

Abstract
The blood–brain barrier (BBB) is located within a unique anatomic interface and has functional ramifications to most of the brain and blood cells. In the past, the BBB was considered a pharmacokinetic impediment to antiepileptic drug penetration into the brain; nowadays it is becoming increasingly evident that targeting of the damaged or dysfunctional BBB may represent a therapeutic approach to reduce seizure burden. Several studies have investigated the mechanisms linking the onset and sustainment of seizures to BBB dysfunction. These studies have shown that the BBB is at the crossroad of a multifactorial pathophysiologic process that involves changes in brain milieu, altered neuroglial physiology, development of brain inflammation, leukocyte–endothelial interactions, faulty angiogenesis, and hemodynamic changes leading to energy mismatch. A number of knowledge gaps, conflicting points of view, and discordance between clinical and experimental data currently characterize this field of neuroscience. As more pieces are added to this puzzle, it is apparent that each mechanism needs to be validated in an appropriate clinical context. We now offer a BBB-centric view of seizure disorders, linking several aspects of seizures and epilepsy physiopathology to BBB dysfunction. We have reviewed the therapeutic, antiseizure effect of drugs that promote BBB repair. We also present BBB neuroimaging as a tool to correlate BBB restoration to seizure mitigation. Add-on cerebrovascular drug could be of efficacy in reducing seizure burden when used in association with neuronal antiepileptic drugs.
Keywords: Blood–brain barrier, Brain-periphery axis, Brain homeostasis, Inflammation, Glucocorticosteroids
A healthy brain requires a healthy blood-brain barrier (BBB) (Zlokovic, 2008; Abbott et al., 2010; Neuwelt et al., 2011). The BBB is a microanatomic system of capillaries functionally coupled with parenchymal brain cells. The BBB controls the blood-to-brain exchange of nutrients, xenobiotics, blood components, and cells, ultimately maintaining the optimal brain milieu necessary for physiologic neuronal function. BBB dysfunction results from a multicellular failure, endothelial cells and astrocytes being among the most studied cells in BBB pathology (Abbott et al., 2010; Neuwelt et al., 2011).
Disturbance of the blood-to-brain equilibrium can be a cause or consequence of central nervous system diseases (Zlokovic, 2008). The unique location of the BBB makes it susceptible to both brain and peripheral pathologic triggers. Taking into account the extent and the complexity of the blood–brain interface and the anatomic proximity of BBB microvessels to neurons, it has long being suspected that BBB failure may affect neuronal firing (Cornford & Oldendorf, 1986). This hypothesis has transitioned from being an experimental observation to a clinically accepted fact. Although BBB damage has been demonstrated to trigger and sustain seizures, a great deal of controversy still surrounds the attempt to assign an etiologic priority to the BBB in the transition from a normal to an ictal state (Fig. 1).
Figure 1.
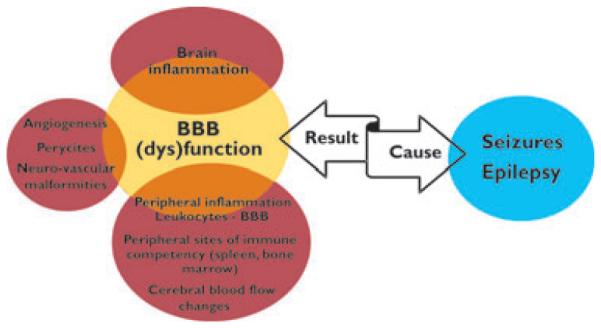
Blood–brain barrier (BBB) dysfunction in seizure disorders. The BBB represents the main vascular interface between brain and blood. Owing to its anatomic location and its unique physiologic properties, the BBB controls brain metabolism and cell- to-cell communication, and it maintains the composition of the extracellular brain milieu. BBB dysfunction has direct effects on virtually all brain cells; BBB dysfunction determines deviation from the physiologic crosstalk between the periphery and the brain. BBB dysfunction can be a cause or the result of seizure activity.
Epilepsia © ILAE
What Is (Not) Known about BBB Mechanisms of Seizures and Epilepsy?
Human studies
Although epilepsies affect approximately 1% of the population, seizures occur in a larger number of subjects, with different clinical implications and management existing between the two. If repetitive seizure activity is a hallmark of epilepsy, sporadic seizures are caused by a combination of central nervous system (CNS) and systemic pathologic triggers. The distinction between seizure and epilepsy is thus not only semantic but germane to the role of BBB dysfunction. Although the occurrence of seizure does not necessarily grant development of epilepsy, seizures represent a nonspecific response of neurons to a broad variety of prodromic stimuli.
A BBB response to seizures was proposed by Cornford in 1986 (Cornford & Oldendorf, 1986). These pioneering studies “forecasted an increased interest and understanding of the ultrastructural events associated with capillaries in seizure states.” Electron microscopy studies showed increased pinocytotic activity, tight junction aberration, and a thickening of the capillary basement membrane in epileptic brains (Cornford, 1999). Shortly thereafter, a link between the BBB, glucose, and brain ion homeostasis was demonstrated. Evidence obtained from human cortical brain resections indicated faulty expression of the BBB glucose transporter GLUT1, whereas [18F]-deoxyglucose positron emission tomography studies showed decreased brain glucose uptake and hypometabolism in the epileptic area. It is accepted that lack of GLUT1 at the BBB pathologically interferes with brain energy levels and function, thereby promoting seizures (Janigro, 1999).
More recent evidence directly demonstrates that BBB damage or dysfunction (1) promotes seizures, (2) contributes to epileptogenesis, and (3) favors seizure recurrence in epileptics (Oby & Janigro, 2006; Marchi et al., 2007; van Vliet et al., 2007; Tomkins et al., 2008; Raabe et al., 2012). A role for BBB dysfunction was also proposed for super refractory status epilepticus (Shorvon & Ferlisi, 2011). Although sudden opening of the BBB facilitates seizure onset (Marchi et al., 2007), long-lasting BBB breakdown is a hallmark of cortical injury associated with altered electroencephalography (EEG) activity (Tomkins et al., 2008). EEG frequency and activity are increased in areas of BBB disruption following traumatic brain injury (TBI). A correlation between focal BBB damage and abnormal neuronal activity was made evident by Gd++ T1-weighted spin-echo scans in subjects where, prior to scan, seizures were not recorded. (Tomkins et al., 2008). Prevention of epileptogenesis after brain trauma remains a clinical challenge (Pitkanen & Lukasiuk, 2011).
Epilepsies manifest with variable extents of BBB dysfunction. Symptomatic and drug-resistant epilepsies are associated with well-defined brain pathologies (e.g., malformations of cortical development, tuberous sclerosis, brain neoplasms, hippocampal sclerosis) where altered BBB morphology, leaky vessels, and aberrant neurovascular structure are observed. The broad hypothesis linking BBB damage to seizures is in agreement with histologic studies showing albumin accumulation in the human epileptic brain (van Vliet et al., 2007; Marchi et al., 2010; Raabe et al., 2012).
