pRC/CMV (0.26 ± 0.05). Each column represents mean ± SEM of three independent measurements.
DISCUSSION
The methods that are currently used to measure MMR activity can basically be grouped into two categories. The first one is an in vitro method using cell-free extracts (35,39). Nuclear or cytoplasmic extracts from various cells are incubated with mismatched DNA substrates, such as mismatched M13mp2 phage DNA, and MMR activity is measured either directly by endonuclease restriction analyses and/or DNA sequencing or indirectly by further transforming MMR− bacteria with the repaired phage DNA mixture. The need for a relatively large number of cells for the preparation of the cell-free extract makes this type of assay expensive. Furthermore, it cannot be utilized to measure the dynamics of MMR activity in live cells in response to the changes in the intracellular and/or extracellular environment. For the second category, mismatched or heteroduplex DNA, including both plasmid and viral DNA, is introduced into cells and then later retrieved for analysis (40–43). In this case, the retrieval process is usually laborious and tedious. The method we describe here can effectively differentiate MMR+ cells from MMR− cells and is relatively fast and inexpensive. More importantly, it can be used to monitor the regulation of MMR activity in live cells.
In our equation for the calculation of the relative EGFP expression, we incorporated the mean intensity of green fluorescence. This is because multiple copies of the heteroduplex plasmid might be transfected into a single cell and because the MMR− cells transfected with the heteroduplex plasmid consistently displayed a small number of green cells with low fluorescence intensity (Table 1). We assumed that an MMR+ cell should correct more mismatched copies of the heteroduplex plasmid than an MMR− cell if they are transfected with multiple copies of the heteroduplex plasmid. We also assumed that fluorescence intensity is proportional to the copy number of the corrected EGFP gene. Therefore, by using an intensity-weighted formula, the calculated MMR efficiency reflects the percentage of correctly repaired heteroduplex DNA copies instead of the percentage of cells that correctly repaired the heteroduplex DNA.
Theoretically, the MMR efficiency of MMR− cells should be around zero. However, our study as well as previous studies using in vitro assays for MMR− cells (34,44) or embryo fibroblasts from MMR gene knockout mouse (45) have detected a significant residual MMR activity. With our method, one contributing factor was that some EGFP expression was independent of the start codon. When a modified EGFP expression plasmid with a fixed mutation at its start codon was transfected into HeLa and HCT116 cells, a small number of green cells with low intensity were observed. After flow cytometry analysis, we found that the AUG-independent expression accounted for 5% of the calculated relative EGFP expression. As such, ∼25–50% of MMR activity measured with our method in HCT116 cells was apparently due to AUG-independent expression of the EGFP because the relative EGFP expression in HCT116 cell is ∼10–20% as shown in the figures. Another possibility for the residual MMR activity may be due to other cellular DNA repair mechanisms such as base excision repair, nucleotide excision repair or even an unknown repair system (46,47). However, it is likely that these other repair mechanisms are less effective than the MMR system. In support of this idea, when a heteroduplex DNA was incubated with cell extract for a short duration of 15 min, the repair efficiency of cell extract from MMR-proficient cells was found to be 10-fold or greater than that of cell extract from MMR-deficient cells (34,48). In the current study, the transfected heteroduplex was inside the cell for ∼24 h to allow for the repair of the mismatch and subsequent expression of EGFP. While this relatively long period of time was optimal for maximal repair and expression of EGFP in the MMR-proficient cells, an appreciable amount of the heteroduplex was apparently also being repaired by systems other than MMR during this time period in the MMR-deficient cells. Altogether, both AUG-independent GFP expression and alternative repair systems could account for the limited difference of MMR efficiency (3-fold) between MMR-proficient and MMR-deficient cells.
A key feature of the MMR system is that repair is directed to the newly synthesized strand (1). In the repair assay with cell extracts, a nick can act as the strand-discrimination signal (35). Unexpectedly, nicked and intact plasmid DNAs were repaired with equal efficiency in our in vivo assay. One possible explanation for this observation is that the transfected nicked plasmid is rapidly ligated and the resultant intact circular plasmid subsequently acted upon by the MMR pathway. In support of this model, in vivo MMR was more efficient in the DNA ligase III-deficient Chinese hamster ovary cell line, EM9. The observation that DNA ligase III—but not DNA ligase I—deficiency increases in vivo MMR efficiency is consistent with a study showing that DNA ligase III is the predominant DNA nick-joining ligase in proliferating mammalian cells (49).
