

Abstract
R-Ras is a Ras family small GTPase highly expressed in mature functional blood vessels in normal tissues. It inhibits pathological angiogenesis and promotes vessel maturation and stabilization. Previous studies suggest that R-Ras affects cellular signaling in endothelial cells, pericytes, and smooth muscle cells to regulate vessel formation and remodeling in adult tissues. R-Ras suppresses VEGF-induced endothelial permeability and vessel sprouting while promoting normalization of pathologically developing vessels in mice. R-Ras attenuates VEGF receptor-2 (VEGFR2) activation by inhibiting internalization of the receptor upon VEGF ligand binding, leading to significant reduction of VEGFR2 autophosphorylation. Here, we show that R-Ras strongly suppresses VEGF-dependent activation of stress-activated protein kinase-2/p38 mitogen-activated protein kinase (SAPK2/p38MAPK) and phosphorylation of downstream heat shock protein 27 (HSP27), a regulator of actin cytoskeleton organization, in endothelial cells. The suppression of p38MAPK activation and HSP27 phosphorylation by R-Ras concurred with altered actin cytoskeleton architecture, reduced membrane protrusion, and inhibition of endothelial cell migration toward VEGF. Silencing of endogenous R-Ras by RNAi increased membrane protrusion and cell migration stimulated by VEGF, and these effects were offset by p38MAPK inhibitor SB203580. These results suggest that R-Ras regulates angiogenic activities of endothelial cells in part via inhibition of the p38MAPK-HSP27 axis of VEGF signaling.
Introduction
VEGF plays multiple important roles in development and maintenance of blood vessels. It is a potent mitogen for endothelial cells (ECs) and also promotes migration and survival of these cells [1]. VEGF-A disrupts endothelial barrier function and strongly increases vessel permeability [2, 3]. Chronically elevated level of VEGF in tissues causes abnormal angiogenesis and vessel leakiness. One of the three VEGF receptors, VEGFR2, is the key mediator of VEGF signaling in vascular endothelium. The activation of VEGFR2 by VEGF results in tyrosine phosphorylation of VEGFR2, triggering transduction of downstream signaling pathways. The p38MAPK-Hsp27 signaling downstream of VEGFR2 Tyr 1214 phosphorylation leads to actin cytoskeleton reorganization and migration of ECs to drive angiogenic sprouting of ECs. While VEGF signaling potently induces sprouting and elongation of new vessels, the formation of functional vessels requires additional cellular signaling to promote vessel maturation. The vessel sprouting and maturation processes must be precisely regulated by balancing pro- and anti-angiogenic signals [4, 5]. The imbalanced signals due to excessive VEGF stimuli leads to overproduction of immature and defective vessels with chaotic organization as seen in pathological angiogenesis in tumors and diabetic retinopathy [6, 7]. In peripheral arterial disease, imbalanced angiogenesis appears to be a crucial reason for the failure of clinical trials for VEGF gene therapy that were conducted in attempts to reperfuse ischemic tissues with VEGF-induced new vessels [8–10].
The small GTPase R-Ras plays an important role in attenuating the response of ECs to VEGF and inhibiting pathological angiogenesis [11]. Using mouse genetic models, we previously demonstrated that R-Ras inhibits excessive vessel sprouting [12] while promoting maturation of nascent vessels in mouse tumors [11]. The R-Ras deficiency increased tumor vessel density but exaggerated the deformation and functional abnormalities of tumor vessels. On the other hand, gain-of-function of R-Ras normalized the vessel structure and function in VEGF-induced angiogenesis in Matrigel implants. R-Ras is highly expressed in differentiated vascular smooth muscle cells, ECs, and pericytes of mature blood vessels in normal adult tissues [11, 12]. In contrast, R-Ras is strongly downregulated during proliferative regeneration and remodeling of blood vessels [12]. R-Ras can activate PI3-kinase, and through it, Akt [13]. However, unlike closely related Ras proteins such as K-Ras and H-Ras, R-Ras does not activate the Raf-MAP kinase pathway or transform rat fibroblasts [13, 14]. R-Ras enhances integrin adhesion to extracellular matrix [11, 15] and also enhances endothelial barrier function by stabilizing VE-cadherin in adherens junction [11]. Genetic studies in mice demonstrated that R-Ras inhibits vascular cell proliferation and invasion, and promotes vascular quiescence displaying its activity to counterbalance vessel activation and promote normalization of pathologically regenerating blood vessels [11, 12]. The vascular normalization effect of R-Ras appears to be independent of hypoxia sensing via PHD2/HIF-2 pathway [11, 16]. The mechanism of R-Ras-mediated blood vessel regulation is incompletely understood.
R-Ras is a Ras protein that negatively regulates angiogenic endothelial sprouting, and hence, it is functionally contrasting to other closely related Ras homologs such as H-Ras and Rap1, which positively regulate this process [17–20]. The unique effect of R-Ras on vessel regulation may be at least in part due to its ability to attenuate VEGF signaling in ECs. We have previously showed that R-Ras inhibits clathrin-dependent internalization of VEGFR2 upon VEGF stimulation that is required for the full activation of the receptor [21]. This effect leads to significant downregulation of receptor autophosphorylation at tyrosine phosphorylation sites including the Tyr 1214 residue. In this study, we focused on the effect of R-Ras on the p38MAPK-HSP27 signaling downstream of VEGFR2 Tyr 1214 phosphorylation and investigated how R-Ras affects actin reorganization, membrane dynamics, and motility of ECs regulated by this VEGF pathway.