The link between BBB damage and seizures is currently considered a “puzzle of the chicken and the egg” (Friedman, 2011). In the case of new onset, iatrogenic acute seizures, the causative role of BBB damage may be easier to recognize. Seizures can be elicited during iatrogenic interventions affecting BBB function, such as cardiopulmonary bypass or during endarterectomy (Okamura et al., 2010). A systemic-intravascular inflammation, occurring as a result of the surgical intervention, is one of the proposed mechanisms underlying BBB damage. Long-term cognitive impairment may follow these procedures; BBB damage, leading to EEG abnormalities, may be one of the factors contributing to brain damage (Okamura et al., 2010). However, direct effects on cerebral perfusion cannot be ruled out. Therefore, stroke and cerebrovascular accidents account for a significant portion of newly diagnosed seizures (Balami et al., 2011). Recent evidence has shown that spreading depression (a wave of cortical, neuronal depolarization), occurring in patients suffering from stroke or traumatic brain injury, provokes changes in the cerebrovascular tone (Dreier, 2011; Dreier et al., 2012). The latter leads to cerebral perfusion alterations, contributing to lesion progression and postischemic seizures (Dreier et al., 2012).
Acute seizures may also result from poorly controlled diabetes or blood osmolality changes. BBB damage may be involved in the generation of seizures in diabetic subjects who are affected by nonketotic hyperglycemia (Kim et al., 2011). Pharmacologic intervention targeting the BBB may therefore be an important tool to prevent acute, comorbid seizures occurring after TBI, stroke, and iatrogenic vascular interventions.
Basic and translational experimental evidence
The etiologic role of BBB damage in seizure disorders is supported by experimental evidence produced using various models of BBB damage (Ivens et al., 2007; Marchi et al., 2007; Rigau et al., 2007; Tomkins et al., 2007; van Vliet et al., 2007; Marchi et al., 2010; Morin-Brureau et al., 2011). Hypersynchronous epileptiform activity was recorded following both acute and long-lasting disruption of the endothelial tight junctions (Seiffert et al., 2004; Marchi et al., 2007). Acute osmotic opening of the BBB causes electrographic seizures characterized by spike-wave complexes and synchronization of neuronal activity. A similar approach was used to demonstrate the role of BBB damage in the progression of epileptogenesis in rats (van Vliet et al., 2007). Evidence also favors a role for BBB failure in cortical-spreading depression or “Jacksonian march” (Tomkins et al., 2007).
A plethora of mechanisms have been suggested to explain how BBB damage facilitates seizure occurrence. Seizurepromoting serum components (e.g., albumin, glutamate, and K+) reach the neurons in conditions of damaged BBB. The passage of serum albumin across the damaged BBB into the brain parenchyma alters neuronal excitability (Ivens et al., 2007). Direct brain exposure to serum albumin is associated with down-regulation of inward-rectifying potassium channels in astrocytes and reduced buffering capacity. Transforming growth factor beta (TGF-b) signaling mediates this effect (Ivens et al., 2007; Tomkins et al., 2007; Friedman et al., 2009). Faulty K+ buffering affects brain energy and homeostasis by way of an ATP-dependent mechanism. In rodent and human epileptic brains, K+ buffering is compromised due to a reduced Kir 4.1 expression.
In addition to K+ channels, brain expression and function of aquaporin-4 (AQP4) was recently shown to be altered in human temporal lobe epilepsy and in experimental models of seizures (Binder & Steinhauser, 2006; Friedman, 2011). Aquaporins (AQPs) are water channels expressed in various body organs including the brain (Amiry-Moghaddam et al., 2010). AQP4 is expressed by astrocyte end-feet at the BBB. AQP4 parenchymal expression is increased in sclerotic hippocampi but reduced at the perivascular side of BBB. Lack of perivascular BBB AQP4 contributes to water flux and K+ buffering impairment.
In conclusion, human studies suggestive of a role of BBB dysfunction in seizure disorders are supported by data obtained using animal models. From an electrophysiologic point of view, BBB damage allows for serum albumin penetration into the brain parenchyma, loss of water, K+ and glutamate management, ultimately altering the extracellular brain milieu and neuronal functions.
Imaging of the BBB in the Epileptic Brain: Current Status and Challenges
Human studies
Specific imaging routines are used to localize brain lesion associated with drug-resistant seizures and to define ictal perfusion and metabolic changes. Although routines may vary among hospitals, magnetic resonance imaging (MRI), positron emission tomography (PET), single photon emission computed tomography (SPECT), and in some cases CT, and their colocalization with scalp EEG, subdural grids, or depth recordings are of fundamental diagnostic value. A clear pathophysiologic distinction exists between remote and local periictal imaging findings (Cole, 2004). Although the pathophysiology of a lesion occurring remotely from the location of ictal activity is not well understood, the basis for local periictal imaging findings is easier to interpret. When considering local periictal imaging abnormalities, the following are observed: hippocampal swelling, focal lesion, BBB breakdown, and altered neurovascular coupling.
The MRI modalities suitable for the evaluation of brain edema derive from the stroke arena. The evaluation of brain diffusion of water molecules can give an indirect indication of BBB status. T2 diffusion-weighted imaging (DWI), apparent diffusion coefficient (ADC) maps, and T2 fluidattenuated inversion recovery (FLAIR) are commonly used to determine stroke volume in patients (Leiva-Salinas & Wintermark, 2010). Diffusivity, or ADC, is an intrinsic property of the brain tissue reflective of local brain water content. DWI detects changes in brain water diffusion relative to both cytotoxic and vasogenic edema (Helpern & Huang, 1995; Doelken et al., 2007; Leiva-Salinas & Wintermark, 2010). Cytotoxic edema develops in response to energy mismatch, leading to brain cell swelling and reduction of the interstitial parenchymal space. Under this condition, the BBB is also progressively impaired as a result of the ionic/water imbalance, further contributing to edema. On the other hand, vasogenic edema occurs after BBB damage (Leiva-Salinas & Wintermark, 2010). The exact margins between the two types of edema remain blurred, since water fluxes across the endothelial and parenchymal cells membranes are interdependent. Vasogenic edema is routinely detected by T2-FLAIR, whereas cytotoxic edema is imaged by T2-DWI. Finally, dynamic contrast enhanced (DCE) MRI allows for the detection of BBB abnormalities and leakage in an array of brain pathologies, including multiple sclerosis, dementia, and cerebral microvascular disease (Armitage et al., 2011; Jelescu et al., 2011). DCE-MRI was shown to be more sensitive to smaller signal changes compared to other MRI sequences (Kassner & Thornhill, 2011). The use of DCE-MRI in patients with epilepsy could be instrumental in characterizing BBB lesions.
Clinical data support the presence of periictal MRI abnormalities and vasogenic edema correlating with tonic–clonic, grand mal, and generalized seizures, including those of unknown etiology (Cole, 2004; Hong et al., 2004; Doelken et al., 2007; Focke et al., 2009; Alvarez et al., 2010; Canas et al., 2010; Tanabe et al., 2011). Depending on the prevalence of the cytotoxic or vasogenic component, the resulting diffusion abnormalities can lead to ADC reduction (Szabo et al., 2005) or increase (Scott et al., 2006), respectively. DWI-T2 and ADC signals were increased in the left temporoparietal area in a case of partial status epilepticus characterized by repetitive right focal motor seizures. The localization of the ictal activity was confirmed by EEG monitoring (Hong et al., 2004). Topographic overlap between EEG and DWI was observed in patients with partial status epilepticus (Di Bonaventura et al., 2009). In addition, gadolinium-enhanced T1 is used to measure BBB damage and vasogenic edema; contrast enhancement is, however, less sensitive than FLAIR (Helpern & Huang, 1995; Hong et al., 2004; Salmenpera & Duncan, 2005; Di Bonaventura et al., 2009; Leiva-Salinas & Wintermark, 2010).
