

Abstract
Age-related macular degeneration (AMD) is a major cause of blindness among the elderly in the developed world. Genetic analysis of AMD has identified 34 high-risk loci associated with AMD. The genes at these high risk loci belong to diverse biological pathways, suggesting different mechanisms leading to AMD pathogenesis. Thus, therapies targeting a single pathway for all AMD patients will likely not be universally effective. Recent evidence suggests defects in mitochondria (mt) of the retinal pigment epithelium (RPE) may constitute a key pathogenic event in some AMD patients. The purpose of this study is to determine if individuals with a specific genetic background have a greater propensity for mtDNA damage. We used human eyebank tissues from 76 donors with AMD and 42 age-matched controls to determine the extent of mtDNA damage in the RPE that was harvested from the macula using a long extension polymerase chain reaction assay. Genotype analyses were performed for ten common AMD-associated nuclear risk alleles (ARMS2, TNFRSF10A, CFH, C2, C3, APOE, CETP, LIPC, VEGF and COL10A1) and mtDNA haplogroups. Sufficient samples were available for genotype association with mtDNA damage for TNFRSF10A, CFH, CETP, VEGFA, and COL10A1. Our results show that AMD donors carrying the high risk allele for CFH (C) had significantly more mtDNA damage compared with donors having the wild-type genetic profile. The data from an additional 39 donors (12 controls and 27 AMD) genotyped for CFH alleles further supported these findings. Taken together, these studies provide the rationale for a more personalized approach for treating AMD by uncovering a significant correlation between the CFH high risk allele and accelerated mtDNA damage. Patients harboring this genetic risk factor may benefit from therapies that stabilize and protect the mt in the RPE.
Keywords: Age-related macular degeneration, Complement Factor H, eyebank tissue, mitochondria, mtDNA, haplogroups, inflammation
1. Introduction
Age-related macular degeneration (AMD) is a leading cause of blindness among older adults in the developed world (Friedman et al., 2004). The disease afflicts over 10 million people in the US and this number is expected to double by 2050 (Rein et al., 2009). Considering the staggering social and financial burden, the impending epidemic of vision loss due to AMD creates an urgent need to develop prevention and treatment strategies. Anti-VEGF therapies have revolutionized the treatment of “wet” or neovascular AMD, which includes ~10% of AMD patients and manifests as the abnormal growth of blood vessels from the choroid into the subretinal region. However, treatment options for the more prevalent “dry” or atrophic AMD are limited to a special formulation of vitamins and minerals that at best, slows down disease progression in only a subset of patients (Chew et al., 2013). Atrophic AMD presents clinically with drusen and changes in the retinal pigment epithelium (RPE). In the advanced form, the loss of RPE and photoreceptors, referred to as geographic atrophy, can occur.
Development of rational therapeutic interventions for AMD requires greater understanding of AMD disease mechanism(s) and underlying cellular pathways. Discovering the mechanism(s) has been challenging due to the multifactorial nature of AMD, where both the environment and genetic factors must be considered. Nuclear-encoded genes at 34 loci involved in several different biological pathways, including complement, cell survival, lipid transport and processing, extracellular matrix remodeling, and angiogenesis, have been associated with AMD (Fritsche et al., 2015; Fritsche et al., 2014; Montezuma et al., 2007; Ratnapriya and Chew, 2013). Additionally, the mitochondrial genome is shown to influence AMD susceptibility. For example, while mtDNA haplogroups J, T and U are associated with AMD, the H haplogroup has a protective effect (Jones et al., 2007; Kenney et al., 2013a; Mueller et al., 2012; Udar et al., 2009). The apparent defects in multiple biological pathways leading to similar clinical phenotypes suggest that a single treatment for all AMD patients may not be universally effective. Thus, we need to develop more refined ways for a priori predictions to design additional efficacious sites of intervention for individual patients.
The aim of the current study was to identify a specific population with defects in the RPE mitochondria (mt). We focused on the RPE since mtDNA damage increases with AMD severity in the RPE but not the neural retina (Terluk et al., 2015). Additionally, proteomic and histological analyses suggest that RPE mt dysfunction contributes to AMD pathology (Feher et al., 2006; Nordgaard et al., 2006, 2008). An early role for mitochondria in AMD pathology is indicated by enhanced mtDNA damage observed in human donor RPE at stages of AMD preceding macular degeneration and vision loss (Karunadharma et al., 2010; Terluk et al., 2015). The mtDNA damage is presumably due to increased production of reactive oxygen species, which cause damage to mt structures and alter redox signaling. In addition, mtDNA damage could also accumulate due to defects in processes required to maintain mt homeostasis, including mt biogenesis, fission/fusion, and autophagy, and/or due to replication errors by DNA polymerase gamma (Kennedy et al., 2013).
To examine the association of nuclear risk loci with mtDNA damage, we used human donor retinas graded for the presence and severity of AMD and genotyped for 10 common AMD risk variants (ARMS2, TNFRSF10A, CFH, C2, C3, APOE, CETP, LIPC, VEGFA, COL10A1). Mitochondrial DNA damage was measured in the macular RPE cells and the relationship of the extent of damage was compared with genotype. In addition, we assessed the mtDNA for the frequency of the JTU haplogroup cluster and H haplogroup and examined potential additive associations. Our studies demonstrate that AMD donors harboring the risk allele for CFH exhibited significantly more mtDNA damage in macular RPE.
2. Materials and Methods
2.1 Human tissue procurement and grading
Donor eyes were obtained from the Minnesota Lions Eye Bank (Minneapolis, MN, USA) with the consent of the donor or donor’s family for use in medical research in accordance with the tenets of the Declaration of Helsinki. After enucleation, eyes were maintained in a moist chamber at 4°C until photographing and dissection. Dissection included a trephine punch (5 mm) of RPE cells centered over the macula. RPE were gently dislodged from Bruch’s Membrane/choroid, flash frozen in liquid nitrogen, and stored at −80 ºC until further processing.
Evaluation of the donor’s stage of AMD was determined by a Board Certified Ophthalmologist/Retina Specialist from stereoscopic fundus photographs of the RPE using the criteria established by the Minnesota Grading System (MGS) for eyebank eyes (Decanini et al., 2007; Olsen and Feng, 2004). MGS 1 represents the control group with no clinically observable eye disease. MGS 2, 3, 4 are early, intermediate, and advanced stages of AMD, respectively. Advanced AMD (MGS 4) includes both dry AMD (central geographic atrophy) and wet AMD (choroidal neovascularisation). Exclusion criteria for the present study include a history of diabetes or glaucoma, clinical symptoms of diabetic retinopathy, advanced glaucoma, and myopic degeneration. Records from the Minnesota Lions Eye Bank provided demographics of the donors including age, race, gender, time and cause of death, and a family report of a limited medical and ocular history (Table 1). These donors were part of two previous studies (Karunadharma et al., 2010; Terluk et al., 2015), which tested the relationship between the extent of mtDNA damage and AMD severity. Analysis of an additional 39 donors, age 60 years and older, is included in this study. Donor characteristics for the new donor samples are provided in Table 3.