Method
Antibodies and reagents
Antibodies for following proteins were purchased from Cell Signaling Technology, Inc. (Danvers, MA): p38MAPK, phospho-p38MAPK Thr180/Tyr182, HSP27, phospho-HSP27 Ser15, phospho-HSP Ser78 antibodies. Anti-phospho-VEGFR2 Tyr1214 was purchased from EMD Millipore (Billerica, MA). Antibodies for R-Ras were obtained from Cell Signaling Technology or Abcam (Cambridge, MA). Alexa Fluor® 594 phalloidin was purchased from Life Technology (Carlsbad, CA). Recombinant human VEGF-A165 was purchased from R&D Systems (Minneapolis, MN).
Cell culture, lentivirus transduction and siRNA transfection
Human umbilical cord vein endothelial cells (HUVECs) and growth media EGM-2 were purchased from Lonza (Basel, Switzerland). Cells were transduced with a constitutively active form of R-Ras (R-Ras38V), or insertless control using pLenti6 lentivirus expression vector (Invitrogen) as described before [12]. R-Ras knockdown was carried out by lentivirus transduction of shRNA that targets the R-Ras sequence 5'- GGA AAT ACC AGG AAC AAG A-3' as described previously [11]. The negative control (NC) shRNA, which does not target any known sequence of the human, mouse, rat, or zebrafish origin, was obtained from COSMO BIO co., ltd (Tokyo, Japan) [11]. To evaluate the stimulation of VEGF, cells were starved for growth factors overnight with 2% horse serum in EBM-2 basal media, stimulated with VEGF, and lysed in cell lysis buffer (0.5% SDS phosphate buffer, pH7.4) at various time points for western blot analyses. For the treatment of p38MAPK inhibitor, growth factor starved cells were incubated with or without 10 nM SB203580 for 2 hours and stimulated with 50 ng/ml VEGF. For actin filament staining, cell were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton-X/PBS and stained with Alexa Fluor® 594 phalloidin.
Immunofluorescence staining of tumor vessels
R-Ras KO mice have been described previously [12]. All animal experiments were performed in accordance with protocols approved by the institutional animal care and use committees of the Sanford-Burnham-Prebys Medical Discovery Institute. B16F10 melanoma and Lewis lung carcinoma (LLC) cells (1×106 cells) were subcutaneously injected into the flank of R-Ras KO or wild type control mice [11]. Ten days (B16F10) or 14 days (LLC) later, tumors were collected. Histological frozen sections were stained with anti phospho-HSP27 Ser78 antibodies, and CD31 antibodies and imaged using Nikon Eclipse 90i microscope. The area of phospho-HSP27 and CD31 double positive staining was quantified and normalized to total CD31 area to analyze phosphorylation of HSP27 in the tumor vessels. Two animals were used for each group. The total number of vessels analyzed for B16F10 tumors was 258 (WT) and 442 (KO). For LLC tumors, 1239 (WT) and 1774 (KO).
Cell projection and retraction measurement
Cells were fluorescently leveled with CellTracker™ Green CMFDA (Life Technologies), and plated on Nunc™ Lab-Tek™ II Chamber Slide™ (Nalge Nunc International, Penfield, New York). After overnight starvation for growth factors, live image was captured by Nikon Eclipse Ti (Nikon insturument Inc.) equipped with Photometrics Coolsnap HQ2 14bit CCD camera (Photometrics) with or without stimulation by 50 ng/ml VEGF-A. For the inhibition of p38MAPK, cells were treated with 10 nM SB203580 (Cell Signaling Technology) for 2 hours before VEGF stimulation. The cell projection and retraction activities were determined by time-laps microscopy for the changes of the cell shape between 10 and 15 min post VEGF stimulation. The areas of projecting or retracting cell membrane were determined by NIS-Elements (Nikon Instrument Co.)
Cell migration assay
Cells were starved for serum in EBM -2 media (Lonza) for 8 hours. Cells were then seeded onto the upper compartment of 8µm-Trasnwell® chamber (Corning) and cultured for 1.5 hour to let the cells to adhere. Then, VEGF-A (5 ng/ml) was added into lower compartment, and cells were further incubated for 2 hours. The upper surface of Transwell membrane was scraped by cotton swab to remove non-migrated cells. After fixing with 4% paraformaldehyde, migrated cells on the lower surface were stained with crystal violet and quantified by counting. The average counting of triplicated chambers is presented.
Statistics
Statistics were performed using 1-way ANOVA with Bonferroni’s multiple comparison test. Student's t-test was used for tumor vessel analyses. Error bars represent S.E.M.
Results
R-Ras attenuates p38MAPK activation induced by VEGF
The activation of VEGFR2 by VEGF results in tyrosine phosphorylation of VEGFR2, triggering transduction of downstream signaling pathways. The p38MAPK-Hsp27 signaling downstream of VEGFR2 Tyr 1214 phosphorylation leads to actin cytoskeleton reorganization, cell membrane spreading, and migration of ECs. R-Ras significantly reduces VEGFR2 phosphorylation though the inhibition of clathrin-mediated internalization of the receptor [21]. We investigated the effect of R-Ras on p38MAPK activation by VEGF. Cultured ECs were transduced with constitutively active mutant of R-Ras (R-Ras38V) or insertless vector (mock) control. These cells were stimulated with VEGF following overnight culture in low-serum containing media. The expression of R-Ras38V inhibited phosphorylation of VEGFR2 at Tyr1214 as we reported previously (Figure 1A) [21]. VEGF strongly increased the phosphorylation of p38MAPK (Thr180/Tyr182) in the mock control ECs with the peak activation at 15 min post VEGF stimulation (Figure 1A – C). In contrast, we observed a strong reduction of p38MAPK phosphorylation in the ECs expressing R-Ras38V (Figure1A – C) demonstrating a significant inhibitory effect of R-Ras on the p38MAPK activity downstream of VEGFR2. R-Ras does not seem to have a direct effect on VEGF-independent p38MAPK activity as we did not observe obvious difference between the mock and R-Ras38V-transduced ECs for the basal levels of p38MAPK phosphorylation without VEGF stimulation (Figures 1A–C). In addition, R-Ras did not have effect on the total expression level of p38MAPK (Figure 1A).