In a case of reversible posterior leukoencephalopathy syndrome (RPLS) associated with seizures, T2-FLAIR abnormalities were found at presentation and resolved after treatment (Doelken et al., 2007). Severe hypertension and vasogenic edema are the accepted mechanisms of RPLS. FLAIR-T2 and DWI–ACD were also used to determine the nature and the extent of brain edema in cases of febrile seizures (Scott et al., 2006). In a case series of eight patients with lateralized or focal neurologic dysfunction consistent with Creutzfeldt-Jakob disease (CJD), EEG focal waves colocalized with FLAIR hyperintensity (Cambier et al., 2003). A decrease in FLAIR hyperintensity was recently observed in a cohort of children with drug-resistant epilepsy who benefited from add-on glucocorticosteroid therapy (Marchi et al., 2011a).
BBB damage (detected by T2-FLAIR) represents a risk during the surgical placement of depth electrodes, triggering iatrogenic seizures. Conversely, iatrogenic seizures did not occur in patients where depth electrodes placement did not provoke BBB damage (Khoury et al., 2011). The prophylactic administration of glucocorticosteroids was efficacious in reducing cerebral swelling and edema resulting from the surgical placement of electrodes. Noteworthy, after glucocorticosteroid administration seizure frequency was also reduced, requiring longer monitoring periods to detect seizures (Araki et al., 2006). These data suggest that BBB damage favors seizures, whereas prevention of BBB damage decreases spontaneous activity.
Experimental evidence
When compared to clinical practice, the use of MRI and CT in animal models of epilepsy is in its infancy. In a recent special issue of Epilepsia one of the introductory remarks was “…this explosion of imaging capabilities at the clinical level has not yet been paralleled in the laboratory.” Increasing evidence supports of the use of MRI and CT for the detection of brain changes during acute seizures and epileptogenesis, and at time of chronic seizures. The study of epileptogenesis using experimental models provides the opportunity to evaluate the pathologic progression of the epileptic disorder. In the pre-MRI era, an extensive body of work described changes occurring at the parenchymal and BBB levels during the silent period and spontaneous seizures. It is in this time frame that MRI finds its best applicability, allowing for a longitudinal monitoring within the same animal. These research findings have been comprehensively reviewed elsewhere (please see Grohn & Pitkanen, 2007).
Initial MRI studies were conducted using the pilocarpine and kainic acid models of seizure (Grohn & Pitkanen, 2007; Grohn et al., 2011). Several postacquisition MRI sequences have been applied to animal models, including T2- and T1-weighted imaging, functional MRI, DWI and diffusion tensor imaging (DTI) (Grohn et al., 2011).
The use of T2-DWI in animal models of seizure was initially addressed by (Helpern & Huang, 1995). Cytotoxic edema was observed a few hours after the induction of status epilepticus, as a result of the abrupt onset of seizures and cell death. Vasogenic edema developed shortly after. Increase in ADC (vasogenic edema) is observed during epileptogenesis and at the onset of spontaneous seizures (see Grohn & Pitkanen, 2007; Grohn et al., 2011 for details). These findings are in agreement with a role of BBB damage in epileptogenesis and in the sustainment of seizures.
Variations of volume of specific brain areas (e.g., hippocampus, cortical areas, lateral ventricles) following stimulation of the amygdala, lithium-pilocarpine induced, or kainic acid induced seizures has also been reported. T1 and T2 MRI were performed in rats where status epilepticus was induced by lithium-pilocarpine. T1–Gd++ revealed a focal BBB breakdown shortly after status. Other studies have shown that the MRI changes induced by status epilepticus are reversible. Secondary T2-weighted changes (increased ACD), indicative of vasogenic edema, are visible a few weeks after status, when spontaneous seizures develop (Roch et al., 2002; Grohn et al., 2011). Reports showed neurovascular and glial pathologic changes detectable by T2 following status epilepticus and prolonged febrile seizures (Grohn & Pitkanen, 2007; Grohn et al., 2011).
Fluorescent tracers and serum markers of BBB integrity
Interpretation of MRI data can benefit from the correlation with postmortem microscopic brain examination. The use of fluorescent tracers, such as fluorescein isothiocyanate (FITC) or Evans Blue labeled serum proteins, allows the localization of structural and functional BBB changes (Fig. 2B,C). Edematous brain regions, as detected by T2-DWI or T2-FLAIR, can, after surgery, be histologically reanalyzed, confirming the cytotoxic or vasogenic origin of the edema. Immunodetection of albumin or immunoglobulin (Ig)Gs is used to study BBB leakage in postmortem animals or in resected human brain specimens; this approach, however, does not allow correlation of the time at which protein extravasation into the brain occurred.
Figure 2.

Measurement of BBB integrity in animal models and patients with drug-resistant epilepsy (DRE). (A) MRI can be used to detect BBB damage. Contrast enhanced (Gd++) T1 allows for the localization of cerebrovascular failure during periictal stages or at onset of seizures, when BBB damage is likely to be prominent. T2-DWI and FLAIR are more sensitive and can be used to localize brain edema during interictal periods. In addition, serum markers (e.g., S100B) can be used to evaluate BBB permeability (see panel C). Postmortem brain specimens can be analyzed for the presence of serum protein extravasates (e.g., IgG and albumin). The dotted line in (A) indicates the predicted evolution of BBB damage during ictal, periictal, and interictal stages. (B) FITC-albumin brain extravasation is observed in rats at time of seizures, indicating BBB damage. (C) Blood S100B levels are elevated during the ictal stages in patients with DRE (*p < 0.05, analysis of variance [ANOVA]). Brain extravasates of serum IgG (arrow heads and dotted line) are detected in brain specimens obtained from patients with DRE. (D) T2-FLAIR shows reduction of edema (arrow heads) in a patient with DRE who responded to add-on glucocorticosteroid therapy.
Epilepsia © ILAE
Although BBB damage allows radiologic and fluorescent tracer entry into the brain, brain-specific molecules or proteins can, conversely, gain access in the bloodstream under conditions of BBB opening. The astrocytic protein S100B has been used as a reporter of BBB integrity in brain tumors and traumatic brain injury (Vogelbaum et al., 2005). Increased S100B blood levels correlate with radiologic indexes of BBB function, such as those detected by MRI. Measurement of serum S100B has a diagnostic value in establishing a temporal relation between BBB failure and seizures monitored by EEG (Fig. 2C).
Blood–Brain Barrier, Inflammation, and Epilepsy
Activation of glial and microglial cells is a crucial, non-neuronal pathologic event contributing to seizure activity. Although a comprehensive evaluation of the literature inherent to brain inflammation in epilepsy goes beyond the scope of this review (Vezzani et al., 2011), herein we summarize the available evidence for BBB dysfunction occurring in conjunction with a misguided systemic inflammation or after inflammation in the CNS.
Brain inflammation
Glial and microglial cells respond to seizures by expressing cytokines and chemokines (Vezzani et al., 2011). Toll-like receptors (e.g., TLR-4) are also involved (Maroso et al., 2010). High-mobility group protein B1 (HMGB1) and TLR4 immunoreactivity was found in the hippocampi of patients with temporal lobe epilepsy and in animal models of seizure. Remarkably, the use of antagonists of HMGB1 and TLR4 reduced seizure burden in mice where intrahippocampal injections of kainate were delivered to induce seizures; TLR4-deficient mice are resistant to kainate-induced seizures. TLR-4 also plays an important role in the innate immune system, recognizing xenopathogens, and it is expressed by circulating monocytes/macrophages, myeloid dendritic cells, mast cells, and B lymphocytes (Rivest et al., 2000).