Table 1.
Donor Characteristics and Clinical InformationA
| MGSB Grade | Sample C (n) | Sex
|
Age
|
Cause of Death (n)D | |
|---|---|---|---|---|---|
| M | F | Mean ± SD | |||
| 1 | 42 | 24 | 18 | 75 ± 9.1E | ACE(4), ALS(1), ARDS(2), Brain Abscess(1), Cancer(15), CHF(4), COPD(3), CVA(1), GI bleed(1), organ failure(1), pneumonia(2), and sepsis(7). |
| 2 | 33 | 16 | 17 | 80 ± 8.6 | ACE(5), Cancer(6), CHF(1), COPD(2), CVA(4), GI bleed(1), Guillan Barre Syndrome(1), head injury(1), organ failure(2), pneumonia(4), sepsis(5), subdural hematoma(1). |
| 3 | 28 | 14 | 14 | 82 ± 8.0 | ACE(6), ARDS(2), aspiration hypoxia(1), Bowel obstruction(1), Cancer(5), CHF(2), COPD(3), surgical complications(1), metabolic acidosis(1), organ failure(2), pneumonia(2), and sepsis(2). |
| 4 | 15 | 5 | 10 | 81 ± 9.7 | Cancer(4), CHF(1), CVA(3), dementia(1), organ failure(3), pneumonia(1), and sepsis(2). |
ACE = acute myocardial event; ALS=amyotrophic lateral sclerosis; ARDS = acute respiratory disease syndrome; CHF = congestive heart failure. COPD = chronic obstructive pulmonary disease; CVA = cerebrovascular accident (stroke); GI bleed = gastrointestinal bleed; Organ failure=kidney and/or multiple organ failure.
Information supplied by Minnesota Lions Eye Bank.
Minnesota Grading System (MGS) was used to evaluate the stage of AMD in eye bank eyes (Olsen and Feng, 2004).
Sample number indicates the total donors used in the current study for each MGS stage. Numbers in Figure 1 indicate the donors from two previous studies (Karunadharma et al., 2010; Terluk et al., 2015) used in the current study.
The number of donors for each cause of death is indicated in parentheses.
MGS1 is significantly younger than MGS 2 and 3 (p<0.05).
Table 3.
Characteristics and Clinical Information for Added Donors A
| MGSB Grade | Sample C (n) | Sex
|
Age
|
Cause of Death (n)D | |
|---|---|---|---|---|---|
| M | F | Mean ± SD | |||
| 1 | 12 | 6 | 6 | 75 ± 5.9E | ACE(3), Anoxic brain injury (1) Cancer(5), CHF(1), CVA(1), pneumonia(1). |
| 3 | 13 | 3 | 10 | 80 ± 4.8 | ACE(3), Cancer(5), ICB/ICH (1), respiratory failure(3), sepsis(1). |
| 4 | 14 | 5 | 9 | 84 ± 6.1 | ACE (4), Cancer(1), Cardiomyopathy (1), CHF(1), CVA(4), GI bleed(1), ICB/ICH (2). |
ACE = acute myocardial event; CHF = congestive heart failure. CVA = cerebrovascular accident (stroke); GI bleed = gastrointestinal bleed; ICB/ICH = Intracranial bleed/intracranial hemorrhage.
Information supplied by Minnesota Lions Eye Bank.
Minnesota Grading System (MGS) was used to evaluate the stage of AMD in eye bank eyes (Olsen and Feng, 2004).
Sample number indicates the total donors used in the current study for each MGS stage.
The number of donors for each cause of death is indicated in parentheses.
MGS1 is significantly younger than MGS 4 (p<0.01).
2.2 Determining mtDNA damage in donor macula RPE
Total genomic DNA was isolated from a 5 mm trephine punch of RPE cells from the macula. mtDNA lesions were determined by long extension-polymerase chain reaction (PCR) assay using primers and conditions as described (Karunadharma et al., 2010; Terluk et al., 2015). Analysis of new samples (n=39) was performed using AccuPrimeTM Taq Polymerase High Fidelity (Life Technologies, Grand Island, NY). The PCR amplification profile for Regions I-IV included an initial denaturation for 30 seconds at 94°C, followed by 21 cycles of 94°C denaturation for 30 seconds for region I, and 25 cycles for regions II, III and IV. Annealing/extension was performed for 30 seconds at 57°C for regions I, II, and IV and at 53°C for region III. The final extension was performed at 68°C for 4 minutes for regions I and II, 6 minutes for region III, and 2 minutes and 30 seconds for region IV. The amplification profile for 222 bp mtDNA fragment (used to provide a measure of mtDNA content) included an initial denaturation for 30 seconds at 94°C, followed by 21 cycles of 94°C denaturation for 30 seconds, and annealing/extension at 55°C for 30 seconds and final extension at 30 seconds. Half template samples and no DNA controls were run on each plate to verify linearity and as a negative control, respectively. Lesion frequency per 10kb per genome (both strands) of the mtDNA was calculated by dividing sample amplification (relative to mtDNA content) by the maximal relative amplification from the control donors (MGS1). Lesion frequency for all samples was then normalized to the average lesion frequency of MGS1. Data are presented as mtDNA damage relative to donors without AMD (MGS1).
2.3 DNA extraction and allelic determination for nuclear genes
Genomic analysis in the Kenney lab was determined from the DNA isolated from donor neural retina. Frozen retinal tissue was incubated in 500μl of STE (100 mM NaCl, 25 mM Na2EDTA, 10 mM Tris-HCl, pH 8.0 overnight at 50°C in 0.5% SDS and 15 ug/ml Proteinase K (Invitrogen, Carlsbad, CA). The RNA was digested for 1 hour at 37°C with 5 μg/ml ribonuclease A followed by phenol extraction (Invitrogen). The DNA was precipitated with 0.5 volume of 7.5 M ammonium acetate and 2 volumes ethanol. The DNA pellet was resuspended in TE and stored at −20°C. The primers for allelic discrimina tion were synthesized by ABI Assay-by-Design based on the Reference SNP (rs) number. The samples were run at GenoSeq, the UCLA Genoytyping and Sequencing Core, on an ABI 7900HT. The data was analyzed with Sequence Detection Systems software from ABI.