For the G–G mismatch, we obtained relative EGFP expression of 65 and 70% for HeLa and SW480 cells, respectively. However, it decreased to 48 and 47% for HeLa and SW480, respectively, when they were transfected with the T–G heteroduplex. Since previous studies have shown that both G–G and T–G mismatches are optimal substrates for the MMR system (23,35,50), it is not likely that the repair of T–G mismatch was less efficient than the repair of G–G mismatch in these two cell lines. The difference may be explained by the fact that the T–G mismatch can be repaired by both MMR and a DNA glycosylase-initiated repair system (50). When we constructed the T–G mismatch, we deliberately engineered the G in the antisense (template) strand. Since the T–G DNA glycosylase always converts T–G to C–G, its repair of the T–G mismatch would create a fixed mutation in the EGFP gene that prevents expression. We suspect that the T–G glycosylase competes with the MMR system for the T–G substrates, leaving the MMR system with fewer substrates to act upon than in the case of G–G mismatch. Thus, while both the T–G and G–G mismatched substrates can be used to distinguish MMR+ from MMR− cells, the calculated relative EGFP expression from G–G mismatch may be a more reliable indicator of the MMR activity in live cells.
The low MMR activity in the hMLH1-deficient cell line HCT116 was complemented after introduction of a normal human chromosome 3, which carries the hMLH1 gene (37). Our in vivo MMR assay reiterated this result. However, it has not been unequivocally demonstrated that the correction of the MMR defect in HCT116 cells is due to the expression of hMLH1 alone or hMLH1 plus additional genes on the chromosome 3. Investigators have attempted to transfect and re-express hMLH1 cDNA to complement the MMR deficiency. In one study, stable expression of hMLH1 protein in Mlh1-deficient mouse embryonic fibroblasts was shown to complement DNA mismatch repair defects (51). However, when transfecting hMLH1 or hMSH2 into MMR− human cell lines, investigators have difficulties in obtaining stably transfected clones. This is apparently due to the fact that overexpression of hMSH2 or hMLH1 can induce apoptosis in both MMR+ and MMR− cells (52), and overexpression of hMLH1 can also suppress cell growth (38). Since MMR activity is scored in individual, live cells within a relatively short period of time using our method, we reasoned that we should be able to detect the effect of transiently expressed hMLH1 on MMR efficiency. Indeed, the MMR efficiency was significantly elevated 48 h after transient co-transfection of hMLH1 expression plasmid with the heteroduplex plasmid into HCT116 cells. Thus, our method can detect the restoration of MMR activity in the MMR-deficient cell.
Since the method we describe here can quantitatively measure MMR activity in a live single cell, it offers two potential utilities. First, it can be used to monitor changes of MMR activity in live cells as they are subjected to varying growth conditions. Hence, it should aid the study of whether and how MMR activity is regulated under different physiological and/or pathological conditions. Second, it may be used clinically for the diagnosis of MMR status in tumor cells. It appears highly feasible that a small number of tumor cells from a primary culture can be microinjected or transfected with the homoduplex or heteroduplex plasmid and the MMR activity in each injected/transfected cell be scored by quantitative fluorescence microscopy. We are currently exploring these possibilities.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Michael Brattain at Roswell Park Cancer Institute for the HCT116 cells, Dr Minoru Koi at the National Institute of Environmental Health Sciences for the HCT116+ch3 and HCT116+ch5 cell lines, Dr Michael Liskay at Oregon Health Science University for pCMV-MLH1 plasmid. For Y.Z., this work was performed in partial fulfillment of the requirements for the Ph.D. degree in the Department of Pharmacology, University of Kentucky College of Medicine. This work was in part supported by DOD Breast Cancer Research Program DAMD17-01-1-0413 and NIH Grant CA75253 to L-Z.S.