During epileptogenesis, an increase in interleukin (IL)-1b and IL-1 receptor brain expression was found at time of BBB damage (Ravizza et al., 2008). Although it is unclear whether inflammation preceded BBB damage or vice versa, it is obvious that cerebrovascular dysfunction, gliosis, and reactive microglia occur as a part of the same proinflammatory reaction. This concept is a departure from the orthodoxy dictating that, in epilepsy, brain cells are the sole players in the inflammatory process leading to seizures. This is perhaps not surprising given that, by definition, inflammation consists of increased vascular permeability and extravasation of serum components; this holds true for peripheral nervous system as well as CNS tissue.
Cyclooxygenase and prostaglandins can also affect neuronal excitability. Whether cyclooxygenase 2 (COX-2) plays a beneficial or detrimental effect in the epileptic brain remains controversial (Serrano et al., 2011). Recent data suggest that neuronal COX-2 promotes an early neuroprotection and a delayed neurodegeneration of pyramidal cells, thereby exacerbating the inflammatory process and BBB damage (Serrano et al., 2011). A faulty angiogenesis was demonstrated to occur during seizures (Rigau et al., 2007; Morin-Brureau et al., 2011). Proangiogenic factors were up-regulated, allowing for proteolysis processes and contributing to BBB damage. Recent data have dissected out the role of vascular endothelial growth factor (VEGF) in determining BBB damage via zona occludens (ZO-1) down-regulation. The selective targeting of this pathway may represent a therapeutic option to preserve BBB integrity and reduce seizure burden.
Peripheral inflammation
Specific BBB dysfunctions occur when circulating leukocytes bind at the intraluminal side of the endothelium. This causes increased BBB permeability to ions and proteins, brain edema, and changes in the brain milieu. In this scenario, gliosis and microglial activation are an epiphenomenon of seizures (Rivest et al., 2000; Riazi et al., 2010). Several indications support the existence of a seizure-promoting communication between the periphery and the brain in response to proinflammatory stimuli. Systemic injection of lipopolysaccharide (LPS), a component of the outer wall of gram-negative bacteria, lowers seizure threshold to pentylenetetrazol (Sayyah et al., 2003). LPS causes up-regulation of monocyte chemotactic protein-1 (MCP-1), IL-6, and TLR-4 at the BBB, in circumventricular organs, choroid plexus, and leptomeninges. Long-term administration of LPS causes an increase in BBB permeability (Stolp et al., 2011). Recent experimental evidence indicates that inflammatory colitis lowers seizure threshold. The peripheral inflammatory process was accompanied by microglial activation and tumor necrosis factor α (TNF-α) increase in the hippocampus (Riazi et al., 2010).
Activation of blood leukocytes and BBB damage is one of the mechanisms of pilocarpine and lithium-pilocarpine seizures. Pilocarpine (or lithium) activates circulating leukocytes, which in turns causes BBB disruption thereby facilitating status epilepticus (Marchi et al., 2009). Remarkably, only one tenth of pilocarpine is required when rats are pretreated with lithium, confirming that systemic inflammation induced by lithium lowers seizure threshold in a manner similar to other systemic proinflammatory treatments (e.g., LPS). Lithium activates CD3+ T lymphocytes and neutrophil/monocytes (CD3), increased CD11b+). Changes in the T-cell CD4/CD8 ratio were observed in the blood in patients with epilepsy (Eeg-Olofsson et al., 1985). Recent evidence also indicates a significant elevation of circulating natural killer (NK) and T CD8+ cells in epileptic subjects during ictal to postictal transition (Bauer et al., 2008). Similar data were found using the pilocarpine model of epilepsy where an increase of peripheral–splenic NK 1.1 and CD8+ cells was found. In this model, BBB damage was observed, whereas the brain inflammatory cell profile remained unchanged (Marchi et al., 2011b).
Determinants of Brain-to-Blood Crosstalk during Seizures
The evidence reviewed so far supports the notion that the symptoms of epilepsy and seizures are not only initiated by CNS cells but can also be caused by altered immune responses in the periphery causing havoc in the brain. This may be potentiated by the activation of sympathetic and parasympathetic neurons and pathways (Glaser & Kiecolt-Glaser, 2005). A cortical stress, such as seizures, may promote activation of the hypothalamic-pituitary-adrenal and the sympathetic adrenal-medullary axes, triggering peripheral leukocyte activation and production of proinflammatory cytokines (Glaser & Kiecolt-Glaser, 2005). Upon activation, blood leukocytes may exacerbate BBB damage. Such brain-to-periphery crosstalk may extend to peripheral organs of immunologic competence such as lymph nodes, the spleen, and bone marrow. Whether seizure activity is sufficient to mobilize immune or precursor cells from the spleen (or other immune-peripheral sites, i.e., bone marrow) needs to be further investigated. Nevertheless, under these circumstances the role of BBB remains crucial, since it resides at the anatomic interface between the periphery and the brain, responding to stimuli and cells coming from both sides.
Recent evidence suggests that leukocyte–BBB interactions facilitate seizures (Fabene et al., 2008). The sequela of events describing this interaction (leukocyte rolling, tethering, and so on) is strikingly similar to what is observed in multiple sclerosis (MS) and ischemic stroke. Similar to MS, perivascular BBB adhesion molecules play a role in the cascade of events leading to seizures. The MS drug natalizumab, an antibody against as specific integrin, reduces pilocarpine seizures by a mechanism encompassing prevention of BBB–T-lymphocyte interaction (Fabene et al., 2008). Similar results were found when using systemically administered glucocorticosteroids (e.g., dexamethasone) or a specific IL-1β receptor antagonist (IL-1RA) (Marchi et al., 2011a). Dexamethasone is a potent antiinflammatory drug that reduces the expression of adhesion molecules, whereas IL-1RA targets both peripheral and brain IL-1β receptors, thereby preventing the increase of vascular permeability and inflammation.
In the setting of seizure disorders, what could be the initiating event for misguided leukocyte-BBB interactions? The exchange of immunologic information between the brainand the periphery encompasses changes in the expression of BBB adhesion molecules (e.g., integrins, p and e selectins, vascular cell adhesion molecule [VCAM]). Recent evidence has shown that, in an experimental model, selectins are overexpressed at the luminal side of the BBB in response to abnormal electrical activity (Librizzi et al., 2007). Chemokines and their receptors (CXCL12 or SDF1 and its receptor CXCR4; CCL2 or monocyte chemotactic protein-1, MCP-1 and its receptor CCR2) are up-regulated at the time of seizures by endothelial and glial cells. Induced expression of MCP-1 in hippocampal blood vessels correlates with BBB permeability changes during epileptogenesis (Foresti et al., 2009; Xu et al., 2009; Hartman et al., 2010). Interference with the leukocyte chemokine receptor CCR5 affects seizure threshold (Louboutin et al., 2011). Seizure activity induced the expression of the CCR5 ligands macrophage inflammatory protein 1 (MIP-1) and RANTES (also known as chemokine ligand 5, CCL5), facilitating BBB leakage. A decrease of CCR5 in circulating blood cells protects from BBB failure and facilitates neurogenic repair (Louboutin et al., 2011). In the pilocarpine model, intravenous injection of “nonepileptic” mononuclear bone marrow cells decreased seizure frequency and duration (Costa-Ferro et al., 2012). This effect was accompanied by a decrease in blood and brain proinflammatory cytokines. These findings support the notion that seizure activity may extend its effect outside of the brain to include peripheral, immune organs, and cells.
Do Circulating Blood Cells Enter the Epileptic Brain?