Analysis of the CFH variant was performed for additional donors (n=39) in the Swaroop lab. The genomic DNA was extracted from inferior retina samples and the quantification was done using Nanodrop. A custom made TaqMan assay was used to genotype CFH_Y402H variant (rs1061170). Four duplicates, as well as four no template controls, were included in all assays. Allelic discrimination analysis was performed on 7900HT Fast Real-Time PCR System using TaqMan® Genotyper Software version 1.3.
2.4 Identification of mtDNA haplogroups
Total DNA was extracted from retinal samples of each individual and the mtDNA haplogroups were identified using either PCR with restriction enzyme digestion (Kenney et al., 2010) and/or allelic discrimination with primers synthesized by ABI Assay-by-Design (Applied Biosystems, Grand Island, NY). Samples were run at GenoSeq, the UCLA Genotyping and Sequencing Core, on an ABI 7900HT. Data were analyzed with Sequence Detection Systems software from ABI.
2.5 Statistics
Statistical analysis included one way ANOVA and Tukey’s post-hoc test to determine if there was a significant difference in donor age (Tables 1 and 3), mtDNA damage with AMD progression (Figure 1B), and in comparing mtDNA damage based on genotype (Figure 3). For the comparison of mtDNA damage for five genes, significance was set at p=0.04 following the Benjamini-Hochberg correction for multiple comparisons (20% FDR). Linear regression was performed of mtDNA damage for CFH genotyped donors to determine if the extent of damage correlated with risk allele frequency (TT=0, TC=1, CC=2) (Figure 4). A one-tailed Student’s t-test was used to test the hypothesis that the presence of the C risk allele for CFH resulted in increased mtDNA damage (Figure 4). An unpaired Student’s t-test was performed to compare the H and JUT cluster haplogroups (Figure 5). Analyses were performed using the statistical software in Origin 9.1 (Originlab Corp., Northhampton, MA). Data are reported as mean ± SEM for each group.
Figure 1. RPE mtDNA damage increases with AMD progression.

Long-extension PCR was used to determine mtDNA lesion frequency in RPE from human donor macula. Data were normalized to the mean of the non-disease age-matched controls (MGS1). Sample size for each group is provided on the graph. (A) Summary of donors from previous studies (Karunadharma et al., 2010; Terluk et al., 2015) used in the current analysis. (B) Graph of mtDNA damage for donors without AMD (MGS1) and donors with progressively more severe stages of AMD using donors from the combined data set. MGS groups were compared by one-way ANOVA and Tukey’s post hoc test. Significance was set at p<0.05. *P<0.001; MGS1 was less than MGS2, MGS3 and MGS4. Data are mean + SEM.
Figure 3. RPE mtDNA damage and AMD-associated risk variants.

Donors were separated into genotypes for five risk variants that are associated with cell survival (TNFRSF10A), the complement pathway (CFH), lipid transport/metabolism (CETP), angiogenesis (VEGFA) and remodeling of the extracellular matrix (COL10A1). Graphs show the amount of mtDNA damage (relative to the average damage of MGS1 control donors) for donors within each genotype. Genotypes are shown above the gene name; the homozygous risk alleles (dark grey bars) are underlined. The number of donors in each group is shown within the bars. mtDNA damage was compared between non-risk, heterozygous risk, and homozygous risk for each gene by one-way ANOVA and Tukey’s post-hoc test when required. The probability values are provided in the graph for each comparison. Significance was set at p=0.04 following the Benjamini-Hochberg correction for multiple comparisons. * Tukey’s post-hoc test showed CC is significantly higher than TT for CFH.
Figure 4. Association Between mtDNA Damage and the CFH Risk Allele. (A).

Refined analysis of data for CFH from Figure 3 shows the distribution of AMD donors (MGS2, 3, 4) for each CFH genotype (left panel) and the mtDNA damage measured in control (n=42) and AMD donors (n=76) (middle panel). A significant linear increase in mtDNA damage correlated with content of the C risk allele in AMD donors (p=0.049) but not in control donors (p=0.59). AMD donors harboring 1 or 2 high risk alleles had significantly higher mtDNA damage compared with AMD donors homozygous for the low risk alleles (TT) (p=0.043; right panel). (B) Distribution of AMD donors (MGS 3 and 4 only) for each genotype (left panel) includes 43 donors from previous studies (Karunadharma et al., 2010; Terluk et al., 2015) and 27 new donors. The extent of mtDNA damage for AMD and control donors for each genotype is shown in the middle panel. While there was a 20% increase in mtDNA damage in donors carrying the C allele, a linear relationship was not observed (p=0.11). Regression analysis of mtDNA damage for control donors (including 12 new donors) was not significant (p=0.09). T-test analysis (right panel) showed AMD donors harboring 1 or 2 high risk alleles had significantly higher mtDNA damage compared with AMD donors homozygous for the low risk alleles (TT) (p=0.022). Total number of donors in each group is indicated on the graph. Data shown are mean ± SEM.
Figure 5. No Correlation Between mtDNA Haplogroups and mtDNA Damage.
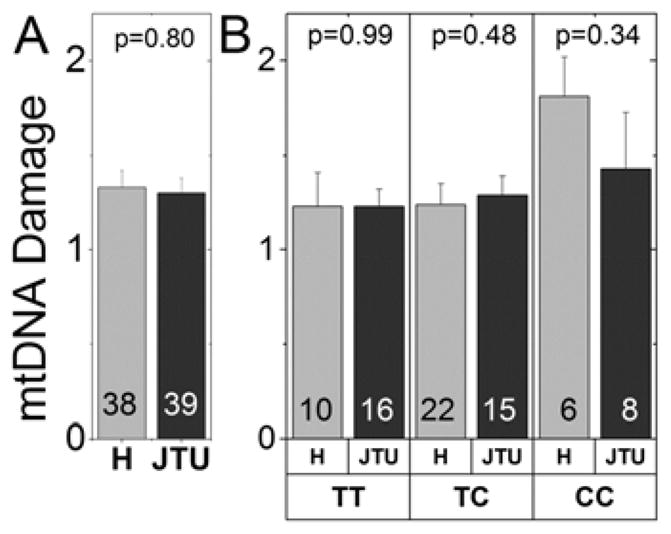
The number of donors for each comparison is provided in the bars. The p value for the t-test comparison is shown at the top of each graph. (A) Comparison of mtDNA damage for donors with H (light grey) versus JTU haplogroup (dark grey) cluster was not significantly different. (B) No additive risk for mtDNA damage was found when comparing the CFH alleles on the H and JTU cluster backgrounds.