Figure 1. R-Ras reduces VEGF-dependent phosphorylation of p38MAPK.
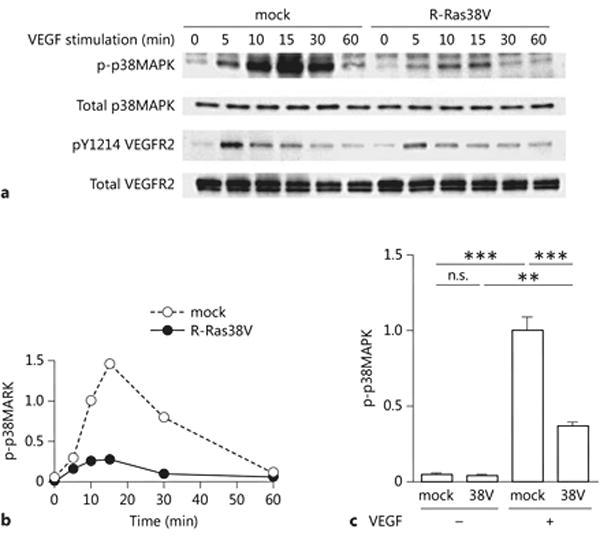
(A) Time course analysis of p38MAPK activation. The constitutively active form of R-Ras, R-Ras38V or control empty vector (mock) was transduced in endothelial cells. Cells were starved for serum and growth factors overnight, and stimulated with 50 ng/ml VEGF. Cell lysate was collected at various time points to determine the phosphorylation of p38MAPK (Thr 180/Tyr 182) and VEGFR2 (Tyr 1214) as well as the total levels of these proteins by western blotting. (B) The level of phospho-p38MAPK (Thr 180/Tyr 182) was quantified from the western blot at each time point, normalized to total p38MAPK, and presented as relative values compared with the mock-transduced control cells at 10 min. A representative result from three independent experiments is shown. (C) Cells were stimulated with 50 ng/ml VEGF for 15 min, and cell lysate was analyzed for phosphorylation of p38MAPK. The graph shows one set of the representative data from three independent experiments. Error bars, SEM. n=3, **p<0.01, ***p<0.001, ns: non-significant difference.
To further investigate the effect of R-Ras, we knocked down endogenous R-Ras by shRNA and stimulated ECs with VEGF. R-Ras knockdown enhanced phosphorylation of p38MAPK (Figure 2A and B), demonstrating the role of R-Ras in negatively regulating the VEGF-p38MAPK signaling.
Figure 2. R-Ras silencing enhances p38MAPK phosphorylation induced by VEGF.

(A) Endogenous R-Ras was knocked down by R-Ras shRNA. Cells were stimulated with VEGF at the indicated doses for 15 min. (B) Cells were stimulated with or without 50 ng/ml VEGF. The level of phospho-p38MAPK was quantified, normalized to total p38MAPK, and presented as relative values compared with the VEGF-stimulated control cells. NC, negative control shRNA. Error bars, SEM. n=3. *p<0.05, ns: non-significant difference.
R-Ras attenuates VEGF-dependent HSP27 phosphorylation
The heat shock protein 27 (HSP27) is a molecular chaperon and an actin cytoskeleton modulator implicated in cancer progression [22]. In ECs, HSP27 is downstream of the VEGFR2-p38MAPK pathway and phosphorylated by MAPK-activated protein kinase 2 (MK2) [23]. HSP27 is expressed in ECs and it is an essential mediator of actin reorganization induced by VEGF [24]. Since we observed a strong attenuation of VEGF-dependent p38MAPK activity by R-Ras, we examined the effect of R-Ras on HSP27 by analyzing its phosphorylation at two serine sites, Ser 15 and Ser 78 residues (Figure 3A and B). VEGF increased phosphorylation of both serine sites of HSP27 in mock control cells. The phosphorylation at Ser 78 was particularly robust. The expression of R-Ras38V reduced the phosphorylation of Ser 15 by approx. 40% and Ser 78 by 70% in VEGF-stimulated ECs. Thus, R-Ras38V nearly nullified the VEGF-dependent phosphorylation of HSP27 at Ser 15 to the unstimulated basal level and also substantially reduced the phosphorylation at Ser 78. Similar to the observation in the analysis of p38MAPK (Figures 1A–C), R-Ras did not seem to show direct effect on the VEGF-independent HSP27 phosphorylation as we did not observe significant differences between mock and R-Ras38V-transduced ECs in the basal levels of HSP27 phosphorylation before VEGF stimulation (Figures 3A–B). We also observed that R-Ras did not have effect on the total expression level of HSP27 (Figure 3A). The shRNA-mediated knockdown of endogenous R-Ras significantly enhanced phosphorylation of both serine residues upon VEGF stimulation of ECs, but not before VEGF stimulation (Figure 3C), further demonstrating the role of R-Ras in suppressing HSP27 phosphorylation induced by VEGF. These results are in agreement with the effect of R-Ras on VEGFR2 and p38MAPK activation upstream of HSP27 (Figures 1 and 2).
Figure 3. R-Ras regulates VEGF-dependent phosphorylation of HSP27.