REFERENCES
- 1.Modrich P. and Lahue,R. (1996) Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem., 65, 101–133. [DOI] [PubMed] [Google Scholar]
- 2.Fishel R. (1998) Mismatch repair, molecular switches, and signal transduction. Genes Dev., 12, 2096–2101. [DOI] [PubMed] [Google Scholar]
- 3.Umar A. and Kunkel,T.A. (1996) DNA-replication fidelity, mismatch repair and genome instability in cancer cells. Eur. J. Biochem., 238, 297–307. [DOI] [PubMed] [Google Scholar]
- 4.Kolodner R.D. and Marsischky,G.T. (1999) Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev., 9, 89–96. [DOI] [PubMed] [Google Scholar]
- 5.Modrich P. (1997) Strand-specific mismatch repair in mammalian cells. J. Biol. Chem., 272, 24727–24730. [DOI] [PubMed] [Google Scholar]
- 6.Palombo F., Gallinari,P., Iaccarino,I., Lettieri,T., Hughes,M., D′Arrigo,A., Truong,O., Hsuan,J.J. and Jiricny,J. (1995) GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science, 268, 1912–1914. [DOI] [PubMed] [Google Scholar]
- 7.Drummond J.T., Li,G.M., Longley,M.J. and Modrich,P. (1995) Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science, 268, 1909–1912. [DOI] [PubMed] [Google Scholar]
- 8.Palombo F., Iaccarino,I., Nakajima,E., Ikejima,M., Shimada,T. and Jiricny,J. (1996) hMutSbeta, a heterodimer of hMSH2 and hMSH3, binds to insertion/deletion loops in DNA. Curr. Biol., 6, 1181–1184. [DOI] [PubMed] [Google Scholar]
- 9.Leung W.K., Kim,J.J., Wu,L., Sepulveda,J.L. and Sepulveda,A.R. (2000) Identification of a second MutL DNA mismatch repair complex (hPMS1 and hMLH1) in human epithelial cells. J. Biol. Chem., 275, 15728–15732. [DOI] [PubMed] [Google Scholar]
- 10.Li G.M. and Modrich,P. (1995) Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proc. Natl Acad. Sci. USA, 92, 1950–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raschle M., Marra,G., Nystrom-Lahti,M., Schar,P. and Jiricny,J. (1999) Identification of hMutLbeta, a heterodimer of hMLH1 and hPMS1. J. Biol. Chem., 274, 32368–32375. [DOI] [PubMed] [Google Scholar]
- 12.Lipkin S.M., Wang,V., Jacoby,R., Banerjee-Basu,S., Baxevanis,A.D., Lynch,H.T., Elliott,R.M. and Collins,F.S. (2000) MLH3: a DNA mismatch repair gene associated with mammalian microsatellite instability. Nat. Genet., 24, 27–35. [DOI] [PubMed] [Google Scholar]
- 13.Toft N.J. and Arends,M.J. (1998) DNA mismatch repair and colorectal cancer. J. Pathol., 185, 123–129. [DOI] [PubMed] [Google Scholar]
- 14.Prolla T.A. (1998) DNA mismatch repair and cancer. Curr. Opin. Cell Biol., 10, 311–316. [DOI] [PubMed] [Google Scholar]
- 15.Eshleman J.R. and Markowitz,S.D. (1996) Mismatch repair defects in human carcinogenesis. Hum. Mol. Genet., 5 Spec No, 1489–1494. [DOI] [PubMed] [Google Scholar]
- 16.Kolodner R.D., Tytell,J.D., Schmeits,J.L., Kane,M.F., Das,G., Weger,J., Wahlberg,S., Fox,E.A., Peel,D., Ziogas,A. et al. (1999) Germ-line msh6 mutations in colorectal cancer families. Cancer Res., 59, 5068–5074. [PubMed] [Google Scholar]
- 17.Wu Y., Berends,M.W., Mensink,R.J., Kempinga,C., Sijmons,R.H., van der Zee,A.J., Hollema,H., Kleibeuker,J.H., Buys,C.M. and Hofstra,R.W. (1999) Association of hereditary nonpolyposis colorectal cancer-related tumors displaying low microsatellite instability with MSH6 germline mutations. Am. J. Hum. Genet., 65, 1291–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cunningham J.M., Christensen,E.R., Tester,D.J., Kim,C.Y., Roche,P.C., Burgart,L.J. and Thibodeau,S.N. (1998) Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res., 58, 3455–3460. [PubMed] [Google Scholar]