3. Results
3.1 Donor Characteristics
Genotyping was performed on a total of 118 donors, age 60 years and older, that were graded for the stage of AMD using the Minnesota Grading System (MGS) (Decanini et al., 2007; Olsen and Feng, 2004). Donors in MGS1 (n=42) represent the age-matched control group with no clinically obvious eye disease. Donors in MGS2 (n=33), MGS3 (n=28), and MGS4 (n=15) represent progressively more severe stages of the disease. A summary of donor demographics and clinical information for each MGS group used in this study is provided in Table 1. Average age for MGS1 (75 ± 9.1; mean ± SD) was significantly younger compared with MGS2 (80 ± 8.6) and MGS 3 (80 ± 8.6) (p<0.05) but not MGS 4 (81± 9.7). We previously showed that there was no statistically significant difference between the extent of mtDNA damage comparing younger (ages 60–72) and older (ages 75–91) donors without disease (Terluk et al., 2015), hence the slight difference in age between MGS1 and donors with AMD used in this study should not generate any bias in our results.
In the current study, we evaluated the relationship between mtDNA damage and donors with specific genotypes utilizing samples from two previous studies (Figure 1A) where we had measured mtDNA damage in the macular RPE cells (Karunadharma et al., 2010; Terluk et al., 2015). Results for mtDNA damage of our combined data sets (Figure 1B), including 76 donors with AMD (MGS2-4) relative to 42 non-diseased controls (MGS1), demonstrated a significant, progressive increase in mtDNA damage with AMD disease severity (p<0.001).
3.2 Genotype Association with AMD Risk Alleles
Recent genome-wide association studies (GWAS) have identified 34 loci associated with AMD (Fritsche et al., 2015). These multiple genetic loci suggest that several putative biological pathways are involved in AMD pathogenesis (Fritsche et al., 2014; Ratnapriya and Chew, 2013). In the current study, donors were genotyped for 10 common risk variants from pathways likely associated with the stress response or cell survival (ARMS2, TNFRSF10A), complement activation (CFH, C2, C3), lipid transport and metabolism (APOE, CETP, LIPC), angiogenesis (VEGFA), and remodeling of extracellular matrix (COL10A1) (Table 2). Risk allele frequencies of our donor samples were generally comparable to values reported in other studies that analyzed a much larger database (Chen et al., 2010; Fritsche et al., 2013; Holliday et al., 2013; Seddon et al., 2009; Yu et al., 2011). Therefore, our donor samples are representative of the general population.
Table 2.
AMD-associated genes and the risk allele frequency for donors in this study.
| SNP 1 | Gene | Alleles 2 (NRA / RA) | RA Frequency 3 (Con / AMD) | Function |
|---|---|---|---|---|
| rs10490924 | ARMS2 | G / T | 0.19 / 0.22 | Cell survival |
| rs13278062 | TNFRSF10A | G / T | 0.49 / 0.51 | Cell survival |
| rs1061170 | CFH | T / C | 0.34 / 0.42 | Complement pathway |
| rs9332739 | C2 | G / C | 0.09 / 0.05 | Complement pathway |
| rs2230199 | C3 | G / C | 0.24 / 0.25 | Complement pathway |
| rs7412 | APOE | C / T | 0.11 / 0.04 | Lipid transport/metabolism |
| rs3764261 | CETP | C / A | 0.32 / 0.24 | Lipid transport/metabolism |
| rs920915 | LIPC | G / C | 0.73 / 0.79 | Lipid transport/metabolism |
| rs4711751 | VEGFA | C / T | 0.57 / 0.49 | Angiogenesis |
| rs3812111 | COL10A1 | A / T | 0.69 / 0.65 | Remodeling extracellular matrix |
SNP reference numbers and pathway/function for each gene were as provided (Ratnapriya and Chen, 2013; Fritsche et al., 2014).
NRA, non-risk allele; RA, risk allele
Risk allele frequency for non-disease control and donors with AMD in the current study.
Abbreviations
AMD, donors with AMD; Con, non-diseased controls; NRA, non-risk allele; RA, risk allele
The distribution of control (MGS1) and AMD (MGS2-4) donors genotyped for homozygous non-risk, heterozygous risk, or homozygous risk alleles within each gene are provided in Figure 2. Of note, the absence or low number of donors in genotype categories for ARMS2, C2, C3, APOE and LIPC limits our ability to derive conclusive results for these AMD-associated genes. Therefore, these risk factors were eliminated from further consideration.
Figure 2. Distribution of control and AMD donors for each genotype.

Donors were genotyped for 10 risk variants associated with (A) cell survival (ARMS2, TNFRSF10A), the complement pathway (CFH, C2, C3), (B) lipid transport/metabolism (APOE, CETP, LIPC), angiogenesis (VEGFA) and remodeling of the extracellular matrix (COL10A1). Shaded area of each bar shows the percent of control (MGS1, light grey) and AMD (MGS2, hatched, MGS3, medium grey, MGS4, dark grey) donors that are homozygous non-risk, heterozygous risk, or homozygous risk (underlined) for each gene. The total number of donors in each risk category is provided at the base of each bar.
3.3 Genotype and mtDNA Damage
The extent of mtDNA damage was compared for donors genotyped for homozygous non-risk, heterozygous risk, or homozygous risk for TNFRSF10A, CFH, CETP, VEGFA, and COL10A1 (Figure 3). The analysis shows that only donors harboring the homozygous risk alleles for CFH (CC > TT; p=0.035) had significantly higher levels of mtDNA damage. This single nucleotide polymorphism (SNPs, rs1061170) causes an amino acid substitution of histidine for tyrosine at position 402 (Y402H) in the CFH protein.
The analysis presented in Figure 3 includes both control and AMD donors for each genotype. To determine the relative contribution of these two donor populations for the CFH genotypes, mtDNA damage was plotted separately for both control and AMD donors (Figure 4A). Consistent with our previous findings, mtDNA damage was lower in age-matched controls versus donors with AMD for each genotype. Linear regression analysis comparing the risk allele frequency (TT=0, TC=1, CC=2) with the extent of mtDNA damage showed a significant correlation for donors with AMD (p=0.049) but not for controls (p=0.59). These results are consistent with a co-dominant multiplicative genetic model for CFH developed from epidemiological studies, whereby each C allele increases the odds of AMD by approximately 2.5-fold (Thakkinstian et al., 2006). To test if the presence of the C allele increased the extent of mtDNA damage, we compared homozygous non-risk donors versus donors carrying the high risk allele. Our data show donors with one or two C alleles had significantly higher mtDNA damage (p=0.043).