(A) Western blot analysis of HSP27 phosphorylation. ECs were stimulated with 50 ng/ml VEGF for 15 min. The phosphorylation at Ser15 and Ser78 of HSP27 were analyzed by western blot using phospho-specific antibodies. (B) The level of phosphorylation was quantified and normalized to the total HSP27. The fold differences were presented in relative values with control mock cells with VEGF stimulation. (D) R-Ras knockdown enhances phosphorylation of HSP27. Error bars, SEM. n=3. *p<0.05, **p<0.01, ***p<0.001, ns: non-significant difference.
We next investigated the role of R-Ras in regulating HSP27 phosphorylation in vivo. B16F10 melanoma or Lewis lung carcinoma cells were subcutaneously transplanted to wild-type or R-Ras knockout (KO) mice, and tumor vessels were analyzed by immunostaining of tumor sections for CD31 and phospho-HSP27 Ser 78. We have previously shown that R-Ras KO mice exhibit enhanced angiogenesis and chaotic vascular structure in these tumor models [11, 12]. In the current study, the phosphorylation of HSP27 was found elevated in the ECs of tumor vessels developing in R-Ras KO mice compared with the ECs of tumor vessels in wild-type control mice and correlated with increased tumor vascularity in the R-Ras KO mice (Figure 4). This observation is consistent with the elevated phosphorylation of VEGFR2 at Tyr 1214 observed in the tumor endothelium of R-Ras KO mice [21] as well as the inhibitory effect of R-Ras on HSP27 phosphorylation observed in cultured ECs (Figure 3A–C). Taken together, these observations suggest a significant effect of R-Ras on the VEGF-HSP27 pathway in pathologically regenerating blood vessels.
Figure 4. Genetic ablation of R-Ras increases phosphorylation of HSP27 in the tumor endothelium.

B16F10 melanoma (A) or LLC (B) tumors were grown for 10 or 14 days, respectively, in R-Ras KO or wild-type control mice. The tumor endothelium was analyzed for HSP27 phosphorylation by co-immunostaining of tumor sections with anti-CD31 and phospho-HSP27 Ser 78 antibodies followed by microscopy. Scale bar, 100µm. (C) Quantification of HSP27 Ser 78 phosphorylation. The area of phospho-HSP27+CD31+ double staining was quantified and normalized to total CD31 area to analyze phosphorylation of HSP27 in the tumor vessels. The tumor vascularity in each group was determined by total CD31 area and vessel density in tumor sections. Data are presented as fold increase relative to the wild-type control animals. *p<0.05, ***p<0.001. Two animals per group. The total number of vessels analyzed for B16F10 tumors, 258 (WT) and 442 (KO). For LLC tumors, 1239 (WT) and 1774 (KO). Error bars, SEM.
R-Ras alters reorganization of actin cytoskeleton induced by VEGF
Phosphorylation of HSP27 modifies the interaction of HSP27 with actin and facilitates actin polymerization [25–27]. VEGF strongly induces reorganization of actin cytoskeleton structures in ECs, in which HSP27 phosphorylation downstream of VEGFR2-p38MAPK signaling plays a major role [28–30]. To examine the effect of R-Ras on the VEGF-induced actin cytoskeleton reorganization, cells were stimulated with VEGF and stained with phalloidin to visualize actin filaments. Unstimulated mock-transduced control cells exhibited actin filaments localized mainly at the cell periphery in a confluent EC culture (Figure 5A). Subconfluent culture was only weakly stained with phalloidin (Figure 5B). As previously reported [31], the stimulation of ECs with VEGF strongly induced the formation of tight bundles of actin filaments that constitute stress fibers running across the cytoplasm in confluent and subconfluent cultures (Figure 5A–C). These stress fibers were assembled in irregular thickness. In contrast, thinner but well-defined stress fibers were found accumulated along the membrane periphery of unstimulated R-Ras38V-transduced cells. Long thin stress fibers were also observed across the cytoplasm of these cells. These stress fibers were oriented in parallel to each other in a well-organized fashion. Such an orientation of actin fibers was independent on VEGF but dependent on R-Ras activity as it was found before VEGF stimulation in R-Ras38V-transduced cells but not in the mock-transduced control cells (Figure 5A–C) or R-Ras-silenced cells (data not shown). VEGF enhanced transcytoplasmic stress fibers in R-Ras38V-expressing ECs in both confluent and subconfluent cultures (Figure 5A–C). However, these stress fibers did not form thick bundles and remained relatively thin, and they were formed in a uniform thickness, which are contrasting to the bundles of stress fibers with irregular length and thickness found in the control cells upon VEGF stimulation. The stress fibers in R-Ras38V-expressing cells are evenly positioned in parallel with constant distance between the fibers. In confluent culture, this positional arrangement of the stress fibers continues with the stress fibers formed in adjacent cells, producing the orderly architecture of actin network in a sheet of EC monolayer (Figure 5A). Interestingly, a similar parallel pattern of actin cytoskeleton organization is found in confluent ECs under laminar shear stress [32].
Figure 5. R-Ras influences actin cytoskeleton organization.

Cells were starved for growth factors overnight and stimulated with or without 50 ng/ml VEGF for 30 min. Confluent monolayer culture (A and D) and subconfluent cultures (B and C) were fixed and stained with Alexa Fluor 594® Phalloidin (green). The boxed areas in B were magnified (C). Fluorescence intensity was equally enhanced in the left panels of C in both pictures (VEGF-, mock and RRas38V) in order to better visualize the actin filaments in unstimulated cells. Next, endogenous R-Ras was silenced by shRNA, and growth factor-starved cells were treated with SB 203580 for 2 hours. Cells were then stimulated with 50ng/ml VEGF for 30 min and stained with phalloidin (D). Experiment was repeated at least 3 times. Representative images were shown. Scale bar, 50 µm.