- 19.Herman J.G., Umar,A., Polyak,K., Graff,J.R., Ahuja,N., Issa,J.P., Markowitz,S., Willson,J.K., Hamilton,S.R., Kinzler,K.W. et al. (1998) Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc. Natl Acad. Sci. USA, 95, 6870–6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Veigl M.L., Kasturi,L., Olechnowicz,J., Ma,A.H., Lutterbaugh,J.D., Periyasamy,S., Li,G.M., Drummond,J., Modrich,P.L., Sedwick,W.D. et al. (1998) Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc. Natl Acad. Sci. USA, 95, 8698–8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kane M.F., Loda,M., Gaida,G.M., Lipman,J., Mishra,R., Goldman,H., Jessup,J.M. and Kolodner,R. (1997) Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res., 57, 808–811. [PubMed] [Google Scholar]
- 22.Meyers M., Theodosiou,M., Acharya,S., Odegaard,E., Wilson,T., Lewis,J.E., Davis,T.W., Wilson-Van,P.C., Fishel,R. and Boothman,D.A. (1997) Cell cycle regulation of the human DNA mismatch repair genes hMSH2, hMLH1, and hPMS2. Cancer Res., 57, 206–208. [PubMed] [Google Scholar]
- 23.David P., Efrati,E., Tocco,G., Krauss,S.W. and Goodman,M.F. (1997) DNA replication and postreplication mismatch repair in cell-free extracts from cultured human neuroblastoma and fibroblast cells. J. Neurosci., 17, 8711–8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donohue P.J., Feng,S.L., Alberts,G.F., Guo,Y., Peifley,K.A., Hsu,D.K. and Winkles,J.A. (1996) Fibroblast growth factor-1 stimulation of quiescent NIH 3T3 cells increases G/T mismatch-binding protein expression. Biochem. J., 319, 9–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pierce A.J., Johnson,R.D., Thompson,L.H. and Jasin,M. (1999) XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev., 13, 2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sattler U., Frit,P., Salles,B. and Calsou,P. (2003) Long-patch DNA repair synthesis during base excision repair in mammalian cells. EMBO Rep., 4, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fisher C.L. and Pei,G.K. (1997) Modification of a PCR-based site-directed mutagenesis method. Biotechniques, 23, 570–1, 574. [DOI] [PubMed] [Google Scholar]
- 28.Brattain M.G., Levine,A.E., Chakrabarty,S., Yeoman,L.C., Willson,J.K. and Long,B. (1984) Heterogeneity of human colon carcinoma. Cancer Metastasis Rev., 3, 177–191. [DOI] [PubMed] [Google Scholar]
- 29.Mulder K.M. and Brattain,M.G. (1989) Effects of growth stimulatory factors on mitogenicity and c-myc expression in poorly differentiated and well differentiated human colon carcinoma cells. Mol. Endocrinol., 3, 1215–1222. [DOI] [PubMed] [Google Scholar]
- 30.Zar J.H. (1984) Multisample hypothesis: the analysis of variance. In Zar,J.H. (ed.), Biostatistical Analysis. Prentice-Hall, Inc., Englewood Cliffs, NJ, pp. 162–184. [Google Scholar]
- 31.Branch P., Hampson,R. and Karran,P. (1995) DNA mismatch binding defects, DNA damage tolerance, and mutator phenotypes in human colorectal carcinoma cell lines. Cancer Res., 55, 2304–2309. [PubMed] [Google Scholar]
- 32.Bronner C.E., Baker,S.M., Morrison,P.T., Warren,G., Smith,L.G., Lescoe,M.K., Kane,M., Earabino,C., Lipford,J. and Lindblom,A. (1994) Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature, 368, 258–261. [DOI] [PubMed] [Google Scholar]
- 33.Papadopoulos N., Nicolaides,N.C., Wei,Y.F., Ruben,S.M., Carter,K.C., Rosen,C.A., Haseltine,W.A., Fleischmann,R.D., Fraser,C.M. and Adams,M.D. (1994) Mutation of a mutL homolog in hereditary colon cancer. Science, 263, 1625–1629. [DOI] [PubMed] [Google Scholar]
- 34.Umar A., Boyer,J.C., Thomas,D.C., Nguyen,D.C., Risinger,J.I., Boyd,J., Ionov,Y., Perucho,M. and Kunkel,T.A. (1994) Defective mismatch repair in extracts of colorectal and endometrial cancer cell lines exhibiting microsatellite instability. J. Biol. Chem., 269, 14367–14370. [PubMed] [Google Scholar]
- 35.Holmes J.J., Clark,S. and Modrich,P. (1990) Strand-specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proc. Natl Acad. Sci. USA, 87, 5837–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caldecott K.W., Tucker,J.D., Stanker,L.H. and Thompson,L.H. (1995) Characterization of the XRCC1-DNA ligase III complex in vitro and its absence from mutant hamster cells. Nucleic Acids Res., 23, 4836–4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koi M., Umar,A., Chauhan,D.P., Cherian,S.P., Carethers,J.M., Kunkel,T.A. and Boland,C.R. (1994) Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N′-nitro-N- nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. [Erratum (1995) Cancer Res., 55, 201.] Cancer Res., 54, 4308–4312. [PubMed] [Google Scholar]