We have shown that mtDNA damage progressively increases with AMD severity (Figure 1B). For the CFH genotypes, there is a higher prevalence of donors with intermediate and late AMD (MGS3-4) in the CC group compared with both TT and TC groups (Figure 4A, left panel). To address the possible bias caused by the unbalanced distribution of AMD severity for each genotype, we compared only MGS3 and 4 donors in each genotype. For this analysis, we added 39 new donors (12 MGS1, 13 MGS3, and 14 MGS4) to the controls and AMD (MGS3 and 4 only) from the previous data set so that we would have sufficient statistical power for the comparison. As shown in Figure 4B (left panel), the combined groups now have an even distribution of donors in intermediate and late stage AMD. Table 3 provides the demographics of the new group of donors, which were all age 60 and older. Plots of the combined data set for age-matched controls and for AMD donors are shown in Figure 4B (middle panel). Controls showed no linear relationship with C allele frequency (p=0.09). For AMD donors, there was a 20% increase in mtDNA damage for donors carrying the high risk allele. However, these data did not follow a linear relationship (p=0.11). Testing the effect of the C allele (Figure 4B, right panel) showed donors harboring one or two copies of the high risk allele had significantly higher mtDNA damage compared with low risk donors (p=0.022). Taken together, data from a total of 103 donors (Figures 4A and 4B) with AMD support the hypothesis that the presence of the CFH risk allele makes mtDNA in the RPE macula more susceptible to damage.
3.4 Mitochondrial Haplogroup and mtDNA Damage
The mtDNA haplogroups have been shown to be either protective (H halogroup) or high risk for AMD (JTU haplogroups) (Jones et al., 2007; Kenney et al., 2013a; Mueller et al., 2012; Udar et al., 2009). The mtDNA haplogroups for each retinal DNA sample were characterized by allelic discrimination and/or PCR along with restriction enzyme digestion. In our population of donors, 34% were J, T, or U haplotype, which reflects the large percentage of Minnesotans of Northern European ancestry from which these haplogroups originated (www.census.gov/prod/2004pubs/c2kbr-35.pdf). The protective H haplogroup with ancestral ties to Southern Europe were present in 32% of our population. When comparing the extent of mtDNA damage for the H protective group with the J, T, and U susceptible haplogroups, we found no significant difference (Fig. 5A). To determine if there was an additive effect of the CFH risk allele (C) with the background mtDNA haplogroup H or the JTU cluster, we compared the extent of mtDNA damage in these subgroups. There were no significant differences in the numbers of mtDNA lesions between CFH + H haplogroup or CFH + JTU cluster groups (Fig. 5B). These findings indicate that the degree of mtDNA lesions was not related to the mtDNA haplogroup profile of each individual.
4. Discussion
AMD is a challenging disease to study due to the unique characteristics of the human eye, including the presence of a macula found only in primates, as well as the lengthy timeframe of >60 years to develop. Importantly, no animal model can faithfully recapitulate all of these features, further limiting our capacity to study disease mechanisms. The use of human donor eyes graded for the stage of AMD captures key features of the disease that not only help define disease mechanisms, but also can identify potential therapeutic targets. Previous results from our proteomic analysis and measurements of mtDNA damage in human donor eyes have led to our focus on the RPE mt as an early site of defect and a potential target for intervention (Karunadharma et al., 2010; Nordgaard et al., 2006, 2008; Terluk et al., 2015).
As a follow-up to these earlier studies, the goal of current research was to establish the rationale for treating AMD patients of a specific genotype with therapies aimed at protecting or improving mt function. Our results show that donors harboring the high risk allele for CFH had significantly more mtDNA damage, suggesting the mt as a novel site of intervention for this patient subpopulation. The absence of higher mtDNA damage in age-matched control donors carrying the risk allele (Figure 4) suggests mt injury is not a direct consequence of the CFH risk variant. Rather, retinal changes associated with the onset of disease coupled with the presence of the risk allele create cellular conditions conducive for accelerated mt damage to occur. While mt damage may not be the singular event initiating AMD, mitochondria’s key role in multiple cellular processes suggests its damage may augment AMD progression. Therefore, interventions aimed at protecting mt function and slowing disease progression would still be highly beneficial.
In considering the limitations of the study design (i.e., low sample number, incomplete genotyping for all known AMD risk genes) our results are likely a conservative estimate of the genetically defined patient populations with higher mt damage. None the less, the potential impact of finding an effective treatment for slowing down AMD progression in patients carrying the CFH risk allele is immense when considering both risk conferred (Odds Ratio ~3.0) and the high percent of patients (30–50% of all AMD patients) harboring this variant (Fritsche et al., 2014; Schaumberg et al., 2007).
CFH is a key regulator of the alternative complement pathway, which is part of the innate immune system that promotes clearance of debris and dead cells and also kills invading pathogens. The role of CFH is to protect host cells from inappropriate complement activation by downregulating the complement cascade, thereby reducing the potential for “by-stander damage” to healthy cells and chronic inflammation. CFH performs this function by binding to a variety of ligands, such as the acute phase C-reactive protein, malondialdehyde (MDA), apoptotic/necrotic cells, and heparin sulfate present in the extracellular matrix (Clark et al., 2010). Biochemical analysis of the Y402H mutant arising from the CFH SNP (rs1061170) has demonstrated reduced binding to these ligands (Laine et al., 2007; Ormsby et al., 2008; Sjoberg et al., 2007; Weissmann et al., 2011; Clark et al., 2006). Reduced CFH binding can lead to the accumulation of toxic debris and drusen, chronic inflammation within the retina, and consequent tissue damage due to aberrant complement activation. For example, CFH is recruited to the surface of dead cells or debris via their MDA adducts whose covalent attachment to proteins creates a danger signal that is recognized by the innate immune system (Weissmann et al., 2011). MDA adducts also stimulate the production and secretion of the pro-inflammatory molecule IL-8 by the RPE. CFH binding to MDA neutralizes the inflammatory properties of MDA and halts complement activation. Importantly, the CFH variant Y402H has reduced capacity to bind MDA-modified proteins, thus allowing for an exaggerated inflammatory response that has been associated with AMD (Weissmann et al., 2011).
The combination of age-related related changes in retinal heparin sulfate content along with the impaired binding of the CFH Y402H variant provides another plausible mechanism behind the increased development of AMD in patients carrying the C genotype (Clark et al., 2010a). Heparin sulfate is a polysaccharide decorating the extracellular matrix of all cells (Clark et al., 2006). Sulfation is usually abundant around Bruch’s membrane but is reduced by 50% between the fourth to ninth decades of life (Keenam et al., 2014). Coupled with this age-related reduction in Bruch’s membrane sulfation, the impaired binding of the Y402H variant to heparin sulfate (Clark et al., 2006) and consequent reduced content of CFH in Bruch’s membrane of Y402H donors (Clark et al., 2010b), could result in increased complement activation and chronic, focal inflammation in individuals harboring the CFH risk allele.