We next treated ECs with a specific inhibitor of p38MAPK, SB203580, to examine whether direct inhibition of p38MAPK produces a similar effect to the effect of R-Ras38V on actin cytoskeleton in VEGF-treated ECs. The control and R-Ras-silenced ECs treated with SB203580 both exhibited the loss of thick bundles of transcytoplasmic stress fibers induced by VEGF (Figure 5D). However, SB203580 did not produce an effect similar to the effect of R-Ras38V to induce orderly organization of parallel stress fibers (Figures 5A–C). This observation indicates that the effect of R-Ras on actin cytoskeleton architecture is not solely due to the inhibition of VEGF-induced p38MAPK activation and its downstream actin polymerization although this effect on p38MAPK may still be important for controlling the characteristics of stress fibers and other actin structures to be assembled in ECs.
R-Ras attenuates membrane dynamics induced by VEGF
We next investigated the effect of R-Ras on cell membrane dynamics stimulated by VEGF. VEGF induces actin reorganization facilitating lamellipodia formation and plasma membrane protrusion necessary for propelling and guiding the migration of ECs (Gerhardt et al., 2003; Lamalice et al., 2007). Activation of p38MAPK has been known to enhance lamellipodia and migration of ECs in a HSP27-dependent manner (McMullen et al., 2005; Rousseau et al., 1997). In our study, VEGF enhanced formation of lamellipodia in subconfluent culture of control cells as expected (Figure 5C arrows). In contrast, the formation of lamellipodia was significantly diminished in the subconfluent culture of R-Ras38V-expressing ECs that are treated with VEGF, indicating that R-Ras inhibits VEGF-dependent lamellipodia formation (Figure 5C). It has been shown that inhibition of endogenous R-Ras in ECs by dominant negative R-Ras significantly enhances the dynamic changes of cell shape in growth factor-rich media [11]. Therefore, we next analyzed how R-Ras affects the effect of VEGF on membrane extension and examined whether this is a p38MAPK-mediated effect. For this purpose, we conducted time-lapse microscopy in which endogenous R-Ras is silenced in ECs by shRNA transduction, and analyzed the cell membrane dynamics with or without SB203580 (Figure 6). Consistent with the role of VEGF in promoting actin polymerization, VEGF increased membrane extension at the edge of the mock-transduced control cells (Figure 6A–C). R-Ras silencing significantly increased the VEGF-dependent membrane extension (Figure 6C). The fraction of retracting membrane area was reduced because the cell membrane was mostly extending in the first 10–15 min of VEGF stimulation. The treatment of cells with SB203580 abolished VEGF-dependent membrane protrusion activity to a similar level in the control and R-Ras-silenced ECs, demonstrating that p38MAPK inhibition offsets the effect of R-Ras silencing (Figure 6C). Based on these results, we conclude that the activity of p38MAPK is responsible for the enhanced membrane extension caused by the R-Ras deficiency. These observations demonstrate that endogenous R-Ras attenuates EC membrane dynamics upon VEGF stimulation via downregulation of VEGF-dependent p38MAPK activity. This observation is consistent with the effect of R-Ras on VEGFR2 Tyr1214 phosphorylation upstream of p38MAPK.
Figure 6. R-Ras silencing enhances VEGF-induced cell membrane projection.

(A) ECs transduced with shRNA for R-Ras or negative control shRNA (NC) were treated with or without 10 nM p38MAPK inhibitor SB203580 for 2 hours. Cells were then stimulated with 50 ng/ml VEGF and observed by time-laps microscopy for the changes of the cell shape between 10 and 15 min post VEGF stimulation (indicated by arrows). (B) Cell shape changes with the areas of projecting (red) and retracting (blue) cell membrane are indicated. (C) The fraction of projecting (red) and retracting (blue) membrane area versus total cell area (% area change) was determined. **p < 0.01, p***<0.001.
R-Ras inhibits migration of ECs toward VEGF
Lamellipodia extension and membrane protrusion driven by actin polymerization are essential to cell migration [31]. Since we observed the effect of R-Ras on VEGF-induced membrane dynamics, we next analyzed how R-Ras affects directional migration of ECs toward VEGF using a culture-insert system. The expression of R-Ras38V blocked EC migration toward VEGF (Figure 7A). On the other hand, R-Ras silencing increased EC migration toward VEGF (Figure 7B). These results are in agreement with the effect of R-Ras that strongly attenuates VEGFR2 Tyr 1214 phosphorylation, p38MAPK activation, and HSP27 phosphorylation (Figure 1–3) that are necessary for VEGF to promote EC migration.
Figure 7. R-Ras inhibits directional EC migration toward VEGF.

(A and B) R-Ras38V-transduced and control ECs were starved for growth factors for 8 hours and placed into the upper compartment of Transwell® chamber. VEGF (5ng/ml) was added into the lower compartment and incubated for 2 hours. Migration of ECs toward VEGF was quantified. n=5, *p<0.001 (B) R-Ras-silenced ECs were starved for growth factors for 8 hours. Cells were treated with 10 nM SB203580 for 2 hours before migration assay. n=5. *p < 0.05, ns: non-significant difference.
The inhibition of p38MAPK by SB203580 reduced migration of control ECs by 60% underscoring the importance of p38MAPK for VEGF-induced EC migration (Figure 7B). SB203580 reduced migration of R-Ras-silenced cells to the same level as the SB203580-treated control cells without R-Ras silencing. Thus, the two cell types with different R-Ras expression levels did not exhibit any difference in migration activity upon p38MAPK inhibition. This result combined with the strong inhibitory effect of R-Ras on VEGF-p38MAPK signaling (Figure 1–3) suggest that R-Ras inhibits EC migration via downregulation of p38MAPK activation by VEGF.