- 38.Shin K.H., Han,H.J. and Park,J.G. (1998) Growth suppression mediated by transfection of wild-type hMLH1 in human cancer cells expressing endogenous truncated hMLH1 protein. Int. J. Oncol., 12, 609–615. [DOI] [PubMed] [Google Scholar]
- 39.Thomas D.C., Roberts,J.D. and Kunkel,T.A. (1991) Heteroduplex repair in extracts of human HeLa cells. J. Biol. Chem., 266, 3744–3751. [PubMed] [Google Scholar]
- 40.Brown T.C. and Jiricny,J. (1987) A specific mismatch repair event protects mammalian cells from loss of 5-methylcytosine. Cell, 50, 945–950. [DOI] [PubMed] [Google Scholar]
- 41.Folger K.R., Thomas,K. and Capecchi,M.R. (1985) Efficient correction of mismatched bases in plasmid heteroduplexes injected into cultured mammalian cell nuclei. Mol. Cell. Biol., 5, 70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hare J.T. and Taylor,J.H. (1985) One role for DNA methylation in vertebrate cells is strand discrimination in mismatch repair. Proc. Natl Acad. Sci. USA, 82, 7350–7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hare J.T. and Taylor,J.H. (1989) Methylation in eukaryotes influences the repair of G/T and A/C DNA basepair mismatches. Cell Biophys., 15, 29–40. [DOI] [PubMed] [Google Scholar]
- 44.Marra G., Iaccarino,I., Lettieri,T., Roscilli,G., Delmastro,P. and Jiricny,J. (1998) Mismatch repair deficiency associated with overexpression of the MSH3 gene. Proc. Natl Acad. Sci. USA, 95, 8568–8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edelmann W., Cohen,P.E., Kane,M., Lau,K., Morrow,B., Bennett,S., Umar,A., Kunkel,T., Cattoretti,G., Chaganti,R. et al. (1996) Meiotic pachytene arrest in MLH1-deficient mice. Cell, 85, 1125–1134. [DOI] [PubMed] [Google Scholar]
- 46.O'Regan N.E., Branch,P., Macpherson,P. and Karran,P. (1996) hMSH2-independent DNA mismatch recognition by human proteins. J. Biol. Chem., 271, 1789–1796. [DOI] [PubMed] [Google Scholar]
- 47.Huang J.C., Hsu,D.S., Kazantsev,A. and Sancar,A. (1994) Substrate spectrum of human excinuclease: repair of abasic sites, methylated bases, mismatches, and bulky adducts. Proc. Natl Acad. Sci. USA, 91, 12213–12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parsons R., Li,G.M., Longley,M.J., Fang,W.H., Papadopoulos,N., Jen,J., de la Chapelle,A., Kinzler,K.W., Vogelstein,B. and Modrich,P. (1993) Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell, 75, 1227–1236. [DOI] [PubMed] [Google Scholar]
- 49.Tomkinson A.E., Starr,R. and Schultz,R.A. (1993) DNA ligase III is the major high molecular weight DNA joining activity in SV40-transformed human fibroblasts: normal levels of DNA ligase III activity in Bloom syndrome cells. Nucleic Acids Res., 21, 5425–5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bill C.A., Duran,W.A., Miselis,N.R. and Nickoloff,J.A. (1998) Efficient repair of all types of single-base mismatches in recombination intermediates in Chinese hamster ovary cells. Competition between long-patch and G-T glycosylase-mediated repair of G–T mismatches. Genetics, 149, 1935–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buermeyer A.B., Wilson-Van,P.C., Baker,S.M. and Liskay,R.M. (1999) The human MLH1 cDNA complements DNA mismatch repair defects in Mlh1- deficient mouse embryonic fibroblasts. Cancer Res., 59, 538–541. [PubMed] [Google Scholar]
- 52.Zhang H., Richards,B., Wilson,T., Lloyd,M., Cranston,A., Thorburn,A., Fishel,R. and Meuth,M. (1999) Apoptosis induced by overexpression of hMSH2 or hMLH1. Cancer Res., 59, 3021–3027. [PubMed] [Google Scholar]