The retinal inflammation and tissue damage associated with the reduced function of the CFH variant could be responsible for initiating the mtDNA damage observed in AMD donors harboring the high risk allele. The potential mechanism may involve the well-established link between chronic inflammation and oxidative stress, which is generated by immune cells that accumulate around sites of injury. Immune cells make free radicals to kill pathogens and secrete cytokines, which upregulate cellular pathways (e.g., NFkB) that increase intracellular oxidative stress in surrounding tissue. Age-associated changes and environmental insults may also contribute to disease onset in genetically susceptible individuals. For example, the lipofuscin that accumulates in the RPE with aging has been shown to inhibit mt function and activate the complement cascade (Vives-Bauza et al., 2008; Zhou et al., 2009). Smoking, one of the strongest modifiable risk factors for AMD, causes mt damage and also activates complement (Mansoor et al., 2014; Wang et al., 2009; Kunchithapautham et al., 2014). The synergistic effect of age-related changes coupled with environmental insults, such as smoking or high fat diet, lowers the threshold for disease and allows the genetic defect to initiate disease. This concept has been proposed as the multiple-hit “threshold” hypothesis (Fritsche et al., 2014) and provides a plausible explanation for why an individual’s genetic profile alone does not consistently predict susceptibility to disease.
In addition to investigating how mtDNA is impacted by the expression of nuclear genes, we also examined the influence of donor mtDNA haplogroups. The rationale for these investigations are based on ethnicity-dependent differences in the genetic risk profile associated with AMD, as well as how the disease manifests in specific ethnic groups. The evidence suggests an individual’s ancestral origin, as defined by their mtDNA haplogroup, may play a role in disease susceptibility. For example, Caucasian AMD patients mainly exhibit geographic atrophy, whereas Asian patients most often present with choroidal neovascularization or polyploidy choroidal vasculopathy with little evidence of drusen (Laude et al., 2010; Kawasaki et al., 2010). Additionally, the effect size for specific genes is stratified by ancestry (Fritsche et al., 2013). Recent studies have demonstrated that specific mtDNA haplogroups confer differences in susceptibility for AMD; while the H haplogroup is protective, the J, T and U haplogroups have increased risk for AMD (Jones et al., 2007; Kenney et al., 2013a; Mueller et al., 2012; Udar et al., 2009).
In the present study, mtDNA haplogroup was determined for each donor and then compared to the level of mtDNA damage (Figure 5). No correlation was found between the extent of mtDNA damage and the individual’s haplogroups (H or JTU). There was no additive risk for mtDNA lesions when the CFH alleles and mtDNA haplogroups were analyzed together. These findings suggest that the association of mtDNA haplogroup with AMD is through mechanisms other than mtDNA damage. Studies using transmitochondrial cybrids (cytoplasmic hybrids containing identical nuclei but different mitochondria) suggest the haplogroup can influence retrograde signaling between the mt and nuclear genome. For example, cybrids containing either mtDNA haplogroups H (protective for AMD) or J (high risk for AMD) show significant alterations in bioenergetic profile and gene expression patterns of the alternative complement, inflammation, and apoptosis pathways (Kenney et al., 2013b; Kenney et al., 2014). H and J cybrids also had different levels of total global methylation, expression patterns of acetylation and methylation genes, and transcription levels for inflammation, angiogenesis and signaling pathways (Atilano et al., 2015).
The innate immune system has recently been targeted as a potential treatment for atrophic AMD (http://www.amd.org/what-is-macular-degeneration/dry-amd/clinical-trials-for-dry-amd/). However, early results have been mostly disappointing (Black and Clark, 2015; Clark and Bishop, 2015). Based on our studies, we suggest that AMD patients with the CFH high-risk genetic profile would reap the greatest benefit from “mito-therapies” aimed at protecting RPE mitochondria.
Highlights.
MtDNA damage was measured in macular RPE of donors genotyped for AMD risk variants
Increased mtDNA damage is present in donors harboring the CFH high risk allele
mtDNA haplogroup did not correlate with the extent of mtDNA damage
AMD patients with high risk genotype may benefit from therapies that protect RPE mitochondria
Acknowledgments
Funding: This work was supported by the Elaine and Robert Larson Endowed Vision Research Chair (to DAF); Beckman Initiative for Macular Research (#1004, #1303 to DAF); Foundation Fighting Blindness (TA-NMT-0613-0620-UMN to DAF); An Anonymous Donor for Macular Degeneration Research and an unrestricted grant from Research to Prevent Blindness to the Department of Ophthalmology and Visual Neurosciences; National Institutes of Health (T32-AG29796 to MRT); Discovery Eye Foundation (to MCK); Polly and Michael Smith Foundation (to MCK) and the National Eye Institute Intramural Research Program (to AS).
All authors have actively contributed to the development of this work through their participation in research, data analysis and/or preparation of the manuscript. The authors wish to acknowledge the contribution of the Minnesota Lions Eye Bank personnel for their assistance in procuring eyes, Kathy Goode and Sung Lee for photographing and processing eye tissue, and the contribution by Dr. Timothy Olsen, MD, in grading donor eyes prior to 2011.