Discussion
Previous studies have shown the importance of R-Ras for negatively regulating the effect of VEGF on angiogenesis. For instance, R-Ras inhibits tumor angiogenesis and VEGF-dependent neovascularization in Matrigel implants in mice [11, 12]. Consistent with this in vivo role, R-Ras has been shown to inhibit internalization of VEGFR2 upon VEGF stimulation thereby attenuating the activation and autophosphorylation of the receptor [21]. In this study, we investigated how this R-Ras activity impacts the VEGF signaling and EC activities, with a particular focus on the events downstream of VEGFR2 Tyr 1214 phosphorylation. We showed that R-Ras strongly downregulates p38MAPK activation and its downstream HSP27 phosphorylation, concomitantly with the reduction of VEGFR2 Tyr 1214 phosphorylation. It has been well-documented that the VEGFR2-p38MAPK-HSP27 pathway is crucial for mediating the effect of VEGF on actin stress fiber assembly, lamellipodia formation, and migration of ECs [24, 28, 31]. The inhibition of this pathway by R-Ras was associated with a distinct pattern of actin cytoskeleton architecture, reduced lemellipodia formation, and reduced membrane protrusion in VEGF-stimulated ECs. In addition, the directional migration of ECs toward VEGF was blocked by constitutively activated R-Ras and enhanced by silencing of endogenous R-Ras. The inhibition of p38MAPK by SB203580 offset membrane protrusion and migration activities enhanced by R-Ras silencing. R-Ras38V did not affect the basal levels of p38MAPK and HSP27 phosphorylation before VEGF stimulation. Likewise, R-Ras silencing had little effect on the p38MAPK and HSP27 phosphorylation levels before VEGF stimulation. These results demonstrate that the effects of R-Ras on p38MAPK and HSP27 are attributable to the inhibition of the VEGF signaling. Mitogen-activated-protein kinase-activated protein (MAPKAP) kinase-2 forms a molecular complex with p38MAPK and HSP27 [33], and it mediates VEGFR2-p38MAPK signaling [34]. The p38MAPK inhibitor we used in this study, SB203580, suppresses the activation of MAPKAPK-2 by p38MAPK and subsequent phosphorylation of HSP27 [35]. Based on these observations, we conclude that R-Ras inhibits the induction of membrane protrusion and migration by VEGF mainly by downregulating the VEGFR2-p38MAPK-HSP27 axis of VEGF signaling. This finding suggests a mechanism by which R-Ras promotes the quiescence of ECs and attenuates vessel sprouting to prevent excessive angiogenesis. Supporting the relevance of our findings to angiogenesis, we found elevated HSP27 phosphorylation in the endothelium of tumor vasculature formed in R-Ras KO mice, which exhibit exaggerated and defective tumor angiogenesis and VEGF-induced angiogenesis [11, 12].
The phalloidin staining of ECs revealed that R-Ras has profound influence on actin cytoskeleton architecture before and after VEGF stimulation. It is conceivable that, in VEGF-stimulated cells, R-Ras negatively regulates actin polymerization and stress fiber assembly through its inhibitory effect on the p38MAPK axis of VEGF signaling. However, ECs expressing constitutively active R-Ras do show enhanced stress fibers upon VEGF stimulation albeit the structure of these actin fibers is quite distinct from that of wild type ECs. It should be noted that the expression of activated R-Ras did not abolish p38MAPK activation by VEGF. Rather, it strongly attenuated p38MAPK activation: there was a 13 fold increase in Thr 180/Tyr 182 phosphorylation in R-Ras38V-transduced cells as compared with a 30 fold increase in the mock-transduced control cells. The cells expressing activated R-Ras exhibited well-organized uniform and parallel stress fibers. Interestingly, the formation of these stress fibers did not support cell migration. It may have even inhibited migration as cell migration was blocked concomitantly with the formation of such actin structures. The formation of this type of stress fibers could not be reproduced in the control wild type cells by direct inhibition of p38MAPK using a chemical inhibitor. These observations suggest that there are additional mechanism(s) underlying the regulation of endothelial actin cytoskeleton architecture by R-Ras. The role of R-Ras in integrin adhesion is well established [15]. Integrin focal adhesion anchors actin cytoskeleton to ECM and regulates the assembly and organization of actin stress fibers [36]. It is likely that integrin focal adhesion contributes to the actin network organization by R-Ras. The significance of integrin-dependent R-Ras effect should be investigated in additional studies. Of note, similarly to what we observed in the confluent R-Ras-transduced ECs under VEGF stimulation, the confluent ECs under laminar shear stress are known to develop well-organized stress fiber alignment, which is parallel to the long axis of the cells and continuous with the stress fibers in adjacent cells [32]. ECs are exposed to shear flow under physiological conditions. The appropriate laminar flow with high shear stress enhances expression of certain genes and proteins, which stabilize ECs and protect against atherosclerosis [37]. The signaling induced by shear stress and R-Ras signaling may share a common molecular mechanism to maintain vascular homeostasis via actin cytoskeleton organization.