Although the role for circulating cells and BBB expression of adhesion molecules is gaining momentum, it is unclear whether blood leukocytes execute their function at the BBB or whether they also exert parenchymal roles. Immune cells in the brain are commonly observed in specific forms of epilepsy such as West syndrome, Rasmussen’s encephalitis, or epilepsies triggered by viral or bacterial infections. Discordant evidence is available on whether leukocytes enter the CNS in drug-resistant forms of epilepsy that lack a typical inflammatory etiology (Granata et al., 2009). The discrepancy between these results may depend on the models used, the time frame chosen, and the analysis performed to determine location and number of cells (Fabene et al., 2008; Marchi et al., 2010; Zattoni et al., 2011). Recent data showed green fluorescent protein (GFP)-transfected leukocytes and CD45+ cell accumulation at the perivascular BBB space in pilocarpine-treated rodents and in epileptic human brains (Marchi et al., 2010). CD45+ cell parenchymal infiltration was instead prominent in animals where seizures were induced using intrahippocampal injection of kainate (Zattoni et al., 2011). Others found that granulocytes appear transiently in the brain during epileptogenesis, whereas monocytes/macrophages are present in the hippocampus only until chronic seizures develop (Ravizza et al., 2008).
The unpredictable nature of these data suggests that blood leukocytes entry into the brain parenchyma may not be a central event in the pathogenesis of seizures. It is possible that brain inflammation can be triggered by the interactions of activated blood leukocytes with luminal BBB adhesion molecules. This phenomenon may promote the release of diffusible factors (e.g., nitric oxide or prostaglandins) that can, in turn, have pathologic effects on neuronal excitability. Recent evidence has suggested this mode of action to play a role in the earlier stages of peripheral inflammation (Riazi et al., 2008, 2010). In addition, it is possible that the presence of leukocytes in the brain parenchyma could be a temporary event taking place only during specific stages of the progression of the disease. In addition, it is notable that the analysis of human brain tissues is not devoid of confounding factors. For instance, the use of subdural grids and depth electrodes may promote local, brain proinflammatory changes. Postsurgical, histopathologic studies have shown the presence of gliosis and leukocyte infiltrates in the brain regions corresponding to invasive EEG monitoring (Stephan et al., 2001).
New Pharmacological Options: Add-on “BBB Drugs”
Although the etiologic role of BBB failure in epilepsy is accepted, the next challenge is to develop antiseizure approaches aimed at restoring BBB integrity. The use of BBB drugs should be tailored to specific epilepsies and, most likely, not all patients with epilepsy may benefit from this therapeutic approach. BBB drugs are likely to be most effective when used as an add-on to available antiepileptic drugs, therefore targeting both BBB cells and neurons (Granata et al., 2009; Marchi et al., 2011a). Antiinflammatory drugs, such as glucocorticosteroids and adrenocorticotropic hormone, have been used in the treatment of encephalopathies (e.g., West and Lennox-Gastaut syndromes, Rasmussen encephalitis). Brain auto-antibodies are measured in serum and cerebral spinal fluid of patients affected by limbic encephalitis and complex-diffuse encephalopathies. Specific ion channel fragments may access the systemic circulation by way of a leaky BBB, triggering the production of autoantibodies; the latter may reenter the brain parenchyma and cross the brain barriers, thereby exacerbating symptoms. Systemically administered immunotherapies targeting circulating auto-antibodies are used to reduce seizure burden (Vincent et al., 2011).
The experimental and clinical evidence reviewed so far support the notion that BBB-inflammatory mechanisms are relevant to the pathophysiology of seizures even in the absence of a typical immunologic profile. This may open a therapeutic opportunity for BBB drugs such as the empirical use of glucocorticosteroids, which have been used in children with difficult-to-treat epilepsies or status epilepticus. Although most studies included small and heterogeneous series, data suggest that add-on glucocorticosteroids are effective in reducing drug-resistant seizures (Verhelst et al., 2005; Grosso et al., 2008; Granata et al., 2009; Marchi et al., 2011a). The beneficial effect of add-on glucocorticosteroids is associated with restoration of BBB function. A case report has shown the efficacy of natalizumab in a patient with drug-resistant epilepsy (Sotgiu et al., 2010). Natalizumab prevents BBB-leukocyte interaction. The antiseizure effects of systemically administered natalizumab, glucocorticosteroid, and IL-1RA support the notion that BBB drugs, acting on proinflammatory mediators, have a beneficial effect in controlling seizures. Recently, a role for BBB dysfunction in super refractory status epilepticus (SRSE) was proposed (Shorvon & Ferlisi, 2011). Of interest, the clinical protocol for the treatment of SRSE includes the use of intravenous steroids and IgG “for all patients in whom there is no cause identified for the super-refractory status epilepticus” (Shorvon & Ferlisi, 2011). It is possible that persistence of status epilepticus may be in part due to BBB damage and inflammatory processes.
Future Challenges
Before any conclusion can be made, a systematic radiologic evaluation of BBB damage in patients with drug-resistant epilepsy and patients with drug-respondent epilepsy needs to be performed; this will corroborate (or not) the urge for new BBB drugs. As previously discussed, MRI sequences are available to detect brain edema associated with BBB damage and parenchymal cellular dysfunction. Moreover, serum markers of BBB damage may constitute a viable surrogate for detecting BBB damage. However, T2-DWI and FLAIR are mostly performed on subjects affected by drug-resistant seizures, status epilepticus, or traumatic brain injury, and not in patients presenting with a new onset, low frequency or drug-respondent seizures.
A corollary of the BBB hypothesis of epilepsy is the fact that activated leukocytes sustain BBB dysfunction and seizures. A comprehensive study assessing the pattern of blood leukocyte activation in drug-resistant forms of epilepsy is needed as well as comparisons to patients with drug-respondent epilepsy. The neuroanatomic correlates of leukocyte location are also important. Therefore, if systemic-BBB inflammation is a target to treat seizures, then new antiinflammatory drugs may not require penetration into the brain. If conversely, the main targets are glial and microglia cells or extravasated leukocytes, then drug brain penetration needs to be taken into account. Finally, the selective targeting of altered proangiogenic processes occurring in the epileptic brain represents an attractive avenue for the development of new therapeutics aimed at preserving BBB functions and reducing seizures (Rigau et al., 2007; Morin-Brureau et al., 2011).
Conclusions
BBB dysfunction represents a hallmark of seizures and epilepsy. Although the importance of the BBB continues to grow, therapeutic options targeting the dysfunctional BBB are being tested in animal models and, in some cases, administrated to cohort of selected patients. BBB drugs may represent an add-on therapeutic option and could be administrated in association with available neuronal antiepileptic drugs. The continuous refinement of experimental models, new molecules, and imaging techniques is shaping this field of research, creating new possibilities to better control drugresistant forms of epilepsy.
Acknowledgments
The work was supported by Epilepsy Foundation Research Grant to Nicola Marchi. R21 HD057256 to Damir Janigro.
Footnotes
Disclosure
Drs. Marchi and Janigro hold intellectual property on the use of serum markers of BBB integrity. None of the other authors has any conflict of interest to disclose.