Abbreviations
- AMD
Age-related macular degeneration
- CFH
complement factor H
- MDA
malondialdehyde
- MGS
Minnesota Grading System
- mt
mitochondria
- PCR
polymerase chain reaction
- RPE
retinal pigment epithelium
- SNP
single nucleotide polymorphism
- VEGF
Vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atilano SR, Malik D, Chwa M, Cáceres-Del-Carpio J, Nesburn AB, Boyer DS, Kuppermann BD, Jazwinski SM, Miceli MV, Wallace DC, et al. Mitochondrial DNA variants can mediate methylation status of inflammation, angiogenesis and signaling genes. Hum Mol Genet. 2015 doi: 10.1093/hmg/ddv173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JRM, Clark SJ. Age-related macular degeneration: genome-wide association studies to translation. Genetics in Medicine. 2015 doi: 10.1038/gim.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Stambolian D, Edwards AO, Branham KE, Othman M, Jakobsdottir J, Tosakulwong N, Pericak-Vance MA, Campochiaro PA, Klein ML, et al. Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. PNAS. 2010;107:7401–7406. doi: 10.1073/pnas.0912702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew EY, Clemons TE, Agron E, Sperduto RD, Sangiovanni JP, Kurinij N, Davis MD Age-Related Eye Disease Study Research Group. Long-term effects of vitamins C and E, B-carotene, and zinc on age-related macular degeneration: AREDS report no. 35. Ophthalmology. 2013;120:1604–11. doi: 10.1016/j.ophtha.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew EY, Klein ML, Clemons TE, Agrón E, Ratnapryiya R, Edwards AO, Fritsche LG, Swaroop A, Abecasis GR Age-Related Eye Disease Study Research Group. No clinically significant association between CFH and ARMS2 genotypes and response to nutritional supplements: AREDS report number 38. Ophthalmology. 2014;121:2173–2180. doi: 10.1016/j.ophtha.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SJ, Bishop PN. Roles of Factor H and related proteins in regulating complement activation in the macula, and relevance to age-related macular degeneration. J Clin Med. 2015;4:18–31. doi: 10.3390/jcm4010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SJ, Bishop PN, Day AJ. Complement factor H and age-related macular degeneration: the role of glycosaminoglycan recognition in disease pathology. Biochem Soc Trans. 2010a;38:1342–1348. doi: 10.1042/BST0381342. [DOI] [PubMed] [Google Scholar]
- Clark SJ, Higman VA, Mulloy B, Perkins SJ, Lea SM, Sim RB, Day AJ. His-384 allotypic variant of factor H associated with age-related macular degeneration has different heparin binding properties from the non-disease-associated form. J Biol Chem. 2006;281:24713–24720. doi: 10.1074/jbc.M605083200. [DOI] [PubMed] [Google Scholar]
- Clark SJ, Perveen R, Hakobyan S, Morgan BP, Sim RB, Bishop PN, Day AJ. Impaired binding of the age-related macular degeneration-associated complement factor H 402H allotype to Bruch’s membrane in human retina. J Biol Chem. 2010b;285:30192–30202. doi: 10.1074/jbc.M110.103986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decanini A, Nordgaard CL, Feng X, Ferrington DA, Olsen TW. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. Am J Ophthalmol. 2007;143:607–615. doi: 10.1016/j.ajo.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feher J, Kovacs I, Artico M, Cavallotti C, Papale A, Gabrieli CB. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol Aging. 2006;27:983–993. doi: 10.1016/j.neurobiolaging.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Friedman DS, O’Colmain BJ, Munoz B, Tomany SC, McCarty C, De Jong PT, Nemesure B, Mitchell P, Kempen J. Prevalence of age- related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–572. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, Zack DJ, Arakawa S, Cipriani V, Ripke S, et al. Seven new loci associated with age-related macular degeneration. Nature Genetics. 2013;45:433–441. doi: 10.1038/ng.2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche LG, Fariss RN, Stambolian D, Abecasis GR, Curcio CA, Swaroop A. Age-related macular degeneration: Genetics and biology coming together. Annu Rev Genomics Hum Genet. 2014;15:151–171. doi: 10.1146/annurev-genom-090413-025610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche LG, Igl W, Bailey JN, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet. 2015 doi: 10.1038/ng.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday EG, Smith AV, Cornes BK, Buitendijk GHS, Jensen RA, Sim X, Aspelund T, Aung T, Baird PN, Boerwinkle E, et al. Insights into the genetic architecture of early stage age-related macular degeneration: a genome-wide association study met-analysis. PLOS One. 2013;8:e53830. doi: 10.1371/journal.pone.0053830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MM, Manwaring N, Wang JJ, Rochtchina E, Mitchell P, Sue CM. Mitochondrial DNA haplogroups and age-related maculopathy. Arch Ophthalmol. 2007;125:1235–1240. doi: 10.1001/archopht.125.9.1235. [DOI] [PubMed] [Google Scholar]
- Karunadharma PP, Nordgaard CL, Olsen TW, Ferrington DA. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010;51:5470–5479. doi: 10.1167/iovs.10-5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki R, Yasuda M, Song SJ, Chen SJ, Jonas JB, Wang JJ, Mitchell P, Wong TY. The prevalence of age-related macular degeneration in Asians: a systematic review and meta-analysis. Ophthalmology. 2010;117:921–927. doi: 10.1016/j.ophtha.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Keenan TDL, Pickford CE, Holley RJ, Clark SJ, Lin W, Dowsey AW, Merry CL, Day AJ, Bishop PN. Age-dependent changes in heparan sulfate in human Bruch’s membrane: Implications for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2014;55:5370–5379. doi: 10.1167/iovs.14-14126. [DOI] [PubMed] [Google Scholar]
- Kennedy SR, Salk JJ, Schmitt MW, Loeb LA. Ultra-sensitive sequencing reveals an ag-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLOS Genet. 2013;9:e1003794. doi: 10.1371/journal.pgen.1003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney MC, Atilano SR, Boyer D, Chwa M, Chak G, Chinichian S, Coskun P, Wallace DC, Nesburn AB, Udar NS. Characterization of retinal and blood mitochondrial DNA from age-related macular degeneration patients. Invest Ophthalmol Vis Sci. 2010;51:4289–97. doi: 10.1167/iovs.09-4778. [DOI] [PubMed] [Google Scholar]
- Kenney MC, Chwa M, Atilano SR, Falatoonzadeh P, Ramirez C, Malik D, Tarek M, Cáceres-del-Carpio J, Nesburn AB, Boyer DS, et al. Inherited mitochondrial DNA variants can affect complement, inflammation and apoptosis pathways: insights into mitochondrial-nuclear interactions. Hum Mol Genet. 2014 doi: 10.1093/hmg/ddu065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney MC, Chwa M, Atilano SR, Pavlis JM, Falatoonzadeh P, Ramirez C, Malik D, Hsu T, Woo G, Soe K, et al. Mitochondrial DNA variants mediate energy production and expression levels for CFH, C3 and EFEMP1 genes: implications for age-related macular degeneration. PLoS ONE. 2013b;8:e54339. doi: 10.1371/journal.pone.0054339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney MC, Hertzog D, Chak G, Atilano SR, Khatibi N, Soe K, Nobe A, Yang E, Chwa M, Zhu F, et al. Mitochondrial DNA haplogroups confer differences in risk for age-related macular degeneration: a case control study. BMC Med Genet. 2013a;14:4. doi: 10.1186/1471-2350-14-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein ML, Francis PJ, Rosner B, Reynolds R, Hamon SC, Schultz DW, Ott J, Seddon JM. CFH and LOC387715/ARMS2 genotypes and treatment with antioxidants and zinc for age-related macular degeneration. Ophthalmology. 2008;115:1019–1025. doi: 10.1016/j.ophtha.2008.01.036. [DOI] [PubMed] [Google Scholar]