Among many Ras-like small GTPases, R-Ras is, to our best knowledge, the only Ras protein that has been shown to inhibit VEGF signaling and suppresses angiogenic phenotype of ECs. The expression of R-Ras is high in quiescent ECs of mature vessels [12]. In contrast, R-Ras is significantly downregulated in ECs during developmental and pathological vessel formation [12]. This downregulation would allow ECs to fully respond to VEGF and initiate cell migration in order to develop new vessel sprouts as instructed by the angiogenic cue. Nonetheless, ECs must cease migration at appropriate timing to allow new sprouts to stabilize and form functional vessels. Our findings suggest that R-Ras provides a mechanism to prevent unnecessary or excessive angiogenic response to VEGF. Previously, we found that R-Ras is important for structural and functional maturation of regenerating blood vessels and normalization of tumor vasculature [11]. Considering this role of R-Ras, the results of our current study suggests a potential link between vessel maturation/normalization and the attenuation of the VEGFR2-p38MAPK-HSP27 axis. The normalization of tumor vasculature could enhance chemotherapy by improving drug delivery and penetration [38]. Ralimetinib, a p38MAPK inhibitor, in combination with conventional chemotherapy is currently in clinical trial for treating ovarian cancer patients. It would be interesting to investigate how this p38MAPK inhibition therapy alters the tumor vasculature in human cancer and consequently affects the overall outcome of the combination therapy.
Conclusion
Based on our results, we conclude that R-Ras inhibits VEGF-induced membrane protrusion and migration via downregulation of the p38MAPK-HSP27 axis of VEGF signaling. Our study postulates how R-Ras contributes to quiescence of ECs and vessel stabilization through inhibition of overstimulation by VEGF.
Highlights.
Inhibition of a VEGF pathway by a Ras GTPase
R-Ras suppresses the p38MAPK-HSP27 axis of VEGF signaling
R-Ras regulates VEGF-dependent endothelial cell membrane dynamics and migration
This finding suggests a mechanism by which R-Ras attenuates excessive angiogenesis
Acknowledgments
Histological preparations and imaging were done at the Histology and Imaging core facilities of Sanford-Burnham-Prebys Medical Discovery Institute at Lake Nona. This work was supported by the National Cancer Institute Grant CA125255, Florida Department of Health Bankhead-Coley Cancer Research Program 4BB17, National Science Foundation CBET-1403535, and American Heart Association 15POST25700319.
References
- 1.Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cellular signalling. 2007;19:2003–2012. doi: 10.1016/j.cellsig.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara N. Vascular endothelial growth factor. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:789–791. doi: 10.1161/ATVBAHA.108.179663. [DOI] [PubMed] [Google Scholar]
- 3.Gavard J, Gutkind JS. Vegf controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of ve-cadherin. Nature cell biology. 2006;8:1223–1234. doi: 10.1038/ncb1486. [DOI] [PubMed] [Google Scholar]
- 4.Darland DC, D'Amore PA. Blood vessel maturation: Vascular development comes of age. The Journal of clinical investigation. 1999;103:157–158. doi: 10.1172/JCI6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nature reviews Drug discovery. 2011;10:417–427. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- 6.Jain RK. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 7.Behl T, Kotwani A. Exploring the various aspects of the pathological role of vascular endothelial growth factor (vegf) in diabetic retinopathy. Pharmacol Res. 2015 doi: 10.1016/j.phrs.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 8.Said SS, Pickering JG, Mequanint K. Advances in growth factor delivery for therapeutic angiogenesis. Journal of vascular research. 2013;50:35–51. doi: 10.1159/000345108. [DOI] [PubMed] [Google Scholar]
- 9.Mughal NA, Russell DA, Ponnambalam S, Homer-Vanniasinkam S. Gene therapy in the treatment of peripheral arterial disease. The British journal of surgery. 2012;99:6–15. doi: 10.1002/bjs.7743. [DOI] [PubMed] [Google Scholar]
- 10.Giacca M, Zacchigna S. Vegf gene therapy: Therapeutic angiogenesis in the clinic and beyond. Gene therapy. 2012;19:622–629. doi: 10.1038/gt.2012.17. [DOI] [PubMed] [Google Scholar]
- 11.Sawada J, Urakami T, Li F, Urakami A, Zhu W, Fukuda M, Li DY, Ruoslahti E, Komatsu M. Small gtpase r-ras regulates integrity and functionality of tumor blood vessels. Cancer cell. 2012;22:235–249. doi: 10.1016/j.ccr.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komatsu M, Ruoslahti E. R-ras is a global regulator of vascular regeneration that suppresses intimal hyperplasia and tumor angiogenesis. Nature medicine. 2005;11:1346–1350. doi: 10.1038/nm1324. [DOI] [PubMed] [Google Scholar]
- 13.Marte BM, Rodriguez-Viciana P, Wennstrom S, Warne PH, Downward J. R-ras can activate the phosphoinositide 3-kinase but not the map kinase arm of the ras effector pathways. Curr Biol. 1997;7:63–70. doi: 10.1016/s0960-9822(06)00028-5. [DOI] [PubMed] [Google Scholar]
- 14.Lowe DG, Goeddel DV. Heterologous expression and characterization of the human r-ras gene product. Molecular and cellular biology. 1987;7:2845–2856. doi: 10.1128/mcb.7.8.2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Z, Vuori K, Wang H, Reed JC, Ruoslahti E. Integrin activation by r-ras. Cell. 1996;85:61–69. doi: 10.1016/s0092-8674(00)81082-x. [DOI] [PubMed] [Google Scholar]