We confirm that we have read the Journal’s position on issue involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- Alvarez V, Maeder P, Rossetti AO. Postictal blood-brain barrier breakdown on contrast-enhanced MRI. Epilepsy Behav. 2010;17:302–303. doi: 10.1016/j.yebeh.2009.12.025. [DOI] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Hoddevik EH, Ottersen OP. Aquaporins: multifarious roles in brain. Neuroscience. 2010;168:859–861. doi: 10.1016/j.neuroscience.2010.04.071. [DOI] [PubMed] [Google Scholar]
- Araki T, Otsubo H, Makino Y, Elliott I, Iida K, Ochi A, Weiss SK, Chuang SH, Rutka JT, Snead OC., III Efficacy of dexamathasone on cerebral swelling and seizures during subdural grid EEG recording in children. Epilepsia. 2006;47:176–180. doi: 10.1111/j.1528-1167.2006.00384.x. [DOI] [PubMed] [Google Scholar]
- Armitage PA, Farrall AJ, Carpenter TK, Doubal FN, Wardlaw JM. Use of dynamic contrast-enhanced MRI to measure subtle blood-brain barrier abnormalities. Magn Reson Imaging. 2011;29:305–314. doi: 10.1016/j.mri.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balami JS, Chen RL, Grunwald IQ, Buchan AM. Neurological complications of acute ischaemic stroke. Lancet Neurol. 2011;10:357–371. doi: 10.1016/S1474-4422(10)70313-6. [DOI] [PubMed] [Google Scholar]
- Bauer S, Koller M, Cepok S, Todorova-Rudolph A, Nowak M, Nockher WA, Lorenz R, Tackenberg B, Oertel WH, Rosenow F, Hemmer B, Hamer HM. NK and CD4+ T cell changes in blood after seizures in temporal lobe epilepsy. Exp Neurol. 2008;211:370–377. doi: 10.1016/j.expneurol.2008.01.017. [DOI] [PubMed] [Google Scholar]
- Binder DK, Steinhauser C. Functional changes in astroglial cells in epilepsy. Glia. 2006;54:358–368. doi: 10.1002/glia.20394. [DOI] [PubMed] [Google Scholar]
- Cambier DM, Kantarci K, Worrell GA, Westmoreland BF, Aksamit AJ. Lateralized and focal clinical, EEG, and FLAIR MRI abnormalities in Creutzfeldt-Jakob disease. Clin Neurophysiol. 2003;114:1724–1728. doi: 10.1016/s1388-2457(03)00109-3. [DOI] [PubMed] [Google Scholar]
- Canas N, Breia P, Soares P, Saraiva P, Calado S, Jordao C, Vale J. The electroclinical-imagiological spectrum and long-term outcome of transient periictal MRI abnormalities. Epilepsy Res. 2010;91:240–252. doi: 10.1016/j.eplepsyres.2010.07.019. [DOI] [PubMed] [Google Scholar]
- Cole AJ. Status epilepticus and periictal imaging. Epilepsia. 2004;45(Suppl. 4):72–77. doi: 10.1111/j.0013-9580.2004.04014.x. [DOI] [PubMed] [Google Scholar]
- Cornford EM. Epilepsy and the blood brain barrier: endothelial cell responses to seizures. Adv Neurol. 1999;79:845–862. [PubMed] [Google Scholar]
- Cornford EM, Oldendorf WH. Epilepsy and the blood–brain barrier. Adv Neurol. 1986;44:787–812. [PubMed] [Google Scholar]
- Costa-Ferro ZS, Souza BS, Leal MM, Kaneto CM, Azevedo CM, da Silva IC, Soares MB, Ribeiro-Dos-Santos R, Dacosta JC. Transplantation of bone marrow mononuclear cells decreases seizure incidence, mitigates neuronal loss and modulates pro-inflammatory cytokine production in epileptic rats. Neurobiol Dis. 2012;46:302–313. doi: 10.1016/j.nbd.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Di Bonaventura C, Bonini F, Fattouch J, Mari F, Petrucci S, Carni M, Tinelli E, Pantano P, Bastianello S, Maraviglia B, Manfredi M, Prencipe M, Giallonardo AT. Diffusion-weighted magnetic resonance imaging in patients with partial status epilepticus. Epilepsia. 2009;50(Suppl. 1):45–52. doi: 10.1111/j.1528-1167.2008.01970.x. [DOI] [PubMed] [Google Scholar]
- Doelken M, Lanz S, Rennert J, Alibek S, Richter G, Doerfler A. Differentiation of cytotoxic and vasogenic edema in a patient with reversible posterior leukoencephalopathy syndrome using diffusion-weighted MRI. Diagn Interv Radiol. 2007;13:125–128. [PubMed] [Google Scholar]
- Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med. 2011;17:439–447. doi: 10.1038/nm.2333. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Major S, Pannek HW, Woitzik J, Scheel M, Wiesenthal D, Martus P, Winkler MK, Hartings JA, Fabricius M, Speckmann EJ, Gorji A. Spreading convulsions, spreading depolarization and epileptogenesis in human cerebral cortex. Brain. 2012;135:259–275. doi: 10.1093/brain/awr303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eeg-Olofsson O, Prchal JF, Andermann F. Abnormalities of T-lymphocyte subsets in epileptic patients. Acta Neurol Scand. 1985;72:140–144. doi: 10.1111/j.1600-0404.1985.tb00855.x. [DOI] [PubMed] [Google Scholar]
- Fabene PF, Navarro MG, Martinello M, Rossi B, Merigo F, Ottoboni L, Bach S, Angiari S, Benati D, Chakir A, Zanetti L, Schio F, Osculati A, Marzola P, Nicolato E, Homeister JW, Xia L, Lowe JB, McEver RP, Osculati F, Sbarbati A, Butcher EC, Constantin G. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat Med. 2008;14:1377–1383. doi: 10.1038/nm.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focke NK, Bonelli SB, Yogarajah M, Scott C, Symms MR, Duncan JS. Automated normalized FLAIR imaging in MRI-negative patients with refractory focal epilepsy. Epilepsia. 2009;50:1484–1490. doi: 10.1111/j.1528-1167.2009.02022.x. [DOI] [PubMed] [Google Scholar]
- Foresti ML, Arisi GM, Katki K, Montanez A, Sanchez RM, Shapiro LA. Chemokine CCL2 and its receptor CCR2 are increased in the hippocampus following pilocarpine-induced status epilepticus. J Neuroinflammation. 2009;6:40. doi: 10.1186/1742-2094-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman A. Blood–brain barrier dysfunction, status epilepticus, seizures, and epilepsy: a puzzle of a chicken and egg? Epilepsia. 2011;52(Suppl. 8):19–20. doi: 10.1111/j.1528-1167.2011.03227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman A, Kaufer D, Heinemann U. Blood–brain barrier breakdown-inducing astrocytic transformation: novel targets for the prevention of epilepsy. Epilepsy Res. 2009;85:142–149. doi: 10.1016/j.eplepsyres.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol. 2005;5:243–251. doi: 10.1038/nri1571. [DOI] [PubMed] [Google Scholar]
- Granata T, Marchi N, Carlton E, Ghosh C, Gonzalez-Martinez J, Alexopoulos AV, Janigro D. Management of the patient with medically refractory epilepsy. Expert Rev Neurother. 2009;9:1791–1802. doi: 10.1586/ern.09.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohn O, Pitkanen A. Magnetic resonance imaging in animal models of epilepsy-noninvasive detection of structural alterations. Epilepsia. 2007;48(Suppl. 4):3–10. doi: 10.1111/j.1528-1167.2007.01236.x. [DOI] [PubMed] [Google Scholar]
- Grohn O, Sierra A, Immonen R, Laitinen T, Lehtimaki K, Airaksinen A, Hayward N, Nairismagi J, Lehto L, Pitkanen A. Multimodal MRI assessment of damage and plasticity caused by status epilepticus in the rat brain. Epilepsia. 2011;52(Suppl. 8):57–60. doi: 10.1111/j.1528-1167.2011.03239.x. [DOI] [PubMed] [Google Scholar]