- Kunchithapautham K, Atkinson C, Rohrer B. Smoke exposure causes endoplasmic reticulum stress and lipid accumulation in retinal pigment epithelium through oxidative stress and complement activation. J Biol Chem. 2014;289:14534–46. doi: 10.1074/jbc.M114.564674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine M, Jarva H, Seitsonen S, Haapasalo K, Lehtinen MJ, Lindeman N, Anderson DH, Johnson PT, Järvelä I, Jokiranta TS, et al. Y402H polymorphism of complement factor H affects binding affinity to C-reactive protein. J Immunol. 2007;178:3831–3836. doi: 10.4049/jimmunol.178.6.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laude A, Cackett PD, Vithana EN, Yeo IY, Wong D, Koh AH, Wong TY, Aung T. Polypoidal choroidal vasculopathy and neovascular age-related macular degeneration: same or different disease? Prog Retin Eye Res. 2010;29:19–29. doi: 10.1016/j.preteyeres.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Mansoor S, Gupta N, Falatoonzadeh P, Kuppermann BD, Kenney MC. 2-thylpyridine, a cigarette smoke component, causes mitochondrial damage in human retinal pigment epithelial cells in vitro. Indian J Ophthalmol. 2014;62:16–22. doi: 10.4103/0301-4738.126168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montezuma SR, Sobrin L, Seddon JM. Review of genetics in age related macular degeneration. Semin Ophthalmol. 2007;22:229–240. doi: 10.1080/08820530701745140. [DOI] [PubMed] [Google Scholar]
- Mueller EE, Schaier E, Brunner SM, Eder W, Mayr JA, Egger SF, Nischler C, Oberkofler H, Reitsamer HA, Patsch W, et al. Mitochondrial haplogroups and control region polymorphisms in age-related macular degeneration: a case-control study. PLoS One. 2012;7:e30874. doi: 10.1371/journal.pone.0030874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordgaard CL, Berg KM, Kapphahn RJ, Reilly C, Feng X, Olsen TW, Ferrington DA. Proteomics of the retinal pigment epithelium reveals altered protein expression at progressive stages of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2006;47:815–822. doi: 10.1167/iovs.05-0976. [DOI] [PubMed] [Google Scholar]
- Nordgaard CL, Karunadharma PP, Feng X, Olsen TW, Ferrington DA. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:2848–2855. doi: 10.1167/iovs.07-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen TW, Feng X. The Minnesota Grading System of eye bank eyes for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2004;45:4484–4490. doi: 10.1167/iovs.04-0342. [DOI] [PubMed] [Google Scholar]
- Ormsby RJ, Ranganathan S, Tong JC, Griggs KM, Dimasi DP, Hewitt AW, Burdon KP, Craig JE, Hoh J, Gordon DL. Functional and structural implications of the complement factor H Y402H polymorphism associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:1763–1770. doi: 10.1167/iovs.07-1297. [DOI] [PubMed] [Google Scholar]
- Ratnapriya R, Chew EY. Age-related macular degeneration- Clinical review and genetics update. Clin Genet. 2013;84:160–166. doi: 10.1111/cge.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rein DB, Wittenborn BS, Zhang X, Honeycutt AA, Lesesne SB, Saaddine J. Forecasting age-related macular degeneration through the year 2050. Arch Ophthalmol. 2009;127:533–540. doi: 10.1001/archophthalmol.2009.58. [DOI] [PubMed] [Google Scholar]
- Schaumberg DA, Hankinson SE, Guo Q, Rimm E, Hunter DJ. A prospective study of 2 major age-related macular degeneration susceptibility alleles and interactions with modifiable risk factors. Arch Ophthalmol. 2007;125:55–62. doi: 10.1001/archopht.125.1.55. [DOI] [PubMed] [Google Scholar]
- Seddon JM, Reynolds R, Maller J, Fagerness JA, Daly MJ, Rosner B. Prediction model for prevalence and incidence of advanced age-related macular degeneration based on genetic, demographic, and environmental variables. Invest Ophthalmol Vis Sci. 2009;50:2044–2053. doi: 10.1167/iovs.08-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöberg AP, Trouw LA, Clark SJ, Sjölander J, Heinegård D, Sim RB, Day AJ, Blom AM. The factor H variant associated with age-related macular degeneration (His-384) and the non-disease-associated form bind differentially to C-reactive protein, fibromodulin, DNA, and necrotic cells. J Biol Chem. 2007;282:10894–10900. doi: 10.1074/jbc.M610256200. [DOI] [PubMed] [Google Scholar]
- Terluk MR, Kapphahn RJ, Soukup LM, Gong H, Gallardo C, Montezuma SR, Ferrington DA. Investigating mitochondria as a target for treating age-related macular degeneration. J Neurosci. 2015;35:7304–7311. doi: 10.1523/JNEUROSCI.0190-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakkinstian A, Han P, McEvoy M, Smith W, Hoh J, Magnusson K, Zhang K, Attia J. Systemic review and meta-analysis of the association between complementary factor H Y402H polymorphisms and age-related macular degeneration. Hum Molec Genet. 2006;15:2784–2790. doi: 10.1093/hmg/ddl220. [DOI] [PubMed] [Google Scholar]
- Udar N, Atilano SR, Memarzadeh M, Boyer DS, Chwa M, Lu S, Maguen B, Langberg J, Coskun P, Wallace DC, et al. Mitochondrial DNA haplogroups associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50:2966–2974. doi: 10.1167/iovs.08-2646. [DOI] [PubMed] [Google Scholar]
- Vives-Bauza C, Anand M, Shiraz AK, Magrane J, Gao J, Vollmer-Snarr HR, Manfredi G, Finnemann SC. The age lipid A2E and mitochondrial dysfunction synergistically impair phagocytosis by retinal pigment epithelial cells. J Biol Chem. 2008;283:24770–80. doi: 10.1074/jbc.M800706200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AL, Lukas TJ, Yuan M, Du N, Handa JT, Neufeld AH. Changes in retinal pigment epithelium related to cigarette smoke: Possible relevance to smoking as a risk factor for age-related macular degeneration. PLoS One. 2009;4:e5304. doi: 10.1371/journal.pone.0005304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HPN, Issa PC, Cano M, Brändstatter H, Tsimikas S, Skerka C, et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011;478:76–81. doi: 10.1038/nature10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Bhangale TR, Fagerness J, Ripke S, Thorleiffson G, Tan PL, Souied EH, Richardson AJ, Merriam JE, Buitendijk GHS, et al. Common variants near FRK/COL10A1 and VEGFA are associated with advanced age-related macular degeneration. Hum Molec Genet. 2011;20:3699–3709. doi: 10.1093/hmg/ddr270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Kim SR, Westlund BS, Sparrow JR. Complement activation by bisretinoid constituents of RPE lipofuscin. Invest Ophthalmol Vis Sci. 2009;50:1392–99. doi: 10.1167/iovs.08-2868. [DOI] [PMC free article] [PubMed] [Google Scholar]