- 16.Mazzone M, Dettori D, Leite de Oliveira R, Loges S, Schmidt T, Jonckx B, Tian YM, Lanahan AA, Pollard P, Ruiz de Almodovar C, De Smet F, Vinckier S, Aragones J, Debackere K, Luttun A, Wyns S, Jordan B, Pisacane A, Gallez B, Lampugnani MG, Dejana E, Simons M, Ratcliffe P, Maxwell P, Carmeliet P. Heterozygous deficiency of phd2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136:839–851. doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meadows KN, Bryant P, Pumiglia K. Vascular endothelial growth factor induction of the angiogenic phenotype requires ras activation. The Journal of biological chemistry. 2001;276:49289–49298. doi: 10.1074/jbc.M108069200. [DOI] [PubMed] [Google Scholar]
- 18.Meadows KN, Bryant P, Vincent PA, Pumiglia KM. Activated ras induces a proangiogenic phenotype in primary endothelial cells. Oncogene. 2004;23:192–200. doi: 10.1038/sj.onc.1206921. [DOI] [PubMed] [Google Scholar]
- 19.Lakshmikanthan S, Sobczak M, Chun C, Henschel A, Dargatz J, Ramchandran R, Chrzanowska-Wodnicka M. Rap1 promotes vegfr2 activation and angiogenesis by a mechanism involving integrin alphavbeta(3) Blood. 2011;118:2015–2026. doi: 10.1182/blood-2011-04-349282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westenskow PD, Kurihara T, Aguilar E, Scheppke EL, Moreno SK, Wittgrove C, Marchetti V, Michael IP, Anand S, Nagy A, Cheresh D, Friedlander M. Ras pathway inhibition prevents neovascularization by repressing endothelial cell sprouting. The Journal of clinical investigation. 2013;123:4900–4908. doi: 10.1172/JCI70230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sawada J, Li F, Komatsu M. R-ras protein inhibits autophosphorylation of vascular endothelial growth factor receptor 2 in endothelial cells and suppresses receptor activation in tumor vasculature. The Journal of biological chemistry. 2015;290:8133–8145. doi: 10.1074/jbc.M114.591511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katsogiannou M, Andrieu C, Rocchi P. Heat shock protein 27 phosphorylation state is associated with cancer progression. Front Genet. 2014;5:346. doi: 10.3389/fgene.2014.00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi M, Nishita M, Mishima T, Ohashi K, Mizuno K. Mapkapk-2-mediated limkinase activation is critical for vegf-induced actin remodeling and cell migration. The EMBO journal. 2006;25:713–726. doi: 10.1038/sj.emboj.7600973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rousseau S, Houle F, Huot J. Integrating the vegf signals leading to actin-based motility in vascular endothelial cells. Trends in cardiovascular medicine. 2000;10:321–327. doi: 10.1016/s1050-1738(01)00072-x. [DOI] [PubMed] [Google Scholar]
- 25.Rousseau S, Houle F, Kotanides H, Witte L, Waltenberger J, Landry J, Huot J. Vascular endothelial growth factor (vegf)-driven actin-based motility is mediated by vegfr2 and requires concerted activation of stress-activated protein kinase 2 (sapk2/p38) and geldanamycin-sensitive phosphorylation of focal adhesion kinase. The Journal of biological chemistry. 2000;275:10661–10672. doi: 10.1074/jbc.275.14.10661. [DOI] [PubMed] [Google Scholar]
- 26.Huot J, Houle F, Spitz DR, Landry J. Hsp27 phosphorylation-mediated resistance against actin fragmentation and cell death induced by oxidative stress. Cancer research. 1996;56:273–279. [PubMed] [Google Scholar]
- 27.Guay J, Lambert H, Gingras-Breton G, Lavoie JN, Huot J, Landry J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. Journal of cell science. 1997;110(Pt 3):357–368. doi: 10.1242/jcs.110.3.357. [DOI] [PubMed] [Google Scholar]
- 28.Rousseau S, Houle F, Landry J, Huot J. P38 map kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15:2169–2177. doi: 10.1038/sj.onc.1201380. [DOI] [PubMed] [Google Scholar]
- 29.Piotrowicz RS, Hickey E, Levin EG. Heat shock protein 27 kda expression and phosphorylation regulates endothelial cell migration. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1998;12:1481–1490. doi: 10.1096/fasebj.12.14.1481. [DOI] [PubMed] [Google Scholar]
- 30.Huot J, Houle F, Marceau F, Landry J. Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells. Circulation research. 1997;80:383–392. doi: 10.1161/01.res.80.3.383. [DOI] [PubMed] [Google Scholar]
- 31.Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circulation research. 2007;100:782–794. doi: 10.1161/01.RES.0000259593.07661.1e. [DOI] [PubMed] [Google Scholar]
- 32.Noria S, Xu F, McCue S, Jones M, Gotlieb AI, Langille BL. Assembly and reorientation of stress fibers drives morphological changes to endothelial cells exposed to shear stress. The American Journal of Pathology. 2004;164:1211–1223. doi: 10.1016/S0002-9440(10)63209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng C, Lin Z, Zhao ZJ, Yang Y, Niu H, Shen X. Mapk-activated protein kinase-2 (mk2)-mediated formation and phosphorylation-regulated dissociation of the signal complex consisting of p38, mk2, akt, and hsp27. The Journal of biological chemistry. 2006;281:37215–37226. doi: 10.1074/jbc.M603622200. [DOI] [PubMed] [Google Scholar]
- 34.Cote MC, Lavoie JR, Houle F, Poirier A, Rousseau S, Huot J. Regulation of vascular endothelial growth factor-induced endothelial cell migration by lim kinase 1-mediated phosphorylation of annexin 1. The Journal of biological chemistry. 2010;285:8013–8021. doi: 10.1074/jbc.M109.098665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. Sb 203580 is a specific inhibitor of a map kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS letters. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- 36.Parsons JT, Martin KH, Slack JK, Taylor JM, Weed SA. Focal adhesion kinase: A regulator of focal adhesion dynamics and cell movement. Oncogene. 2000;19:5606–5613. doi: 10.1038/sj.onc.1203877. [DOI] [PubMed] [Google Scholar]
- 37.Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiological reviews. 2011;91:327–387. doi: 10.1152/physrev.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jain RK. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer cell. 2014;26:605–622. doi: 10.1016/j.ccell.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]