- Grosso S, Farnetani M, Mostardini R, Cordelli D, Berardi R, Balestri P. A comparative study of hydrocortisone versus deflazacort in drug-resistant epilepsy of childhood. Epilepsy Res. 2008;81:80–85. doi: 10.1016/j.eplepsyres.2008.04.016. [DOI] [PubMed] [Google Scholar]
- Hartman NW, Carpentino JE, LaMonica K, Mor DE, Naegele JR, Grabel L. CXCL12-mediated guidance of migrating embryonic stem cell-derived neural progenitors transplanted into the hippocampus. PLoS ONE. 2010;5:e15856. doi: 10.1371/journal.pone.0015856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helpern JA, Huang N. Diffusion-weighted imaging in epilepsy. Magn Reson Imaging. 1995;13:1227–1231. doi: 10.1016/0730-725x(95)02036-s. [DOI] [PubMed] [Google Scholar]
- Hong KS, Cho YJ, Lee SK, Jeong SW, Kim WK, Oh EJ. Diffusion changes suggesting predominant vasogenic oedema during partial status epilepticus. Seizure. 2004;13:317–321. doi: 10.1016/j.seizure.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, Seiffert E, Heinemann U, Friedman A. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130:535–547. doi: 10.1093/brain/awl317. [DOI] [PubMed] [Google Scholar]
- Janigro D. Blood–brain barrier, ion homeostatis and epilepsy: possible implications towards the understanding of ketogenic diet mechanisms. Epilepsy Res. 1999;37:223–232. doi: 10.1016/s0920-1211(99)00074-1. [DOI] [PubMed] [Google Scholar]
- Jelescu IO, Leppert IR, Narayanan S, Araujo D, Arnold DL, Pike GB. Dual-temporal resolution dynamic contrast-enhanced MRI protocol for blood-brain barrier permeability measurement in enhancing multiple sclerosis lesions. J Magn Reson Imaging. 2011;33:1291–1300. doi: 10.1002/jmri.22565. [DOI] [PubMed] [Google Scholar]
- Kassner A, Thornhill R. Measuring the integrity of the human blood–brain barrier using magnetic resonance imaging. Methods Mol Biol. 2011;686:229–245. doi: 10.1007/978-1-60761-938-3_10. [DOI] [PubMed] [Google Scholar]
- Khoury JA, Noe KH, Drazkowski JF, Sirven JI, Zimmerman RS. Iatrogenic seizures during intracranial EEG monitoring. Epilepsia. 2011;52:e123–e125. doi: 10.1111/j.1528-1167.2011.03161.x. [DOI] [PubMed] [Google Scholar]
- Kim DW, Moon Y, Gee NH, Choi JW, Oh J. Blood–brain barrier disruption is involved in seizure and hemianopsia in nonketotic hyperglycemia. Neurologist. 2011;17:164–166. doi: 10.1097/NRL.0b013e3182173528. [DOI] [PubMed] [Google Scholar]
- Leiva-Salinas C, Wintermark M. Imaging of acute ischemic stroke. Neuroimaging Clin N Am. 2010;20:455–468. doi: 10.1016/j.nic.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librizzi L, Regondi MC, Pastori C, Frigerio S, Frassoni C, de Curtis M. Expression of adhesion factors induced by epileptiform activity in the endothelium of the isolated guinea pig brain in vitro. Epilepsia. 2007;48:743–751. doi: 10.1111/j.1528-1167.2007.01047.x. [DOI] [PubMed] [Google Scholar]
- Louboutin JP, Chekmasova A, Marusich E, Agrawal L, Strayer DS. Role of CCR5 and its ligands in the control of vascular inflammation and leukocyte recruitment required for acute excitotoxic seizure induction and neural damage. FASEB J. 2011;25:737–753. doi: 10.1096/fj.10-161851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Angelov L, Masaryk T, Fazio V, Granata T, Hernandez N, Hallene K, Diglaw T, Franic L, Najm I, Janigro D. Seizure-promoting effect of blood–brain barrier disruption. Epilepsia. 2007;48:732–742. doi: 10.1111/j.1528-1167.2007.00988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Fan Q, Ghosh C, Fazio V, Bertolini F, Betto G, Batra A, Carlton E, Najm I, Granata T, Janigro D. Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiol Dis. 2009;33:171–181. doi: 10.1016/j.nbd.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Teng Q, Ghosh C, Fan Q, Nguyen MT, Desai NK, Bawa H, Rasmussen P, Masaryk TK, Janigro D. Blood–brain barrier damage, but not parenchymal white blood cells, is a hallmark of seizure activity. Brain Res. 2010;1353:176–186. doi: 10.1016/j.brainres.2010.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Granata T, Freri E, Ciusani E, Ragona F, Puvenna V, Teng Q, Alexopolous A, Janigro D. Efficacy of anti-inflammatory therapy in a model of acute seizures and in a population of pediatric drug resistant epileptics. PLoS ONE. 2011a;6:e18200. doi: 10.1371/journal.pone.0018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Johnson AJ, Puvenna V, Johnson HL, Tierney W, Ghosh C, Cucullo L, Fabene PF, Janigro D. Modulation of peripheral cytotoxic cells and ictogenesis in a model of seizures. Epilepsia. 2011b;52:1627–1634. doi: 10.1111/j.1528-1167.2011.03080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, Rossetti C, Molteni M, Casalgrandi M, Manfredi AA, Bianchi ME, Vezzani A. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16:413–419. doi: 10.1038/nm.2127. [DOI] [PubMed] [Google Scholar]
- Morin-Brureau M, Lebrun A, Rousset MC, Fagni L, Bockaert J, de Bock F, Lerner-Natoli M. Epileptiform activity induces vascular remodeling and zonula occludens 1 downregulation in organotypic hippocampal cultures: role of VEGF signaling pathways. J Neurosci. 2011;31:10677–10688. doi: 10.1523/JNEUROSCI.5692-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwelt EA, Bauer B, Fahlke C, Fricker G, Iadecola C, Janigro D, Leybaert L, Molnar Z, O’Donnell ME, Povlishock JT, Saunders NR, Sharp F, Stanimirovic D, Watts RJ, Drewes LR. Engaging neuroscience to advance translational research in brain barrier biology. Nat Rev Neurosci. 2011;12:169–182. doi: 10.1038/nrn2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oby E, Janigro D. The blood–brain barrier and epilepsy. Epilepsia. 2006;47:1761–1774. doi: 10.1111/j.1528-1167.2006.00817.x. [DOI] [PubMed] [Google Scholar]
- Okamura T, Ishibashi N, Kumar TS, Zurakowski D, Iwata Y, Lidov HG, Jonas RA. Hypothermic circulatory arrest increases permeability of the blood brain barrier in watershed areas. Ann Thorac Surg. 2010;90:2001–2008. doi: 10.1016/j.athoracsur.2010.06.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, Lukasiuk K. Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 2011;10:173–186. doi: 10.1016/S1474-4422(10)70310-0. [DOI] [PubMed] [Google Scholar]
- Raabe A, Schmitz AK, Pernhorst K, Grote A, von der Brelie C, Urbach H, Friedman A, Becker AJ, Elger CE, Niehusmann P. Cliniconeuropathologic correlations show astroglial albumin storage as a common factor in epileptogenic vascular lesions. Epilepsia. 2012;53:539–548. doi: 10.1111/j.1528-1167.2012.03405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravizza T, Gagliardi B, Noe F, Boer K, Aronica E, Vezzani A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis. 2008;29:142–160. doi: 10.1016/j.nbd.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Riazi K, Galic MA, Kuzmiski JB, Ho W, Sharkey KA, Pittman QJ. Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc. Natl. Acad. Sci. U.S.A. 2008;105:17151–17156. doi: 10.1073/pnas.0806682105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazi K, Galic MA, Pittman QJ. Contributions of peripheral inflammation to seizure susceptibility: cytokines and brain excitability. Epilepsy Res. 2010;89:34–42. doi: 10.1016/j.eplepsyres.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Rigau V, Morin M, Rousset MC, de Bock F, Lebrun A, Coubes P, Picot MC, Baldy-Moulinier M, Bockaert J, Crespel A, Lerner-Natoli M. Angiogenesis is associated with blood–brain barrier permeability in temporal lobe epilepsy. Brain. 2007;130:1942–1956. doi: 10.1093/brain/awm118. [DOI] [PubMed] [Google Scholar]
- Rivest S, Lacroix S, Vallieres L, Nadeau S, Zhang J, Laflamme N. How the blood talks to the brain parenchyma and the paraventricular nucleus of the hypothalamus during systemic inflammatory and infectious stimuli. Proc Soc Exp Biol Med. 2000;223:22–38. doi: 10.1046/j.1525-1373.2000.22304.x. [DOI] [PubMed] [Google Scholar]
- Roch C, Leroy C, Nehlig A, Namer IJ. Magnetic resonance imaging in the study of the lithium-pilocarpine model of temporal lobe epilepsy in adult rats. Epilepsia. 2002;43:325–335. doi: 10.1046/j.1528-1157.2002.11301.x. [DOI] [PubMed] [Google Scholar]
- Salmenpera TM, Duncan JS. Imaging in epilepsy. J Neurol Neurosurg Psychiatry. 2005;76(Suppl. 3):iii2–iii10. doi: 10.1136/jnnp.2005.075135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayyah M, Javad-Pour M, Ghazi-Khansari M. The bacterial endotoxin lipopolysaccharide enhances seizure susceptibility in mice: involvement of proinflammatory factors: nitric oxide and prostaglandins. Neuroscience. 2003;122:1073–1080. doi: 10.1016/j.neuroscience.2003.08.043. [DOI] [PubMed] [Google Scholar]
- Scott RC, King MD, Gadian DG, Neville BG, Connelly A. Prolonged febrile seizures are associated with hippocampal vasogenic edema and developmental changes. Epilepsia. 2006;47:1493–1498. doi: 10.1111/j.1528-1167.2006.00621.x. [DOI] [PubMed] [Google Scholar]
- Seiffert E, Dreier JP, Ivens S, Bechmann I, Tomkins O, Heinemann U, Friedman A. Lasting blood–brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci. 2004;24:7829–7836. doi: 10.1523/JNEUROSCI.1751-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano GE, Lelutiu N, Rojas A, Cochi S, Shaw R, Makinson CD, Wang D, FitzGerald GA, Dingledine R. Ablation of cyclooxygenase-2 in forebrain neurons is neuroprotective and dampens brain inflammation after status epilepticus. J Neurosci. 2011;31:14850–14860. doi: 10.1523/JNEUROSCI.3922-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorvon S, Ferlisi M. The treatment of super-refractory status epilepticus: a critical review of available therapies and a clinical treatment protocol. Brain. 2011;134:2802–2818. doi: 10.1093/brain/awr215. [DOI] [PubMed] [Google Scholar]
- Sotgiu S, Murrighile MR, Constantin G. Treatment of refractory epilepsy with natalizumab in a patient with multiple sclerosis. Case report. BMC Neurol. 2010;10:84. doi: 10.1186/1471-2377-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan CL, Kepes JJ, SantaCruz K, Wilkinson SB, Fegley B, Osorio I. Spectrum of clinical and histopathologic responses to intracranial electrodes: from multifocal aseptic meningitis to multifocal hypersensitivity-type meningovasculitis. Epilepsia. 2001;42:895–901. doi: 10.1046/j.1528-1157.2001.042007895.x. [DOI] [PubMed] [Google Scholar]
- Stolp HB, Johansson PA, Habgood MD, Dziegielewska KM, Saunders NR, Ek CJ. Effects of neonatal systemic inflammation on blood-brain barrier permeability and behaviour in juvenile and adult rats. Cardiovasc Psychiatry Neurol. 2011;2011:469046. doi: 10.1155/2011/469046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo K, Poepel A, Pohlmann-Eden B, Hirsch J, Back T, Sedlaczek O, Hennerici M, Gass A. Diffusion-weighted and perfusion MRI demonstrates parenchymal changes in complex partial status epilepticus. Brain. 2005;128:1369–1376. doi: 10.1093/brain/awh454. [DOI] [PubMed] [Google Scholar]
- Tanabe T, Hara K, Shimakawa S, Fukui M, Tamai H. Hippocampal damage after prolonged febrile seizure: one case in a consecutive prospective series. Epilepsia. 2011;52:837–840. doi: 10.1111/j.1528-1167.2010.02958.x. [DOI] [PubMed] [Google Scholar]
- Tomkins O, Friedman O, Ivens S, Reiffurth C, Major S, Dreier JP, Heinemann U, Friedman A. Blood–brain barrier disruption results in delayed functional and structural alterations in the rat neocortex. Neurobiol Dis. 2007;25:367–377. doi: 10.1016/j.nbd.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Tomkins O, Shelef I, Kaizerman I, Eliushin A, Afawi Z, Misk A, Gidon M, Cohen A, Zumsteg D, Friedman A. Blood–brain barrier disruption in post-traumatic epilepsy. J Neurol Neurosurg Psychiatry. 2008;79:774–777. doi: 10.1136/jnnp.2007.126425. [DOI] [PubMed] [Google Scholar]
- van Vliet EA, da Costa AS, Redeker S, van Schaik R, Aronica E, Gorter JA. Blood–brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain. 2007;130:521–534. doi: 10.1093/brain/awl318. [DOI] [PubMed] [Google Scholar]
- Verhelst H, Boon P, Buyse G, Ceulemans B, D’Hooghe M, Meirleir LD, Hasaerts D, Jansen A, Lagae L, Meurs A, Coster RV, Vonck K. Steroids in intractable childhood epilepsy: clinical experience and review of the literature. Seizure. 2005;14:412–421. doi: 10.1016/j.seizure.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol. 2011;7:31–40. doi: 10.1038/nrneurol.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol. 2011;10:759–772. doi: 10.1016/S1474-4422(11)70096-5. [DOI] [PubMed] [Google Scholar]
- Vogelbaum MA, Masaryk T, Mazzone P, Mekhail T, Fazio V, McCartney S, Marchi N, Kanner A, Janigro D. S100beta as a predictor of brain metastases: brain versus cerebrovascular damage. Cancer. 2005;104:817–824. doi: 10.1002/cncr.21220. [DOI] [PubMed] [Google Scholar]
- Xu JH, Long L, Tang YC, Zhang JT, Hut HT, Tang FR. CCR3, CCR2A and macrophage inflammatory protein (MIP)-1a, monocyte chemotactic protein-1 (MCP-1) in the mouse hippocampus during and after pilocarpine-induced status epilepticus (PISE) Neuropathol Appl Neurobiol. 2009;35:496–514. doi: 10.1111/j.1365-2990.2009.01022.x. [DOI] [PubMed] [Google Scholar]
- Zattoni M, Mura ML, Deprez F, Schwendener RA, Engelhardt B, Frei K, Fritschy JM. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J Neurosci. 2011;31:4037–4050. doi: 10.1523/JNEUROSCI.6210-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. The blood–brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]